

Abstract
A series of TRPV1 agonists with amide, reverse amide, and thiourea groups in the B-region and their corresponding α-methylated analogues were investigated. Whereas the α–methylation of the amide B-region enhanced the binding affinities and potencies as agonists, that of the reverse amide and thiourea led to a reduction in receptor affinity. The analysis indicated that proper hydrogen bonding as well as steric effects in the B-region are critical for receptor binding.
Keywords: Vanilloid Receptor 1, TRPV1, agonist, capsaicin
Introduction
The transient receptor potential V 1 (TRPV1) is a molecular integrator of nociceptive stimuli expressed predominantly on unmyelinated pain-sensing nerve fibers (C-fibers) and small A δ fibers in the dorsal root, trigeminal, and nodose ganglia.1–3 TRPV1 is structurally a homotetramer and functions as a non-selective cation channel with high Ca2+ permeability. The receptor is activated not only by protons,4 heat,5 and endogenous substances such as anandamide6 and lipoxygenase products7 but also by natural vanilloids such as capsaicin (CAP)8 and resiniferatoxin (RTX)9 or indirectly by bradykinin.10 The activation of TRPV1 by these agents leads to an increase in intracellular Ca2+ resulting in excitation of the primary sensory neurons. The functional blockade of this receptor, by antagonism or by desensitization subsequent to stimulation by agonists, promises considerable therapeutic utility targeting inflammatory and neuropathic pain, cystitis, and bladder hyperreflexia.11–15
Naturally found TRPV1 ligands, resiniferatoxin (RTX) and capsaicin (Figure 1), have a vanilloid moiety (A region) that is connected through either an ester linkage or amide bond (B region) to a diterpene moiety or a long alkyl chain (C region), respectively.16 Recent SAR studies indicated that the modification of the A region, especially the modification of the 4-hydroxyl group, led to a complete loss of agonistic activity,18 and replacing the B region linkage with a reverse amide, hydroxamate or thiourea altered the potency.19
Figure 1.

Previously we have reported that the A region derivatives of TRPV1 antagonists with an α–methyl group demonstrated enhanced binding affinity and potent antagonism for TRPV1.17 As a continuing effort to investigate the SAR of TRPV1 agonists based on the A-, B- and C-regions, we have synthesized, based on our lead agonists (1 and 2), a series of TRPV1 agonists bearing amide, reverse amide and thiourea B-regions together with their corresponding α–methylated analogues in the B-region, and we have analyzed the effect of α–methylation on receptor affinity and agonist potency. We describe here the syntheses and functional TRPV1 activities of the designed ligands and the SAR analysis of the B-region.
Chemistry
The syntheses of the 3-substituted N-(4-t-butylbenzyl)-(4-hydroxyphenyl)acetamide/propionamide analogues were started from commercially available 3-substituted-4-hydroxyphenylacetic acids 3a–c (Scheme 1). The acetamides 4b–c were prepared from 3b–c, respectively, by the coupling with 4-t-butylbenzylamine,. Dibenzylation of 3 followed by alkylation using LHMDS and methyl iodide afforded 5. Compound 5a was debenzylated and then coupled with the amine to provide the 3-methoxy propanamide 8a. On the other hand, compounds 5b–c were hydrolyzed to the acids 7 under basic conditions; the acids were then coupled with the amine and subsequently deprotected to provide the 3-halo propionamides 8b–c, respectivly.
Scheme 1.

Reagents and conditions: (a) BnBr, K2CO3, acetone, reflux, 15 h, 98–99%; (b) LHMDS, CH3I, THF, −78°C, 30 min, 68–70%; (c) LiOH, THF-H2O, r.t., 15 h, 90–92%; (d) 4-t-butylbenzylamine, EDC, DMAP or HOBt, CH2Cl2, r.t., 15 h, 52–80%; (e) Pd/C, H2, CH3OH, r.t., 1 h, 70% for X=OCH3 and 55% for X=F: 1M BCl3 in CH2Cl2, −78°C, 62% for X=Cl
The 4-aminoethoxy acetamides 10a–c were prepared from 3-substituted 4-hydroxyphenyl acetamides (1, 4b–c) in 3 steps (Scheme 2). The 2-azidoethoxy group was introduced by the alkylation of the 4-hydroxyl group with 1-bromo-2-chloroethane and the subsequent displacement reaction with sodium azide to give 9. The azide group of 9 was reduced by either Staudinger reduction or hydrogenation to yield the final amines 10a–c. The 4-aminoethoxy propanamides 14a–c were prepared from 4-hydroxy intermediates 11 in the same manner as shown in scheme 2 (Scheme 3). The C-region analogues of acetamide/propionamide were prepared from the acid intermediates (3a, 6 and 13a) by the coupling with the corresponding amines to yield the final compounds 18–25 (Scheme 4).
Scheme 2.

Reagents and conditions: (a) 1,2-dibromoethane, 40% KOH, Bu4NOH, 93% for X=OCH3: 1-bromo-2-chloroethane, K2CO3, acetone, reflux, 49–60% for X=F, Cl; (b) NaN3, TBAB, KI, toluene, reflux, 71–80%; (c) PPh3, H2O, THF, 14% for Cl and 58% for X=OCH3: Pd/C, H2, CH3OH, r.t., 6 h, 38% for X=F
Scheme 3.

Reagents and conditions: (a) cat. H2SO4, EtOH, reflux, 98–99%; (b) 1-bromo-2-chloroethane, K2CO3, acetone, reflux, 55–65%; (c) NaN3, TBAB, KI, toluene, reflux, 6 h, 90–95%;(d) NaH, CH3I, DMF, 0 °C, 1 h, 65–75 %; (e) NaOH, H2O, reflux, 2 h, 98–99%; (f) 4-t-butylbenzylamine, EDC, DMAP, CH2Cl2, r.t., 15 h, 70–74%; (g) Pd/C, H2, CH3OH, r.t., 80–85%
Scheme 4.

Reagents and conditions: (a) EDC, DMAP, DMF, r.t., 15h, 70–80%; (b) Pd/C, H2, CH3OH, r.t., 80%
The reverse amide analogues were synthesized from the benzylamine 23 or α–methyl benzylamine 28, which was prepared from 4-hydroxy-3-methoxy acetophenone by reductive amination (Scheme 5). The coupling reactions of above benzylamines with commercially available fatty acids provided the C-region analogues with reverse amide 24–36 and α–methyl reverse amide 29–31. α–Methyl thiourea analogue 32 was also prepared from 28 by the coupling with 4-t-butylbenzyl isothiocyanate.
Scheme 5.

Reagents and conditions: (a) RNH2, EDC, DMAP, Et3N, DMF, r.t., 15 h, 65–70%; (b) NH2OH HCl, Pyridine, CH2Cl2, reflux, 95%; (c) Pd/C, cHCl, MeOH, r.t., 15 h, 90%; (d) 4-t-butylbenzyl isothiocyanate, NEt3, CH2Cl2, r.t., 60%
The 4-aminoethoxy thiourea 34 and its α–methyl analogue 36 were readily synthesized from the corresponding benzylamines 23 and 28 by following the routes described in schemes 3 and 5, respectively (Scheme 6).
Scheme 6.

Reagents and conditions: (a) (Boc)2O, NEt3, H2O-Dioxane, r.t., 2 h, 99%; (b) 1,2-dibromoethane, 40% KOH, Bu4NOH, 50 °C, 2 h, 90%; (c) NaN3, DMF, 100 °C, 15h, 74%; (d) TFA, CH2Cl2, 0 °C, 5h; (e) t-4-butylbenzyl isothiocyanate, NEt3, CH2Cl2, r.t., 15 h, 60%; (flPPh3, H2O, THF, 99%; (g) 1-bromo-2-chloroethane, K2CO3, acetone, reflux, 3 h, 94%; (h) NaN3, TBAB, KI, toluene, reflux, 99%; (i) (Boc)2O, Pd/C, H2, EtOH, r.t.; (j) NH2OH HCl, pyridine, 80 °C, 2h; (k) Pd/C, H2, cHCl, EtOH, r.t., 15 h; (l) TFA, CH2Cl2, 0 °C, 1 h, 55%; PPh3, H2O, THF, 99%
Result and Discussion
The binding affinities and agonistic/antagonistic functional activities of the synthesized TRPV1 ligands were determined in vitro by a binding competition assay with [3H]RTX and a functional 45Ca2+ uptake assay using rat TRPV1 heterologously expressed in Chinese hamster ovary (CHO) cells, as previously described.20 The results are summarized in Tables 1–4, together with the potencies of the parent agonists 1 and 2.
Table 1.
Amide B-region with 4-t-butylbenzyl C-region
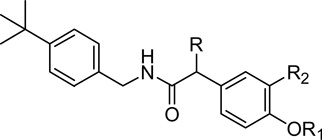 | ||||||
|---|---|---|---|---|---|---|
| R | R1 | R2 | Ki (nM) Binding Affinity |
EC50 (nM) Agonism |
Ki (nM) Antagonism |
|
| 1 | H | H | OCH3 | 667 (±86) | 82 (±34) | NE |
| 4b | H | H | F | 808 (±179) | 911 (±88) | (30%)b |
| 4c | H | H | Cl | 132 (±29) | 206 (±41) | NE |
| 8a | CH3 | H | OCH3 | 308 (±7.0) | 24 (±6.4) | (15%)b |
| 8b | CH3 | H | F | 178 (±33) | 221 (±63) | (20%)b |
| 8c | CH3 | H | Cl | 33.1 (±6.6) | (77%)a | (9%)b |
| 10a | H | CH2CH2NH2 | OCH3 | 1,230 (±230) | 358 (±58) | (12%)b |
| 10b | H | CH2CH2NH2 | F | 10,100 (±1,300) | (63%)a | (18%)b |
| 10c | H | CH2CH2NH2 | Cl | 2,470 (±490) | (36%)a | (56%)b |
| 14a | CH3 | CH2CH2NH2 | OCH3 | 504 (±160) | 238 (±79) | (16%)b |
| 14b | CH3 | CH2CH2NH2 | F | 2,160 (±410) | (91%)a | NE |
| 14c | CH3 | CH2CH2NH2 | Cl | 380 (±110) | (75%)a | (38%)b |
only fractional calcium uptake compared with that induced by 300 nM capsaicin.
only fractional antagonism at 30 µM
Table 4.
Thiourea B-region with 4-t-butylbenzyl C-region
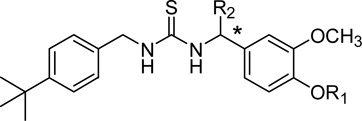 | |||||
|---|---|---|---|---|---|
| R1 | R2 | Ki (nM) Binding Affinity |
EC50 (nM) Agonism |
Ki (nM) Antagonism |
|
| 2 | H | H | 59 (±9.0) | 2.6 (±1.1) | NE |
| 32 | H | CH3 | 194 (±5) | 380 (±160)a | (35%)b |
| 34 | CH2CH2NH2 | H | 84 (±27) | 132 (±34) | NE |
| 36 | CH2CH2NH2 | CH3 | 1,430 (±520) | 5,800 (±1,700) | NE |
only fractional calcium uptake (70%) compared with that induced by 300 nM capsaicin
only fractional antagonism at 30 µM
In order to investigate how the introduction of an α–methyl group into the B region of agonists affects their TRPV1 binding and functional activity as compared to that of the parent compounds, we first fixed the C-region as a 4-tert-butylbenzyl moiety and varied the A-region functionality (Table 1). Compound 8a, an α–methylated analogue of the parent agonist (1), showed enhanced binding affinity and agonism by 2- and 3-fold, respectively. When we replaced the 3-methoxy group with halides to provide 8b and 8c, these analogues bound TRPV1 even more tightly (Ki = 178 nM for 8b, Ki = 33 nM for 8c) and their affinities increased ca. 4.5- and 4-fold compared to 4b and 4c, respectively. However, their functional activity shifted towards partial agonism / partial antagonism (as most evident for the decreased level of agonism of 8c). The SAR pattern in which the halogenations in the A-region shifted the functional activity to antagonism, with increased antagonism as the size of the halogen increased, was established in our previous report.21
Previously, we reported that 2-aminoethylation of the 4-hydroxyl group in the capsainoid improved in vivo stability as well as analgesic activity compared to the parent agonist.22 Thereby, we also examined the effect of α–methylation on the 2-aminoethoxy analogues of the amide agonists. Although all 4-aminoethoxy analogues synthesized were found to be partial agonists/antagonists, the binding affinities of α–methylated analogues 14a–c increased significantly compared to the acetamides 10a–c by 2.5–6.5 fold, respectively.
Next, we swapped the 4-tert-butylbenzyl group of 1 with long alkyl and arylalkyl chains, resembling the C-region of capsaicin, to provide 15–17 and examined the effect of their α-methylation (Table 2). Whereas the α-methylation of acetamide 15 did not affect its receptor activity, α-methylated analogues 19 and 20 showed a 1.5–2 fold enhancement in binding affinity and agonism compared to 16 (nonivamide) and 17. DA-5018 is the 2-aminoethyl analogue of 17. The α-methylation of DA-5018 to generate 22 likewise led to ca. 2-fold increase in receptor activity. The results indicated that α-methylation in the amide B-region might enhance favorable hydrogen bonding with the receptor or provide an additional hydrophobic interaction of a methyl group with the receptor.
Table 2.
Amide B-region with alkyl and arylalkyl C-region
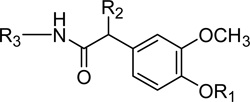 | ||||||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | Ki (nM) Binding Affinity |
EC50 (nM) Agonism |
Ki (nM) Antagonism |
|
| 15 | H | H | (CH2)7CH3 | 848 (±67) | 77 (±18) | NE |
| 16 | H | H | (CH2)8CH3 | 240 (±43) | 61 (±16) | NE |
| 17 | H | H | (CH2)3Ph(3,4-Me2) | 354 (±84) | 41 (±26) | NE |
| 18 | H | CH3 | (CH2)7CH3 | 744 (±32) | 130 (±16) | NE |
| 19 | H | CH3 | (CH2)8CH3 | 159 (±22) | 26.3 (±7.6) | NE |
| 20 | H | CH3 | (CH2)3Ph(3,4-Me2) | 177 (±30) | 21.3 (±2.7) | NE |
| DA-5018 | CH2CH2NH2 | H | (CH2)3Ph(3,4-Me2) | 930 (±170) | 589 (±76) | NE |
| 22 | CH2CH2NH2 | CH3 | (CH2)3Ph(3,4-Me2) | 518 (±110) | 404 (±67) | NE |
To further evaluate the effect of α-methylation in the B-region, we also investigated the reverse amide (Table 3) and thiourea B-regions (Table 4) in a similar fashion. The reverse amide B-region agonists olvanil 24 and 25–26 and their α-methylated analogues 29–30 were prepared for the comparison of receptor activities. The analysis indicated that α-methylation in the reverse amide B-region led to a decrease in receptor activity in which 29–31 showed 2–4 fold reductions in binding affinity compared to the corresponding acetamides. The similar SAR pattern was also observed in the thiourea B-region. The α-methylated thioureas 32 and 36 showed marked loss of binding affinity compared to 2 and 34 by 3- and 17-fold. The result indicated that, different from the amide B-region, the α-methylation of the reverse amide and thiourea might interfere with appropriate hydrogen bonding with the receptor due to steric hindrance.
Table 3.
Reverse amide B-region with alkyl C-region
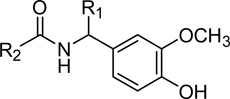 | |||||
|---|---|---|---|---|---|
| R1 | R2 | Ki (nM) Binding Affinity |
EC50 (nM) Agonism |
Ki (nM) Antagonism |
|
| 24 (Olvanil) | H | (CH2)7CH=CH(CH2)7CH3 | 142 (±27) | 202 (±19) | NE |
| 25 | H | (CH2)7CH3 | 1223 (±18) | 223 (±47) | NE |
| 26 | H | (CH2)8CH3 | 864 (±10) | 83 (±16) | NE |
| 29 | CH3 | (CH2)7CH=CH(CH2)7CH3 | 226 (±52) | 678 (±140) | NE |
| 30 | CH3 | (CH2)7CH3 | 4,275 (±99) | 1580 (±290)a | (44%)b |
| 31 | CH3 | (CH2)8CH3 | 3,300 (±610) | 1250 (±140) | (28%)b |
only fractional calcium uptake (72%) compared with that induced by 300 nM capsaicin
only fractional antagonism at 30 µM
In summary, we have synthesized a series of TRPV1 agonists with the amide, reverse amide, and thiourea groups in the B-region along with their corresponding α-methylated analogues. Within the amide analogues, the α–methylation enhanced binding affinity and agonism compared to the parent agonist due to favorable hydrogen bonding interaction or additional hydrophobic interaction of a methyl in the binding site. By contrast, the α–methylation of the reverse amide and thiourea agonists led to the reduction in receptor activity, suggesting that the placement of a methyl group might interfere with appropriate hydrogen bonding with the receptor due to steric hindrance. The SAR analysis of α–methylation implicates hydrogen bonding as well as a steric effect in the B-region as critical for receptor binding. The SAR of the two enantiomers of the α–methylated amide is underway to further explore interactions with the receptor.
Acknowledgement
This research was supported by Grants R11-2007-107-02001-0 from the NRF and was supported in part by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, National Cancer Institute (Project Z1A BC 005270). We thank other members of the Blumberg group for assistance with some of the assays.
References and notes
- 1.Szallasi A, Blumberg PM. Pharmacol. Rev. 1999;51:159. [PubMed] [Google Scholar]
- 2.Nagy I, Santha P, Jancso G, Urban L. Eur. J. Pharmacol. 2004;500:351. doi: 10.1016/j.ejphar.2004.07.037. [DOI] [PubMed] [Google Scholar]
- 3.Alawi K, Keeble J. Pharmacol. Ther. 2010;125:181. doi: 10.1016/j.pharmthera.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 4.Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. Neuron. 1998;21:531. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- 5.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. Nature. 1997;389:816. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 6.Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V, Julius D, Hogestatt ED. Nature. 1999;400:452. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]
- 7.Hwang SW, Cho H, Kwak J, Lee SY, Kang CJ, Jung J, Cho S, Min KH, Suh YG, Kim D, Oh U. Proc. Natl. Acad. Sci. U S A. 2000;97:6155. doi: 10.1073/pnas.97.11.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walpole CS, Wrigglesworth JR. Capsaicin in the study of pain. San Diego, CA: Academic Press; 1993. [Google Scholar]
- 9.Appendino G, Szallasi A. Life Sci. 1997;60:681. doi: 10.1016/s0024-3205(96)00567-x. [DOI] [PubMed] [Google Scholar]
- 10.Julius D, Basbaum AI. Nature. 2001;413:203. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- 11.Gunthorpe MJ, Chizh BA. Drug. Discov. Today. 2009;14:56. doi: 10.1016/j.drudis.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 12.Wong GY, Gavva NR. Brain. Res. Rev. 2009;60:267. doi: 10.1016/j.brainresrev.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Szallasi A, Cortright DN, Blum CA, Eid SR. Nat. Rev. Drug Discov. 2007;6:357. doi: 10.1038/nrd2280. [DOI] [PubMed] [Google Scholar]
- 14.Westaway SM. J. Med. Chem. 2007;50:2589. doi: 10.1021/jm060637e. [DOI] [PubMed] [Google Scholar]
- 15.Krause JE, Chenard BL, Cortright DN. Curr. Opin. Investig. Drugs. 2005;6:48. [PubMed] [Google Scholar]
- 16.Conway SJ. Chem. Soc. Rev. 2008;37:1530. doi: 10.1039/b610226n. [DOI] [PubMed] [Google Scholar]
- 17.Kim YS, Kil MJ, Kang SU, Ryu H, Kim MS, Cho Y, Bhondwe RS, Thorat SA, Sun W, Liu K, Lee JH, Choi S, Pearce LV, Pavlyukovets VA, Morgan MA, Tran R, Lazar J, Blumberg PM, Lee J. Bioorg. Med. Chem. 2012;20:215. doi: 10.1016/j.bmc.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee J, Kang SU, Kil MJ, Shin M, Lim JO, Choi HK, Jin MK, Kim SY, Kim SE, Lee YS, Min KH, Kim YH, Ha HJ, Tran R, Welter JD, Wang Y, Szabo T, Pearce LV, Lundberg DJ, Toth A, Pavlyukovets VA, Morgan MA, Blumberg PM. Bioorg. Med. Chem. Lett. 2005;15:4136. doi: 10.1016/j.bmcl.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 19.Lee J, Kang SU, Choi HK, Lim JO, Kil MJ, Jin MK, Kim KP, Sung JH, Chung SJ, Ha HJ, Kim YH, Pearce LV, Tran R, Lundberg DJ, Wang Y, Toth A, Blumberg PM. Bioorg. Med. Chem. Lett. 2004;14:2291. doi: 10.1016/j.bmcl.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Szabo T, Welter JD, Toth A, Tran R, Lee J, Kang SU, Suh YG, Blumberg PM. Mol. Pharmacol. 2002;62:947. doi: 10.1124/mol.62.4.947. [DOI] [PubMed] [Google Scholar]
- 21.Kang DW, Kim YS, Lim KS, Kim MS, Pearce LV, Pavlyukovets VA, Tao AK, Lang-Kuhs KA, Blumberg PM, Lee J. Bioorg. Med. Chem. 2010;18:8092. doi: 10.1016/j.bmc.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee J, Lee J, Kang M-S, Kim K-P, Chung S-J, Blumberg PM, Yi J-B, Park YH. Bioorg. Med. Chem. 2002;10:1171. doi: 10.1016/s0968-0896(01)00387-x. [DOI] [PubMed] [Google Scholar]