

Abstract
The bis(arylimidazol-2-ylidene)pyridine cobalt methyl complex, (iPrCNC)CoCH3, was evaluated for the catalytic hydrogenation of alkenes. At 22 °C and 4 atm of H2 pressure, (iPrCNC)CoCH3 is an effective pre-catalyst for the hydrogenation of sterically hindered, unactivated alkenes such as trans-methylstilbene, 1-methyl-1-cyclohexene and 2,3-dimethyl-2-butene, representing one of the most active cobalt hydrogenation catalysts reported to date. Preparation of the cobalt hydride complex, (iPrCNC)CoH was accomplished by hydrogenation of (iPrCNC)CoCH3. Over the course of 3 hours at 22 °C, migration of the metal-hydride to the 4-position of the pyridine ring yielded (4-H2-iPrCNC)CoN2. Similar alkyl migration was observed upon treatment of (iPrCNC)CoH with 1,1-diphenylethylene. This reactivity raised the question as to whether this class of chelate is redoxactive, engaging in radical chemistry with the cobalt center. A combination of structural, spectroscopic and computational studies was conducted and provided definitive evidence for bis(arylimidazol-2-ylidene)pyridine radicals in reduced cobalt chemistry. Spin density calculations established that the radicals were localized on the pyridine ring, accounting for the observed reactivity and suggest a wide family of pyridine-based pincers may also be redox active.
Introduction
The hydrogenation of carbon-carbon multiple bonds catalyzed by homogeneous transition metal complexes is one of the most widely employed reactions in the synthesis of commodity and fine chemicals.1 There has been renewed interest in developing catalysts using earth abundant elements as alternatives or even improvements to more commonly used precious metal compounds.2,3 One potential challenge to the discovery of catalysts based on first row metals is the difference in redox properties compared to their second and third row congeners. In catalytic hydrogenation and related processes such as hydrosilylation and hydroboration, important bondbreaking and making processes such as oxidative addition and reductive elimination often rely on two-electron changes.4 First row metals often engage in one-electron chemistry, offering alternative pathways that disrupt desired transformations required for catalytic turnover.5
Two strategies have predominated in overcoming the preference for one-electron redox chemistry associated with base metals to achieve catalysis. The first and most established method is the use of highly covalent, strong field ligands that favor low spin configurations and higher metal oxidation states.6 The rich and continually evolving catalytic chemistry observed with thermal and photoactivated Fe(CO)5 and Fe2(CO)9 supports the viability of this approach.7,8 A second strategy employs redox-active ligands – those that can engage in reversible one-electron chemistry with the transition metal.9,10,11 By enabling cooperative metal-ligand redox events, net two-electron chemistry can be achieved while the metal engages in only one-electron processes.12 This concept, important in the function of many metalloenzymes,13 has emerged as an effective strategy in base metal catalysis,2a,10,12,14,15 small molecule activation and group transfer chemistry.16,17
Aryl-substituted bis(imino)pyridine iron dinitrogen complexes have proven to be effective pre-catalysts for alkene hydrogenation,18,19 hydrosilylation,18,20,21 cycloaddition22,23,24 and hydroboration25,26 reactions. In many cases, the activity and selectivity of these compounds rivals or surpasses established precious metal catalysts.27 Notably, introduction of electron donating substituents into the 4-position of the pyridine ring increased the activity for alkene hydrogenation28 and hydrosilylation.20b For example, (4-Me2N-iPrPDI)Fe(N2)229 exhibited higher turnover frequencies than the first generation pre-catalyst, (iPrPDI)Fe(N2)2 (iPrPDI = 2,6-(2,6-iPr2-C6H3-N=CMe)2C5H3N) in both reaction types (Scheme 1). These findings prompted study of even more electron rich iron bis(dinitrogen) complexes. Danopoulos and coworkers reported the synthesis of (iPrCNC)Fe(N2)2 (iPrCNC = 2,6-(2,6-iPr2-C6H3-imadazol-2-ylidene)2-C5H3N)30 and infrared stretching frequencies of the N2 ligands establish a more electron rich iron center than in the corresponding bis(imino)pyridine compounds. Accordingly, (iPrCNC)Fe(N2)2 and its less sterically protected variants, (MeCNC)Fe(N2)2 and (MesCNC)Fe(N2)2, are extremely active hydrogenation pre-catalysts. The latter compounds exhibit rapid turnover for unactivated tri-and tetrasubstituted alkenes, challenging substrates for even the most active precious metal catalysts.31
Scheme 1.

Bis(imino)pyridine and pyridine N-heterocyclic dicarbene iron dinitrogen complexes that are active pre-catalysts for olefin hydrogenation. The dimers, [(RPDI)Fe(N2)](μ2-N2) (R = Me, Et), are depicted as monomers for simplicity.
Cobalt compounds have also demonstrated considerable promise for alkene hydrogenation. Historically, cobalt-catalyzed alkene hydrogenation has been accomplished using borohydrides or hydrogen as the terminal reductant although activities and selectivities are generally low.32 Budzelaar and coworkers have since reported that four-coordinate, aryl- and alkyl-substituted bis(imino)pyridine cobalt alkyl and hydride complexes, (PDI)CoR,33,34 are active for the hydrogenation of simple, unactivated terminal and gem-alkenes; more sterically hindered trisubstituted olefins are unreactive.35 Our laboratory has prepared enantiopure, C1-symmetric variants of these compounds and observed asymmetric hydrogenation of styrenyl-substituted olefins with high enantioselectivity.36 Hanson and coworkers have recently reported a cobalt(II) alkyl complex supported by a [PNP]-type pincer ligand that is active for the hydrogenation of olefins, ketones, aldehydes and imines37 as well as for the acceptorless dehydrogenation of alcohols.38 This catalyst class offers tolerance to a range of functional groups, although hydrogenation of sterically hindered substrates such as tri- and tetrasubstituted olefins remains a challenge.39
The similarity between bis(imino)pyridines and the bis(arylimidazol-2-ylidene)pyridines and the improved catalytic performance for alkene hydrogenation observed with iron dinitrogen complexes of the latter pincer class28 prompted evaluation of the corresponding cobalt alkyl compounds. Danopoulos and coworkers have previously reported the synthesis of (iPrCNC)CoCH3 by treatment of (iPrCNC)CoBr with methyllithium.40 While this compound was evaluated for ethylene polymerization activity, catalytic alkene hydrogenation studies, to our knowledge, were not reported. Here we describe the catalytic hydrogenation of sterically hindered alkenes using bis(arylimidazol-2-ylidene)pyridine cobalt complexes. The high activity of the cobalt pre-catalysts raised the question as to the electronic structure of the compounds and if this class of pincer ligand engages in radical chemistry with a reduced base metal. A combined spectroscopic and computational study clearly established the presence of ligand-centered radicals and migration of hydrides and alkyls to the 4-position of the pyridine, supporting this view of the electronic structure.
Results and Discussion
I. Evaluation of Catalytic Performance
Our studies commenced with evaluation of the bis(arylimidazol-2-ylidene)pyridine cobalt methyl complex, (iPrCNC)CoCH3 for the catalytic hydrogenation of a series of alkenes. A summary of the observed hydrogenation activity is reported in Table 1. A comparison to previously reported (iPrCNC)Fe(N2)2-catalyzed alkene hydrogenation28 is also presented in the table. Each catalytic hydrogenation was conducted under standard conditions employing 5 mol% of the metal pre-catalyst, 0.916 M substrate in benzene with 4 atm of H2 at 22 °C. The conversion to alkane was determined by gas chromatography and 1H NMR spectroscopy.
Table 1. Catalytic hydrogenation of alkenes with bis(arylimidazol-2-ylidene)pyridine cobalt and iron pre-catalysts.
| Entrya | Substrate | (iPrCNC)CoCH3 | (iPrCNC)Fe(N2)2 |
|---|---|---|---|
| 1b |
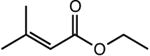
|
>95% (1 hr) | >95% (1 hr) |
| 2b |
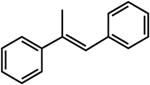
|
>95% (5 hr) | 89% (12 hr) |
| 3c |
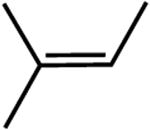
|
>95% (5 hr) | >95% (15 hr) |
| 4b |
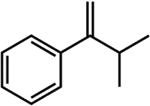
|
>95% (5 hr) | >95% (15 hr) |
| 5b,d |
|
>95% (7 hr) | >95% (8 hr) |
| 6b |
|
95% (13 hr) | >95% (18 hr) |
| 7b |
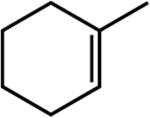
|
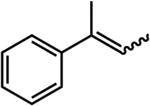
95% (120 hr)e | 20% (24 hr) |
| 8c |
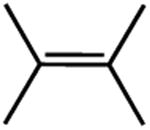
|
15% (24 hr)f | 0% (24 hr) |
All catalytic reactions carried out with 5 mol% of the metal complex (0.032 mmol) in 0.916 M substrate benzene solution with 4 atm of H2 at 22 °C.
Conversions determined by GC-FID.
Conversions determined by 1H NMR spectroscopy.
Substrate used as racemic mixture.
32% conversion at 12 hours.
At 50 °C, conversion is 69% over 24 hours.
The first substrate examined was ethyl 3,3-dimethylacrylate (entry 1) as this alkene has been used as a benchmark for iron-catalyzed olefin hydrogenation.19,28 The cobalt methyl pre-catalyst, (iPrCNC)CoCH3, exhibits comparable activity to (iPrCNC)Fe(N2)2, completing the hydrogenation in one hour under standard conditions. Because of the high activity, only more challenging, unfunctionalized hindered alkenes were examined. The trisubstituted alkenes, trans-methylstilbene (entry 2), 2-methyl-2-butene (entry 3), the isomers of β -methylstyrene (entry 5) and 2,6dimethyl-2-octene(entry 6) were also readily hydrogenated. Each substrate reached complete conversion over the course of hours. Hydrogenation of 1-methyl-1-cyclohexene (entry 7), a traditionally more challenging endo cyclic trisubstituted olefin, was also successful. Approximately 32% hydrogenation to alkane was observed after 12 hours and complete conversion was achieved in 120 hours. Unfortunately, only minimal turnover was observed at 22 °C with the tetrasubstituted olefin, 2,3-dimethyl-2-butene (entry 5). Heating the catalytic reaction to 50 °C resulted in 69% conversion to product after 24 hours. These results demonstrate that (iPrCNC)CoCH3 is a more active pre-catalyst than the corresponding iron dinitrogen complex, (iPrCNC)Fe(N2)2 and is one of the most active base metal alkene hydrogenation catalysts known, proving effective for unactivated, sterically hindered alkenes.41
II. Synthesis, Characterization and Reactivity Studies of a Bis(arylimidazol-2-ylidene)pyridine Cobalt Hydride
The high catalytic hydrogenation activity of the (iPrCNC)CoCH3 pre-catalyst prompted study of the corresponding cobalt hydride, a likely component of the catalytic cycle. Exposure of a 25 mM benzene-d6 solution of (iPrCNC)CoCH3 to 1 atm of hydrogen gas resulted in rapid liberation of CH4 (as judged by 1H NMR spectroscopy) and formation of a new, diamagnetic C2ν symmetric compound identified as the desired cobalt hydride, (iPrCNC)CoH (Scheme 2).
Scheme 2.

Synthesis and reactivity of a bis(arylimidazol-2-ylidene)pyridine cobalt hydride.
The benzene-d6 1H NMR resonances for the chelate in (iPrCNC)CoH are similar to those for (iPrCNC)CoCH3 with the hydrogen in the 4-position of the pyridine located downfield at 11.3 ppm. The Co-H resonance was observed as a very broad (ν1/2 ∼ 148 Hz) signal whose chemical shift is dependent on the amount of excess dihydrogen present. In a rigorously degassed sample, the peak appears at −27 ppm while in the presence of 1 atmosphere of H2 this resonance shifts to −18 ppm with additional broadening. A slight broadening of the 4-pyridine and imidazole positions accompanies the downfield shift, consistent with rapid exchange between the Co-H and free H2 on the NMR timescale.
Addition of D2 to a 25 mM benzene-d6 solution of (iPrCNC)CoCH3 resulted in rapid liberation of an 87:13 ratio of CH3D and CH4 along with formation of (iPrCNC)CoD. The observation of CH4 likely arises from competitive isopropyl aryl group cyclometallation. Continued exposure to excess D2 resulted in selective incorporation of the isotopic label into the 4-position of the pyridine, one of the imidazolylidene C-H bonds (adjacent to the pyridine) and one of the isopropyl methyl groups (Scheme 2). The rate of deuterium incorporation into the isopropyl methyl and imidazolylidene positions was significantly faster than the pyridine 4-position. Complete deuteration of these positions was accomplished after approximately six hours with periodic agitation of the J. Young NMR tube after which time the Co-H was no longer observed.
Allowing a benzene-d6 solution of (iPrCNC)CoH to stand at 22 °C under an N2 atmosphere resulted in conversion to a new C2v symmetric cobalt compound over the course of 3 hours. The toluene solution infrared spectrum exhibited a strong band centered at 2048 cm−1 signaling dinitrogen coordination. Notable resonances in the benzene-d61H NMR spectrum include two triplets centered at 3.57 and 4.31 ppm signaling formation of allylic and vinylic protons due to modification of the pyridine ring. The spectroscopic data in conjunction with structural characterization of related compounds (vida infra) establish the identity of the purple-blue product as (4-H2-iPrCNC)CoN2, arising from migration of the cobalt hydride to the para-pyridine position of the bis(arylimidazol-2-ylidene)pyridine chelate. The assignment of the allylic resonances was also confirmed by isotopic labeling, whereby (iPrCNC)CoD was synthesized and then converted to (4-H,D-iPrCNC)CoN2. The migration of the cobalt hydride to the 4-position of the pyridine ring is suppressed by excess dihydrogen (or dideuterium). In this matter, isotopic labeling experiments could be conducted to determine the fate of the Co-D during formation of (4-H,D-iPrCNC)CoN2. During the course of these investigations, we also discovered that (4-H2-iPrCNC)CoN2 could also be prepared by treatment of (iPrCNC)CoBr with NaBEt3H.
In solution, (4-H2-iPrCNC)CoN2 proved to be unstable, slowly undergoing decomposition over the course of days even at temperatures as low as −35 °C. This instability has thus far inhibited the isolation of single crystals for X-ray diffraction. Nevertheless, solutions of the compound could be generated by hydrogenation of (iPrCNC)CoCH3 and used for subsequent reactivity studies. It was of interest to determine whether the newly formed C-H bond in the 4-position of the pyridine ring was subject to abstraction. Addition of one equivalent of Ph3CCl to a toluene solution of (4-H2-iPrCNC)CoN2 at −35 °C resulted in an immediate color change from deep blue to brown. Analysis of the product mixture by 1H NMR spectroscopy established formation of (iPrCNC)CoCl and a stoichiometric quantity of Ph3CH (Scheme 2). The cobalt complex was isolated in 74% yield and was also characterized by X-ray diffraction. The overall molecular geometry and metrical parameters are similar to those previously reported for (iPrCNC)CoBr.40 A representation of the molecular structure is presented in the Supporting Information and selected bond distances are presented in the electronic structure section of the manuscript. Importantly, the reactivity with Ph3CCl demonstrates the ability of the functionalized bis(arylimidazol-2-ylidene)pyridine chelate to participate in cooperative metal-ligand reactivity where removal of a C-H bond is coupled with formation of a cobalt-chloride bond.
The reactivity of (iPrCNC)CoH with alkenes was explored to gain insight into insertion behavior relevant to catalytic hydrogenation. Addition of a slight excess (3.5 equivalents) of 1-butene or isobutene to benzene-d6 solutions of (iPrCNC)CoH furnished (iPrCNC)CoCH2(CH2)2CH3 and (iPrCNC)CoCH2CH(CH3)2, respectively (eq 1). Both compounds were characterized by 1H and 13C NMR spectroscopy and established preference for the metal-alkyl derived from 1,2-insertion of the alkene into the cobalt-hydride. Over the course of 48 hours under a dinitrogen atmosphere at 22 °C in benzene-d6 solution both compounds decompose to an intractable mixture of cobalt products.The rate of decomposition is accelerated if the samples are stored under vacuum. Notably, there was no evidence for migration of the alkyl group to the 4-position of the pyridine ring.
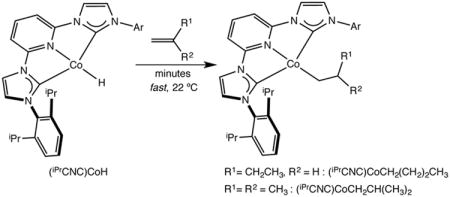 |
(1) |
Insertion studies were also conducted with a more hindered 1,1-disubstituted olefin. Addition of one equivalent of 1,1-diphenylethylene to a 25 mM benzene-d6 solution of (iPrCNC)CoH under an N2 atmosphere resulted in a rapid color change from yellow-brown to deep blue. Analysis of a benzene-d6 solution of the product by IR spectroscopy revealed a strong band centered at 2050 cm−1, again signaling dinitrogen coordination. The benzene-d61H NMR spectrum exhibited the number of resonances consistent with Cs symmetry. As with (4-H2-iPrCNC)CoN2, a doublet and triplet were observed at 4.33 and 4.90 ppm, respectively, signaling modification of the pyridine ring. The combined IR and NMR data support formation of (4-CPh2CH3-iPrCNC)CoN2, arising from migration of the alkyl to the 4-position of the pyridine ring (eq 2).
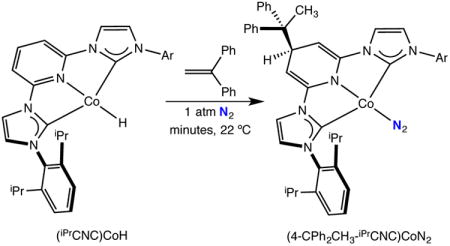 |
(2) |
The modification of the pyridine ring in (4-CPh2CH3-iPrCNC)CoN2 was confirmed by X-ray diffraction. A representation of the molecular structure is presented in Figure 1 and selected bond distances and angles are reported in Table 2. The overall molecular geometry about the metal is best described as idealized planar, as anticipated for a formally Co(I), d8 center. The aryl substituents are nearly orthogonal to the idealized metal-chelate plane and the N(6)-N(7) distance of 1.112(2) Å signals weak activation of the N2 ligand. The 4-position of the central pyridine ring is substituted with an alkyl ligand resulting from a new bond between C(6) and C(36), the more substituted carbon of the alkene forming a quaternary center. The C(5)-C(6) and C(6)-C(7) distances are elongated to 1.513(3) and 1.526(2) Å consistent with formation of an sp3 hybridized carbon in the 4-position although C(6) deviates little from the idealized plane of the heterocycle.
Figure 1.

Solid state molecular structure of (4-CPh2CH3-iPrCNC)CoN2 at 30% probability ellipsoids. Hydrogen atoms, except for the one attached to C(6), and one diethyl ether molecule were omitted for clarity.
Table 2. Selected bond distances (Å) and angles (deg) for (4-CPh2CH3-iPrCNC)CoN2.
| Co(1)–N(3) | 1.8553(16) |
| Co(1)–C(3) | 1.9015(19) |
| Co(1)–C(9) | 1.8980(19) |
| Co(1)–N(6) | 1.7500(17) |
| C(4)–C(5) | 1.333(3) |
| C(7)–C(8) | 1.335(3) |
| C(5)–C(6) | 1.513(3) |
| C(6)–C(7) | 1.526(2) |
| N(1)–C(3) | 1.357(3) |
| C(3)–N(2) | 1.381(2) |
| N(4)–C(9) | 1.377(2) |
| C(9)–N(5) | 1.366(2) |
| N(6)–N(7) | 1.112(2) |
| N(1)–C(3)–N(2) | 103.67(15) |
| N(4)–C(9)–N(5) | 103.28(16) |
One possibility accounting for the formation of (4-CPh2CH3-iPrCNC)CoN2 is 2,1-insertion of the alkene into the cobalt-hydrogen bond followed by migration of the alkyl to the 4-position of the pyridine ring. Migration of alkyl groups to the 4-position of related bis(imino)pyridines has been observed by Cámpora and coworkers in manganese chemistry and has been evolved into a useful synthetic method for the preparation of 4-alkyl substituted variants of these ligands.42 The preference for 2,1 insertion of 1,1-diphenylethylene contrasts the 1,2-insertion preference observed with 1-butene and isobutene. In rhodium chemistry, Jones and coworkers have rationalized the thermodynamic preference between linear and branched metal alkyls as arising from the relative strengths of the newly formed β-C-H bonds.43 It is likely that for the cobalt complexes described here, the preference for formation of a methyl C-H bond over a methine C-H is larger for 1,1-diphenylethylene than for 1-butene (methylene) or isobutene. The resulting tertiary cobalt alkyl is sufficiently sterically pressured to undergo homolysis and the resultant stabilized radical then adds to the 4-position of the pyridine ring. Notably, 1,1-diphenylethylene undergoes catalytic hydrogenation44 and was completely converted to the corresponding alkane in the presence of 5 mol% (iPrCNC)CoCH3 and 1 atm of H2 over the course of 24 hours.
An alternative pathway to account for formation of (4-CPh2CH3-iPrCNC)CoN2 involves H-atom transfer (HAT) from the cobalt-hydride.45 Halpern proposed sequential HAT events from in situ generated (CO)4CoH during the reduction of anthracene46 and in the hydrogenation of α-methylstyrene with (CO)5MnH.47 HAT pathways have also been proposed in alkene reductions with chromium,48,49 tungsten,50 and vanadium51 hydrides. Our current data do not allow for distinction of these pathways. However, the observation of the cobalt-hydride and alkyl migration to the 4-position of the pyridine raised the possibility of radical character in bis(arylimidazol-2-ylidene)pyridine chelates and prompted further study of the electronic structure of (iPrCNC)CoX (X = H, alkyl, halide) complexes.
III. Electronic Structure of Bis(arylimidazol-2-ylidene)pyridines Cobalt Complexes
The superior catalytic activity of (iPrCNC)CoCH3 as compared to (iPrCNC)Fe(N2)2 and related bis(imino)pyridine compounds raised the question as to the electronic structure of the pre-catalyst and the role of the bis(arylimidazol-2-ylidene)pyridine ligand. The observation of hydride and alkyl migration to the 4-position of the chelate also suggested the possibility of radical character in this ligand class. It is now well-established that diamagnetic (iPrPDI1−)CoIICH3 is a low spin Co(II) compound engaged in antiferromagnetic coupling with a bis(imino)pyridine radical anion.52 The similar geometries between (iPrPDI1−)CoIICH3 and (iPrCNC)CoCH3 suggest the possibility of similar electronics. Determining the oxidation state of both the metal and the pincer ligand is essential for understanding the mechanism of turnover and raises the broader question as to whether bis(arylimidazol-2-ylidene)pyridine chelates can engage in radical chemistry with an appropriately reducing base metal. To address these issues, a series of spectroscopic, computational and preparative studies were carried out.
A. Evaluation of the Free Ligand
To determine if bis(arylimidazol-2-ylidene)pyridine chelates are redox-active, studies with the free ligand were initially conducted. Our laboratory has recently reported that cyclic voltammetry is one of the most sensitive evaluators of the electronic properties of aryl-substituted bis(imino)pyridines.53,54,55 Reduction potentials between −2.35 and −2.83 V (vs Fc/Fc+) were observed in THF solution depending on the specific substitution pattern of the bis(imino)pyridine. Performing identical measurements on the free bis(arylimidazol-2-ylidene)pyridine, iPrCNC,56 in THF solution (1 mM iPrCNC in 0.1 M [nBu4]PF6 electrolyte) were unsuccessful as no reversible redox waves were observed, suggesting a less thermodynamically accessible radical anion for this ligand class.
Despite the lack of success with electrochemical reduction, access to the desired bis(arylimidazol-2-ylidene)pyridine radical anion was explored chemically. For spectroscopic reference, alkali metal salts of previously reported aryl-substituted bis(imino)pyridine radical anions57 were initially synthesized to evaluate various synthetic routes as well as to compare EPR spectroscopic signatures. Stirring THF solutions of the free chelates with excess sodium resulted in formation of red-purple solutions identified as Na[iPrPDI] and Na[(iPrBPDI)], respectively (iPrBPDI= 2,6-(2,6-iPr2-C6H3N=CPh)2C5H3N). After dilution with toluene (toluene/THF ∼ 10:1) the samples were investigated by X-band EPR spectroscopy at 298 K (Figure 2). The spectrum obtained for Na[(iPrBPDI)] exhibits a sharp isotropic signal centered at giso = 2.003 with ten resolved lines due to hyperfine coupling. Examples of EPR spectra of bis(imino)pyridine radicals coordinated to diamagnetic metal ions have been reported previously by Gambarotta and coworkers for (iPrPDI)AlMe2,58 our laboratory for (iPrBPDI)VE2 (E = O, S, NPh)59 and (iPrPDI)CoN2.60 These studies established that the major contributions to the hyperfine coupling pattern arise from the nitrogen and hydrogen nuclei constituting the plane of the bis(imino)pyridine ligand with additional contributions from the metal center as expected for a radical delocalized over the conjugated π-system of the chelate. Given these precedents, the EPR spectrum of Na[(iPrBPDI)] was readily simulated assuming hyperfine coupling to the pyridine and imine nitrogen nuclei (Aiso(14Npy) = 6.2 MHz, Aiso(14Nim) = 3.3 MHz), the para-pyridine proton (Aiso(1Hpara) = 14.0 MHz), and the sodium cation (Aiso(23Na) = 8.4 MHz). In agreement with the previous studies and related efforts with pyridine radical anions,61 the largest contributions to the hyperfine coupling arises from coupling to the proton in the 4-position of the pyridine ring, while contributions from the protons in the 3-position are small and not resolved in the recorded spectrum of Na[(iPrBPDI)]. It should be noted that the observed ten line pattern is merely the envelope of a more complex hyperfine pattern that is not further resolved due to line broadening. Assuming only contributions from the H, N, and Na nuclei of the chelate plane, a total of 360 resonances is expected for a complex with Cs symmetry or higher. This number increases for the EPR spectrum of Na[(iPrPDI)] due to contributions from the in plane methyl substituents of the chelate backbone producing a theoretical maximum of 2520 lines. Indeed, the recorded isotropic spectrum centered at giso = 2.003 for Na[(iPrPDI)] exhibits a pattern that is similar to the spectrum of Na[(iPrBPDI)] split by additional hyperfine features. Unfortunately, we have not been able to obtain a satisfactory fit of the data due to the high complexity of the hyperfine pattern.
Figure 2.

THF/Toluene solution EPR spectrum of Na[(iPrBPDI)] (top) and Na[iPrPDI] (bottom) recorded at 298 K. Conditions for (a) Na[(iPrBPDI)]: microwave frequency = 9.3749 GHz, power = 0.63 mW, modulation amplitude = 0.05 mT/100 kHz; (b) Na[(iPrPDI)]: microwave frequency = 9.3746 GHz, power = 0.63 mW, modulation amplitude = 0.05 mT/100 kHz.
Similar studies were conducted with the bis(arylimidazol-2-ylidene)pyridine, iPrCNC. Stirring the free dicarbene with excess sodium metal in THF immediately produced an orange solution. Analysis by EPR spectroscopy established the formation of a paramagnetic product assigned as Na[iPrCNC] (Figure 3). An alternative route, whereby CNC(HBr)262 was treated with excess sodium, proved less reliable and generated unidentified byproducts. Similar to the bis(imino)pyridine radicals, the X-band EPR spectrum of Na[iPrCNC] centered at giso = 2.003 exhibits the complexity expected for an organic radical delocalized over the π-system of the pyridine and NHC rings. Again assuming hyperfine contributions from the in-plane N, H, and Na nuclei of the complex, a maximum number of 16200 resonances are possible, giving rise to the observed multi-line pattern. Computational studies (see below) reveal that the spin density in the [iPrCNC] radical anion are mostly localized on the pyridine ring suggesting that hyperfine contributions from the imidazole nuclei are small compared to contributions from the pyridine ring and the metal cation. However, the multitude of small contributions adds to the line broadening of the spectrum, thus lowering the resolution and complicating simulations of the data.
Figure 3.

Toluene solution EPR spectrum of Na[iPrCNC] (top) and MgBr[(iPrCNC)] (bottom) recorded at 298 K. Conditions for (a) Na[iPrCNC]: microwave frequency = 9.3783 GHz, power = 2.00 mW, modulation amplitude = 0.02 mT/100 kHz; (b) MgBr[(iPrCNC)]: microwave frequency = 9.3710 GHz, power = 2.00 mW, modulation amplitude = 0.1 mT/100 kHz.
In an attempt to simplify the hyperfine structure of the [iPrCNC] radical anion, alternative reductants were explored to replace the sodium cation (100% 23Na, I = 3/2) by a nonmagnetic cation. Stirring a THF slurry of CNC(HBr)2 with excess magnesium powder in the presence of catalytic amounts of anthracene63 generated an orange solution identified as MgBr[(iPrCNC)] (Figure 3, top). The X-band EPR spectrum exhibits an isotropic signal (giso = 2.003) consistent with an S = 1/2 organic radical. As expected, the introduction of the predominantly nonmagnetic magnesium nucleus (90% 24/26Mg, I = 0; 10% 25Mg, I = 5/2) simplifies the data significantly producing a spectrum with a 14-line main feature and additional small signals visible at the edges of the spectrum arising from coupling to 25Mg. Despite the smaller number of potential hyperfine lines (4050) the spectrum of MgBr[(iPrCNC)] covers a slightly wider field range, which is most likely due to small changes in the spin distribution over the π-system induced by Mg as compared to Na resulting in slightly different coupling constants for the N and H nuclei. Unfortunately, a satisfactory simulation could not be obtained even for the simplified spectrum of the magnesium derivative.
B. Synthesis of Additional Bis(arylimidazol-2-ylidene)pyridine Cobalt Complexes
Additional bis(arylimidazol-2-ylidene)pyridine cobalt compounds were synthesized as references for structural and spectroscopic measurements. Previous studies with bis(imino)pyridine cobalt complexes served as a guide for specific cobalt derivatives. Gibson34 and our laboratory60,64 have established that the cationic bis(imino)pyridine cobalt dinitrogen complex, [(iPrPDI)CoN2]+ is a low-spin Co(I), d8 compound with a neutral, redox-innocent chelate. Likewise, one electron oxidation of (iPrPDI1−)CoIICH3 is ligand-based, furnishing [(iPrPDI0)CoIICH3]+, an overall S = 1/2 compound best described as low-spin Co(II) with a neutral bis(imino)pyridine.65 EPR studies on the latter support this view with identification of a cobalt-centered spin. Inspired by these findings, the synthesis of the analogous bis(arylimidazol-2-ylidene)pyridine cobalt cations was explored.
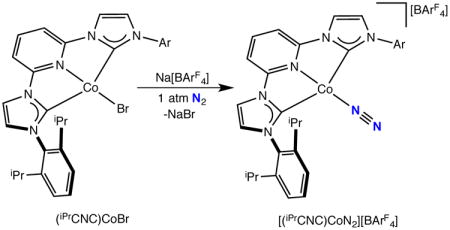
Addition of NaBArF4 to a diethyl ether solution of (iPrCNC)CoBr under a dinitrogen atmosphere followed by recrystallization from pentane-fluorobenzene yielded a bright green diamagnetic solid identified as [(iPrCNC)CoN2][BArF4] (BArF4 = B(3,5-(CF3)2C6H3)4) in 92% yield (eq 3). The toluene solution infrared spectrum of [(iPrCNC)CoN2][BArF4] exhibits a strong band at 2141 cm−1, signaling dinitrogen coordination. The 1H and 13C NMR spectra recorded in fluorobenzene-d5 are consistent with a C2v symmetric molecule. The compound was characterized by single crystal X-ray diffraction and a representation of the molecular structure is presented in Figure 4. Selected metrical parameters for this and all of the bis(arylimidazol-2-ylidene)pyridine cobalt compounds crystallographically characterized for electronic structure elucidation are presented in Table 3. The crystallographic data establishes a planar cobalt dinitrogen complex with no close contacts between the cation and anion (see Supporting Information).
Figure 4.

Solid state molecular structure of [(iPrCNC)CoN2][BArF4] at 30% probability ellipsoids. Hydrogen atoms, one molecule of pentane and BArF4 anion were omitted for clarity. See Supporting Information for full structural representation.
Table 3. Selected bond distances (Å) for (iPrCNC), [(iPrCNC)CoCH3][BArF4], [(iPrCNC)CoN2][BArF4], (iPrCNC)CoCl, (iPrCNC)CoBr and (iPrCNC)CoCH3.
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| (iPrCNC)a | (iPrCNC)CoMe+ | (iPrCNC)CoN2+ | (iPrCNC)CoCl | (iPrCNC)CoBrb | (iPrCNC)CoMeb | |
| Co–Npyr | 1.9193(13) | 1.871(2) | 1.832(2) | 1.840(5) | 1.865(3) | |
| Co–Ccarbene | 1.9479(17) | 1.913(3) | 1.904(3) | 1.918(6) | 1.914(4) | |
| 1.9432(17) | 1.903(3) | 1.907(3) | 1.909(6) | 1.898(4) | ||
| Co–X | 1.9542(18) | 2.2004(8) | 2.3169(8) | 1.951(4) | ||
| Co–N2 | 1.801(3) | |||||
| Npyr–Cipso | 1.327(4) | 1.333(2) | 1.340(4) | 1.357(4) | 1.353(7) | 1.358(5) |
| 1.345(4) | 1.335(2) | 1.339(4) | 1.369(4) | 1.371(7) | 1.357(5) | |
| Cipso–Cm-pyr | 1.362(5) | 1.382(3) | 1.375(4) | 1.367(4) | 1.379(7) | 1.373(7) |
| 1.388(5) | 1.381(2) | 1.383(4) | 1.371(5) | 1.378(8) | 1.374(6) | |
| Cm-pyr–Cp-pyr | 1.387(5) | 1.388(3) | 1.384(5) | 1.399(5) | 1.397(9) | 1.397(7) |
| 1.392(5) | 1.389(3) | 1.389(5) | 1.392(5) | 1.410(9) | 1.416(6) | |
| Cipso–Ncarbene | 1.413(4) | 1.399(2) | 1.396(4) | 1.393(4) | 1.386(7) | 1.394(5) |
| 1.427(4) | 1.403(2) | 1.404(4) | 1.400(4) | 1.387(7) | 1.397(5) | |
| Ncarbene–Ccarbene | 1.372(4) | 1.384(2) | 1.373(3) | 1.383(4) | 1.388(6) | 1.399(5) |
| 1.378(4) | 1.382(2) | 1.379(4) | 1.380(4) | 1.384(7) | 1.400(5) | |
| Ccarbene–Naryl | 1.349(4) | 1.346(2) | 1.352(4) | 1.364(4) | 1.360(7) | 1.375(6) |
| 1.359(4) | 1.347(2) | 1.348(4) | 1.366(4) | 1.364(7) | 1.386(6) | |
Data taken from reference 56.
Data taken from reference 40.
 |
(3) |
A second example of a compound expected to have a neutral, redox innocent bis(arylimidazol-2-ylidene)pyridine was [(iPrCNC)CoCH3][BArF4]. Oxidation of (iPrCNC)CoCH3 with [Cp2Fe][BArF4] in fluorobenzene furnished blue-green crystals identified as the desired cobalt methyl cation, [(iPrCNC)CoCH3][BArF4] in 15% isolated yield (eq 4).
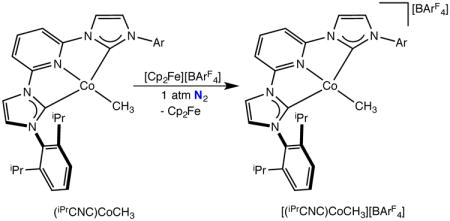 |
(4) |
A solution (Evans method) magnetic moment of 1.8(5) μB was measured at 22 °C, establishing an S = 1/2 ground state. Accordingly, the EPR spectrum (Figure 5) recorded on a solid powder sample at 10 K exhibits a rhombic signal, consistent with the observed doublet state. The spectrum was successfully simulated assuming g values of gx = 3.36, gy = 2.43, gz = 1.85 and hyperfine coupling to 59Co (100%, I = 7/2, Axx = 1062 MHz, Ayy = 581 MHz, Azz = 494 MHz). The remarkable anisotropy of the g tensor and the large hyperfine coupling constants demonstrate a cobalt-centered spin consistent with previous reports on the EPR spectra of square-planar Co(II) complexes.66
Figure 5.

Solid state powder EPR spectrum of [(iPrCNC)CoCH3][BArF4] recorded at 10 K. Conditions: microwave frequency = 9.3825 GHz, power = 0.5 mW, modulation amplitude = 1.0 mT/100 kHz;
The solid-state structure was determined by X-ray diffraction and a representation of the molecule is presented in Figure 6. The crystallographic data confirm the identity of the compound as [(iPrCNC)CoCH3][BArF4] and there are no close contacts between the cation and anion (see Supporting Information). The sum of the angles about the metal center is 359.96(8)°, indicating a idealized planar compound.
Figure 6.

Solid state molecular structure of [(iPrCNC)CoCH3][BArF4] at 30% probability ellipsoids. Hydrogen atoms and BArF4 anion were omitted for clarity. See Supporting Information for full structure representation.
Examples of compounds with the potential of adopting one-electron reduced chelates were of particular interest to determine whether bis(arylimidazol-2-ylidene)pyridines are redox active. Previous studies with bis(imino)pyridine compounds identified (iPrPDI)CoN2 as a low-spin Co(I), d8 compound with a one-electron reduced chelate accounting for the observed S = 1/2 ground state.60 The toluene solution EPR spectrum of the compound recorded at 298 K experimentally verified the ligand-centered radical as the SOMO of the molecule thereby providing definitive evidence for the redox-activity of this ligand class. Accordingly, the bis(arylimidazol-2-ylidene)pyridine analog, (iPrCNC)CoN2 was targeted for synthesis and electronic structure determination.
Attempts to prepare the desired neutral cobalt dinitrogen compound by reduction of (iPrCNC)CoBr with sodium amalgam yielded a mixture of cobalt products with (4-H2-iPrCNC)CoN2 as the only identifiable compound. Performing the reduction with sodium metal in the presence of 5 mol% of naphthalene also furnished a mixture of products from which (4-H2-iPrCNC)CoN2 and Na[iPrCNC] were identified. By contrast, stirring a toluene or benzene solution of [(iPrCNC)CoN2][BArF4] in the presence of one equivalent of (η5-C5Me5)2Co for 10 minutes yielded a blue-green solution. Monitoring the reaction by infrared spectroscopy established that all of the [(iPrCNC)CoN2][BArF4] is consumed with concomitant appearance of a band centered at 2047 cm−1, signaling formation of the neutral cobalt dinitrogen complex, (iPrCNC)CoN2 (eq 5).
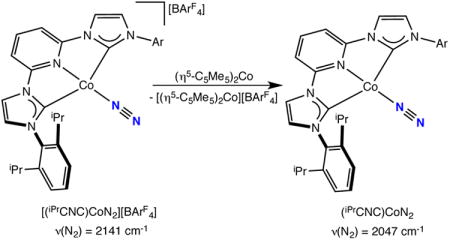 |
(5) |
Analysis of the in situ generated product by EPR spectroscopy in benzene solution at 298 K established an S = 1/2 compound, similar to (iPrPDI)CoN2. The observed isotropic signal at giso = 1.998 exhibits eight partially overlapped hyperfine lines, arising from coupling of the principally ligand-centered radical to the I = 7/2 59Co nucleus (Figure 7). Simulation of the data established an isotropic hyperfine coupling constant, Aiso = 36 MHz. No hyperfine coupling to hydrogen or nitrogen atoms was observed, which can be attributed to increased line broadening due to coordination of the ligand radical to the cobalt center, as observed for the iPrPDI analog.60 The Aiso is similar to that observed in previously reported (iPrPDI)CoN2 (24 MHz), supporting an electronic structure description of a low-spin Co(I) ion with a [iPrCNC]1− chelate radical anion. Unfortunately, (iPrCNC)CoN2 underwent rapid decomposition in solution to an unidentified S = 1/2 cobalt compound. The spectrum was simulated with the presence of an S = 1/2 species centered at giso = 2.00 at 40% ratio. A time course of the decomposition as measured by EPR spectroscopy is reported in the Supporting Information.
Figure 7.

Benzene solution EPR spectrum of (iPrCNC)CoN2 recorded at 298 K. Conditions: microwave frequency = 9.3735 GHz, power = 20.0 mW, modulation amplitude = 0.10 mT/100 KHz. Simulation was performed with the presence of an unidentified S = 1/2 species at g = 2.000. The relative ratio of (iPrCNC)CoN2 and the unidentified species in this spectrum is approximately 3:2. See Supporting Information for subspectra from the simulation and decomposition time course.
C. Analysis of the Metrical Parameters of Structurally Characterized Bis(arylimidazol-2-ylidene)pyridine Cobalt Complexes
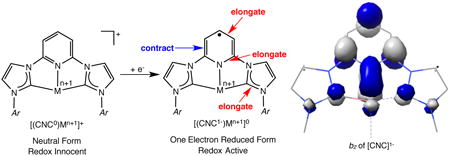
Distortions to the metrical parameters of a chelating ligand has been established as a diagnostic tool for establishing redox activity.67,68 Supporting these observations with computational and spectroscopic studies is useful for determining the spectroscopic oxidation state of redox-active transition metal compounds. Because several bis(arylimidazol-2-ylidene)pyridine cobalt complexes have now been structurally characterized, meaningful analysis can be carried out to establish the bond distances associated with the neutral and one electron reduced forms of the ligand. As will be presented in a subsequent section of the manuscript, the established ranges have also been computationally verified.
Selected bond distances for structurally characterized bis(arylimidazol-2-ylidene)pyridine cobalt complexes are presented in Table 3. Also presented in Table 3 are the metrical parameters of the free chelate as a reference and comparison. Computational studies (vide infra) and resonance structures allow prediction of the distortions to the chelate. Illustrated in the table is the DFT computed b2 molecular orbital of the bis(arylimidazol-2-ylidene)pyridine radical anion that highlights the expected bond elogations and contractions. Reduction of the bis(arylimidazol-2-ylidene)pyridine pincer results in elongations and contractions to the central pyridine ring as well as an elongation of Ccarbene-Nimid distances. Within the pyridine ring, the Npyr-Cipso and Cmeta-Cpara distances are expected to elongate upon addition of an electron while Cipso -Cmeta bonds contract.
In the free bis(arylimidazol-2-ylidene)pyridine (iPrCNC), [(iPrCNC)CoCH3][BArF4], and [(iPrCNC)CoN2][BArF4], the Npyr-Cipso distances range between 1.327(4) and 1.345(5) Å. In the cobalt monohalide and methyl complexes, this length range expands to 1.353(7) to 1.371(7) Å, consistent with one electron reduction of the chelate. The Cmeta-Cpara distances also exhibit distortions, albeit slightly less pronounced, ranging between 1.384(5)-1.392(5) Å in the redox innocent compounds that elongate to 1.392(5)-1.416(6) Å in the (iPrCNC1−)CoIIX derivatives. The anticipated contraction of the Cipso-Cmeta distances is apparent but less definitive. In the redox innocent compounds, values range between 1.362(5)-1.388(5) Å and are altered to 1.367(4)-1.378(8) Å in the (iPrCNC1−)CoIIX examples. It should be noted that the lower limit of the redox innocent range is skewed by one anomalous value in the structure of the free bis(arylimidazol-2-ylidene)pyridine. If this value is discarded, the range for the neutral ligand becomes 1.375(4)-1.388(5) Å. The Ccarbene-Naryl distances also exhibit distortions, ranging between 1.346(2)-1.359(4) Å in redox innocent compounds and elongate to 1.360(7)-1.386(6) Å in the reduced chelate. The distortions in bond distances was also anticipated from examination of the calculated b2 orbital of [CNC]1− chelate, which clearly establishes anti-bonding character at elongated positions. Taken together, the crystallographic data clearly establish that the (iPrCNC1−)CoIIX family of compounds, much like their bis(imino)pyridine counterparts, contain radical bis(arylimidazol-2-ylidene)pyridine chelates and low-spin cobalt(II) centers.
D. X-ray Absorption Studies
The single crystal X-ray diffraction data for both (iPrCNC)CoBr and (iPrCNC)CoCH3 are consistent with low-spin Co(II) complexes with bis(arylimidazol-2- ylidene)pyridine radical anions. To further support this view of the electronic structure, X-ray absorption spectroscopic (XAS) studies were conducted (Figure 8). This study also included (iPrPDI1−)CoIIBr and (iPrPDI1−)CoIICH3, established low-spin Co(II) compounds with chelate radical anions. The rising edge and pre-edge region of all four compounds fall in a similar energy range (Table 4), supporting the same cobalt oxidation state for all members of the series. For (iPrCNC)CoBr and (iPrPDI)CoCl, the first pre-edge features appear at 7709.8 and 7709.3 eV, respectively, values consistent with those previously reported for Co(II) compounds.69 The 0.5 eV shift for the bis(arylimidazol-2-ylidene)pyridine derivative is likely due to the greater π-acidity of this chelate relative to the bis(imino)pyridine. The replacement of Cl by Br in the first coordination sphere, however, may also contribute to a decrease in edge energy. Likewise, the pre-edges for (iPrCNC)CoCH3 and (iPrPDI)CoCH3 were observed at 7709.7 eV, and at 7709.3 eV, respectively. Both of the bis(arylimidazol-2-ylidene)pyridine cobalt compounds exhibit an additional pre-edge feature at slightly higher energy, likely due to a 1s to ligand π* transition.70,71 Collectively, the XAS results support low-spin cobalt(II) compounds with bis(arylimidazol-2-ylidene)pyridine radical anions.
Figure 8.

Normalized Co K-edge X-ray absorption spectra of the rising edge (left) and pre-edge (right) of (iPrCNC)CoBr and (iPrCNC)CoCH3 recorded at 10 K. Also included are the data for (iPrPDI)CoCl and (iPrPDI)CoCH3.
Table 4. Cobalt K-edge parameters for bis(arylimidazol-2-ylidene)pyridine and bis(imino)pyridine cobalt halide and methyl compounds.
| Compound | Pre-edge Energy (eV) | Pre-edge Intensity Aa | Pre-edge Intensity Bb | Temperature (K) |
|---|---|---|---|---|
| (iPrCNC)CoBr | 7709.8 | 33(3) | 42(3) | 10 |
| 7711.8 | 17(4) | 7(1) | 10 | |
| (iPrCNC)CoCH3 | 7709.7 | 29.8(6) | 37.6(8) | 10 |
| 7711.2 | 5.8(8) | 7(2) | 10 | |
| (iPrPDI)CoCl | 7709.3 | 14.1(9) | 18(1) | 10 |
| (iPrPDI)CoCH3 | 7709.3 | 13(1) | 16(2) | 10 |
Fit using method described in reference 72.
Fit using method described in reference 73.
E. 1H NMR Spectroscopic Studies
The NMR spectra of diamagnetic transition metal complexes bearing redox-active ligands has proven valuable in determination of their electronic structure. Budzelaar and coworkers52,74 and subsequently our laboratory60,70,75 have demonstrated the utility of 1H NMR chemical shifts of the in-plane hydrogens of bis(imino)pyridine chelates to be diagnostic of radical chemistry and participation in the overall electronic structure. Deviations of the in-plane chelate hydrogens from their diamagnetic reference values have been attributed to either thermal population of a triplet excited state52 or from temperature independent paramagnetism.75
Because of the simplicity and insightfulness of the technique, the 1H NMR spectrum of (iPrCNC)CoCH3 was examined in detail. As reported by Danopoulos,40 the benzene-d6 1H NMR spectrum of (iPrCNC)CoCH3 at 22 °C exhibits a downfield shifted resonance at 10.65 ppm for the para-pyridine proton. This shift is similar to the value of 10.19 ppm reported for (iPrPDI)CoCH3.76 The spectra of the two compounds are presented in Figure 9.
Figure 9.

Benzene-d6 1H NMR spectra of (iPrPDI)CoCH3 (top) and (iPrCNC)CoCH3 (bottom). The data for (iPrPDI)CoCH3 was previously reported in ref. 76 and (iPrCNC)CoCH3 in ref. 40.
The similarity in the 1H NMR shifts of (iPrCNC)CoCH3 and (iPrPDI)CoCH3 are consistent with the other spectroscopic and metrical data that indicate the former is a low spin cobalt(II) compound with a bis(arylimidazol-2-ylidene)pyridine radical anion. Because thermal population of a triplet excited state has been invoked to explain the anomalous chemical shifts of (iPrPDI)CoCH3,52 variable temperature 1H NMR experiments were performed on a series of (iPrCNC)CoX (Br, CH3, H) compounds in toluene-d8. Stack plots of the spectra are reported in the Supporting Information. For both (iPrCNC)CoBr and(iPrCNC)CoCH3, the chemical shifts are invariant with temperature, changing < 0.1 ppm from -70 to 22 °C. Thus, no change in population of the triplet state was detectable by 1H NMR spectroscopy. The hydride compound, (iPrCNC)CoH is anomalous. Almost all of the resonances are invariant with temperature with the exception of the para-pyridine, which broadens significantly from −40 °C to 22 °C. This broadening is accompanied by an upfield shift of approximately 0.5 ppm upon warming. Recall, this compound undergoes migration of the metal-hydride to the 4-position of the pyridine and exchange with free H2 and thus we are unable to distinguish the changes in the NMR spectrum resulting from a chemical process or an unusual electronic structure.
The observation of temperature independent chemical shifts in the bis(arylimidazol-2-ylidene)pyridine cobalt bromide and methyl compounds raised the question of similar behavior in (iPrPDI)CoCH3. To our knowledge, the variable temperature NMR data for this compound has not been reported. Over the temperature range of −70 to 40 °C, no significant change in chemical shifts was observed (see Supporting Information for stack plots). Budzelaar and coworkers previously calculated a difference of 7.2 kcal for the energy between the singlet and triplet states for (iPrPDI)CoCH3.52 Assuming a Boltzman distribution between the states, the abundance of the triplet state at 22 °C is 4.7 × 10−4%. Cooling the sample to −70 °C, decreases this value to 1.8 × 10−6% while warming to 40 °C increases the population 9.4 × 10−4%. That is to say the triplet state of (iPrPDI)CoCH3 is 260 times more abundant at 22 °C than −70 °C and 526 times more abundant at 40 °C than −70 °C.
If thermal population of a triplet excited state is indeed the origin of the anomalous 1H NMR shifts observed at 22 °C, a 4.7 × 10−4% population of the triplet excited state is responsible for shifting the imine methyl groups approximately 3-4 ppm from their diamagnetic reference values. Cooling the sample should decrease this population significantly and likewise induce a significant shift of the imine methyl group and other in plane hydrogens toward their diamagnetic reference values. Similarly, heating the sample should result in a greater dispersion of these chemical shifts. Because no change was observed over a 110 °C temperature range, we conclude that thermal population of a triplet excited state is not the origin of the anomalous shifts. The more likely explanation is temperature independent paramagnetism, whereby a triplet excited state mixes into the singlet ground state by spin orbit coupling as the resulting 1H NMR chemical shifts are invariant with temperature. Such behavior has been observed in isoelectronic, neutral ligand complexes of bis(imino)pyridine iron.75,77
F. Computational Studies
Broken-symmetry78 DFT computational studies were conducted to corroborate the experimental results and firmly establish the electronic structures of various bis(arylimidazol-2-ylidene)pyridine cobalt compounds. Full molecule calculations and geometry optimizations were conducted in each case at the B3LYP level of DFT. Broken symmetry calculations were performed to describe the open shell singlet configuration of the redox-active cobalt compounds. In the broken symmetry notation BS(m, n) describes a state in which there are m unpaired spin-up electrons and n unpaired spin-down electrons on separate fragments.78,79,80
The first class of compounds examined were those expected to have neutral bis(arylimidazol-2-ylidene)pyridine chelates. The two cationic cobalt complexes, [(iPrCNC)CoN2]+ and [(iPrCNC)CoCH3]+ were initially explored and only the cationic portion of the molecule was calculated with S = 0 and S = 1/2 ground states, respectively. The crystallographic data suggest no participation of the chelate in the electronic structure of the compounds. A comparison between the experimental and computational bond distances for both compounds is reported in the Supporting Information.
Calculations on [(iPrCNC)CoN2]+ were performed using both BS(1,1) and Restricted Kohn-Sham (RKS) formalisms. The BS(1,1) input converged to the close-shell solution identical to the results from the RKS input. A truncated, qualitative molecular orbital diagram is presented in Figure 10 and clearly establishes a low-spin Co(I) complex with a neutral, redox-innocent bis(arylimidazol-2-ylidene)pyridine, consistent with the experimental data. The highest occupied molecular orbital is exclusively cobalt dz2, as expected for a square planar, d8 metal complex. To provide additional support for the accuracy of the DFT result, a vibrational frequency calculation of the N2 stretch was performed. This calculation was carried out with the BP86 functional for computational expediency. This calculation predicts the N2 stretching frequency at 2173 cm−1, in reasonable agreement with the experimental value of 2141 cm−1. Thus both the experimental and computational data support [(iPrCNC0)CoIN2]+ as the best description of the electronic structure.
Figure 10.

Qualitative molecular orbital diagram for [(iPrPDICoN2]+ from a geometry optimized B3LYP calculation.
Computations were also performed on [(iPrCNC)CoCH3]+ using a doublet ground state as established experimentally. Applying a spin-unrestricted formalism, the calculations converged to an open-shell solution and a comparison of computed and experimental bond distances are reported in the Supporting Information. Presented in Figure 11 is a truncated qualitative molecular orbital diagram along with a Mulliken spin density plot. The computational results clearly support a low-spin Co(II) complex with a redox neutral bis(arylimidazol-2-ylidene)pyridine. The ligand oxidation state is similar to that observed with [(iPrCNC)CoN2]+. For [(iPrCNC)CoCH3]+, the SOMO of the molecule is a cobalt dz2 molecular orbital, consistent with the observed EPR spectrum for the compound.
Figure 11.

Qualitative molecular orbital diagram (left) for [(iPrCNC)CoCH3]+ from an UKS calculation at B3LYP level. Spin density plot (right) was obtained from Mulliken population analysis (red, positive spin density; yellow, negative spin density).
Having corroborated the electronic structures of compounds with neutral bis(arylimidazol-2-ylidene)pyridines, cobalt complexes with one electron reduced chelates (i.e. (iPrCNC)CoX) were studied. Geometry optimizations were performed with RKS and BS(1,1) inputs at the B3LYP level of DFT. The BS(1,1) solution was found to be 7 kcal/mol lower in energy than the RKS alternative, supporting a redox-active description. The metrical parameters from both calculations are in excellent agreement with the experimentally determined values and are summarized in the Supporting Information.
Examination of the computed molecular orbitals from the open shell singlet solution (Figure 12) revealed a low spin Co(II) complex with three doubly occupied metal d orbitals and a SOMO of primarily dxz parentage. The SOMO is engaged in an antiferromagnetic coupling interaction (S = 0.58) with an iPrCNC b2 molecular orbital. Examination of the Mulliken spin population analysis also corroborates this view of the electronic structure.
Figure 12.

Qualitative molecular orbital diagram (left) for (iPrCNC)CoBr from a BS(1,1) calculation at B3LYP level. Spin density plot (right) was obtained from Mulliken population analysis (red, positive spin density; yellow, negative spin density).
Similar studies were also conducted on the cobalt methyl complex, (iPrCNC)CoCH3. As with (iPrCNC)CoBr, a BS(1,1) solution was preferred corresponding to a low-spin cobalt(II) complex and a bis(arylimidazol-2-ylidene)pyridine radical anion. A truncated, qualitative molecular orbital diagram and Mulliken spin population analysis are presented in Figure 13. Three doubly occupied metal d orbitals and a SOMO of primarily dxz character are observed. The SOMO is engaged in an antiferromagnetic coupling interaction (S = 0.48) with an iPrCNC b2 molecular orbital. As with the bromide, the ligand-centered radical is largely pyridine localized.
Figure 13.

Qualitative molecular orbital diagram for (iPrCNC)CoCH3 obtained from a BS(1,1) DFT calculation at the B3LYP level of DFT (left). Spin density plot for (iPrCNC)CoCH3 was obtained from a Mulliken population analysis (right).
Presented in Figure 14 is a comparison of the Mulliken spin density of bis(imino)pyridine versus bis(arylimidazol-2-ylidene)pyridine cobalt methyl complexes. It is notable that the radical character on the bis(arylimidazol-2-ylidene)pyridine chelate is largely localized in the pyridine ring with little contribution from the N-heterocyclic carbenes. This feature likely accounts for the observed reactivity where metal-hydrides and stabilized alkyl radicals migrate to the 4-position of the ring. In bis(imino)pyridine compounds, significant delocalization over the entire π-system of the chelate is observed and hence analogous migration processes have not been observed in reduced cobalt chemistry (Figure 14).
Figure 14.

Comparison of the spin density plots of (iPrPDI)CoCH3 (left) and (iPrCNC)CoCH3 (right).
One final set of time dependent DFT calculations were conducted, using methods previously described,81 to further understand the observed XAS spectra. Using 50 roots (Figure 15), both closed-shell (RKS) and BS(1,1) inputs were evaluated for (iPrCNC)CoCH3 to determine whether or not XAS could distinguish redox innocent and redox active possibilities. The closed shell solution resulted in no calculated pre-edge transition while the BS(1,1) solution reproduced one pre-edge transition dominated by a transition to the cobalt dxz orbital. These further corroborate the BS(1,1) electronic structure description for (iPrCNC)CoCH3 and firmly establish bis(arylimidazol-2-ylidene)pyridine as a redox-active ligand.
Figure 15.

Calculated XAS spectra of (iPrCNC)CoBr and (iPrCNC)CoCH3 using the BP86 functional. A broadening of 1.5 eV has been applied to the computed spectra which have not been energy normalized.
IV. Evaluation of the Catalytic Alkene Hydrogenation Activity of Other Bis(arylimidazol-2-ylidene)pyridine Cobalt Precursors
Determination of the electronic structure of various bis(arylimidazol-2-ylidene)pyridine cobalt compounds prompted evaluation of their catalytic alkene hydrogenation activity. Such studies may ultimately allow for a correlation between redox activity of the supporting ligand and catalytic performance. The hydrogenation of trans-methylstilbene was used as a representative catalytic reaction. Each trial was conducted with 5 mol% of the metal complex in a 0.916 M solution of the substrate in the desired solvent under 4 atm of H2 at 22 °C for one hour. The results of these studies are reported in Table 5.
Table 5. Comparison of various bis(arylimidazol-2-ylidene)pyridine cobalt precursors for the catalytic for the hydrogenation of trans-methylstilbene.
| Cobalt Compound | Solvent | Conversiona,b |
|---|---|---|
| (iPrCNC)CoCH3 | C6H6 | 50% |
| (iPrCNC)CoCH3 | C6H5F | 14% |
| (4-H2-iPrCNC)CoN2 | C6H6 | 27% |
| [(iPrCNC)CoN2][BArF4]c | C6H6 | 27% |
| [(iPrCNC)CoN2][BArF4] | C6H5F | 6% |
All catalytic reactions carried out with 5 mol% of the metal complex (0.032 mmol) in 0.916 M substrate solution in the desired solvents with 4 atm of H2 at 22 °C.
Conversions determined by GC-FID.
Generated in situ using (iPrCNC)CoBr and 1.05 eq. NaBArF4.
Because of the poor solubility of [(iPrCNC)CoN2][BArF4] in hydrocarbon solvents such as benzene, the catalytic performance of the compound was initially evaluated in fluorobenzene. Only 6% conversion to alkane was observed under these conditions. For comparison, the catalytic activity of (iPrCNC)CoCH3 was also evaluated in fluorobenzene and the conversion was reduced to 14% as compared to 50% in benzene. Accordingly, a method was developed to generate [(iPrCNC)CoN2][BArF4] in situ in hydrocarbon solvents. A benzene solution of (iPrCNC)CoBr was stirred with a slight excess of NaBArF4 and the catalytic hydrogenation of trans-methyl stilbene was conducted under standard conditions. Using this method, an improved conversion of 27% was observed after one hour but the observed activity remains inferior to the neutral cobalt methyl compound, (iPrCNC)CoCH3. One final example, the neutral cobalt(I) dinitrogen compound, (4-H2-iPrCNC)CoN2 was studied. Only 27% conversion to alkane was observed after one hour under standard conditions, again inferior to the neutral cobalt methyl compound. While it is tempting to correlate electronic structure with catalytic performance, more detailed studies are required to make such assertions as the kinetics of the reaction, number of active sites and stability of the active species may be the origin of the observed differences.
Concluding Remarks
The bis(arylimidazol-2-ylidene)pyridine cobalt methyl complex, (iPrCNC)CoCH3, was found to be one of the most active base metal catalysts for the hydrogenation of unactivated, sterically hindered olefins such as trans-methylstilbene, 1-methyl-1-cyclohexene and 2,3-dimethyl-2-butene. Spectroscopic observation of the catalytically relevant hydride compound, (iPrCNC)CoH established hydride migration to the 4-position of the pyridine ring of the chelate. This reactivity was extended to alkyls upon addition of 1,1-diphenylethylene to (iPrCNC)CoH. The chemical participation of the ligand signaled the possibility of ligand radicals in the electronic structure of (iPrCNC)CoX (X = halide, alkyl, H) compounds. Accordingly, a combination of structural, spectroscopic and computational studies established bis(arylimidazol-2-ylidene)pyridine chelates as redox-active, undergoing one electron reduction in reduced cobalt chemistry. Unlike redox-active bis(imino)pyridines which have a more extended π system, the ligand-centered radical in the bis(arylimidazol-2-ylidene)pyridine is essentially pyridine localized, likely contributing to the observed hydride and alkyl migration chemistry. These studies establish the utility of bis(arylimidazol-2-ylidene)pyridines as excellent support ligands in base metal catalysis and also adds these pincers to the growing number of examples of redox-active tridentate chelates.82 These results also suggest that other pyridine-based pincer ligands may be redox-active when coordinated to an appropriately reducing bas metal. This question and its broader impact on base metal catalysis are currently under scrutiny in our laboratory.
Experimental Section83
Preparation of (4-CPh2CH3-iPrCNC)CoN2
A thick-walled glass vessel was charged with 0.100 g (0.160 mmol) of (iPrCNC)CoCH3 and approximately 10 mL of toluene. The vessel was submerged in a -78 °C bath and the headspace was evacuated and replaced with an atmosphere of H2. The contents of the vessel were warmed to ambient temperature and stirred for 15 minutes. The excess dihydrogen was removed at −78 °C and 0.030 g (0.170 mmol) of 1,1-diphenylethylene was added at 22 °C under an atmosphere of dinitrogen. A rapid color change to deep blue was observed and the reaction mixture was stirred for an additional 15 minutes. The solution was then filtered through Celite and concentrated to dryness. The residue was dissolved in diethyl ether and stored at −35 °C to afford 0.082 g (0.010 mmol, 62% yield) of blue solid identified as (4-CPh2CH3-iPrCNC)CoN2. Analysis for C49H54N7Co: Calc. C, 73.57; H, 6.80; N, 12.26. Found: C, 73.52; H, 6.66; N, 11.93. 1H NMR (benzene-d6, 22 °C): δ = 0.99 (d, 7 Hz, 6H, CH(CH3)2), 1.03 (d, 7 Hz, 6H, CH(CH3)2), 1.37 (d, 7 Hz, 12H, CH(CH3)2), 1. 77 (s, 3H, Ph2CCH3), 2.87 (spt, 7 Hz, 2H, CH(CH3)2), 2.91 (spt, 7 Hz, 2H, CH(CH3)2), 4.33 (d, 4 Hz, 2H, vinyl H), 4.90 (t, 4 Hz, 1H, allyl H), 6.03 (d, 2 Hz, 2H, imidazolylidene backbone), 6.30 (d, 2 Hz, 2H, imidazolylidene backbone), 6.96 (d, 7 Hz, 4H, aryl or phenyl), 7.06 (t, 7 Hz, 4H, aryl or phenyl), 7.18 (d, 7 Hz, 4H, aryl or phenyl), 7.35 (d, 7 Hz, 4H, aryl or phenyl). 13C {1H} NMR (benzene-d6, 22 °C): δ = 22.4 (Ph2CCH3), 23.9 (CH(CH3)2), 24.0 (CH(CH3)2), 24.4 (CH(CH3)2), 24.5 (CH(CH3)2), 28.5 (CH(CH3)2), 28.6 (CH(CH3)2), 45.2 (allyl), 53.1 (Ph2CCH3), 77.5 (vinyl), 113.3 (imidazolylidene backbone), 123.0 (imidazolylidene backbone), 123.7 (aryl), 123.8 (phenyl), 125.6 (aryl), 128.3 (phenyl), 129.8 (phenyl), 136.0 (aryl), 145.9 (aryl or 2-pyr), 146.23 (aryl or 2-pyr), 146.25 (aryl or 2-pyr), 149.0 (phenyl), 194.1 (carbene), one resonance not located. IR (benzene-d6, 22 °C): ν(N2) = 2050 cm−1
Preparation of [(iPrCNC)CoCH3][BArF4] (BArF4 = B(3,5-(CF3)2C6H3)4)
A 20 mL scintillation vial equipped with a magnetic stir bar was charged with (iPrCNC)CoCH3 (0.036 g, 0.059 mmol, 1 equiv.) and approximately 2 mL of fluorobenzene. The solution was chilled in a -35 °C freezer for 10 minutes, then a diethyl ether solution of [Cp2Fe][BArF4] (0.062 g, 0.059 mmol, 1 equiv., in 1 mL diethyl ether) was added dropwise with rapid magnetic stirring. An immediate color change from yellowish-brown to blue-green was observed. The mixture was stirred at 22 °C for 5 minutes, after which the mixture was filtered through Celite and subsequently stored at −15 °C. Over a period of 2 hours, a brown-colored oil condensed on the bottom of the glass container and was separated from the supernatant by carefully decanting the solution into a clean 20-mL scintillation vial. After 3 repeated decanting processes over a period of 6 hours, the solution eventually resumed a teal color, with no further formation of a brown oil. Layering the solution with pentane (∼ 5 mL) and storing the mixture at −15 °C resulted formation of bright green crystals identified as [(iprCNC)CoCH3][BArF4] (0.013 g, 0.009 mmol, 15% yield). Once crystalline, the solid was no longer soluble in benzene, toluene or light hydrocarbons but was highly soluble in diethyl ether and fluorobenzene. Analysis for C68H56BCoF24N5: Calc. C, 55.60; H, 3.82; N, 4.77. Found: C, 55.45; H, 4.11; N, 4.49. μeff (Evans method, 22 °C) = 1.8(5) μB. 1H NMR (benzene-d6, 22 °C): δ = -9.98 (182 Hz, 12H, CH(CH3)2), 4.39 (22 Hz, 12H, CH(CH3)2), 6.57 (12 Hz, 4H, m-BArF4), 7.85 (9 Hz, 10H, o-BArF4 and p-aryl), 8.84 (20 Hz, 4H, CH(CH3)2), 9.35 (17 Hz, 2H, 3-pyr), 12.64 (474 Hz, 3H, CoCH3), 15.61 (76 Hz, 2H, imidazolylidene backbone), 16.07 (94 Hz, 2H, imidazolylidene backbone), 26.25 (33 Hz, 1H, 4-pyr), m-aryl not located.
Supplementary Material
Acknowledgments
PJC thanks the U. S. National Science Foundation and Deutsche Forschungsgemeinschaft for a Cooperative Activities in Chemistry between U. S. and German Investigators grant (CHE-1026084). JMD also acknowledges support from the National Institute of General Medical Sciences (Award Number T32GM008500). SCES thanks NSF for a graduate research fellowship (DGE-0646086) for support. Portions of the research were carried out at the Stanford Synchrotron Radiation Lightsource, a national user facility operated by Stanford University on behalf of the DOE, BES. The SSRL SMB Program is supported by DOE, BER and NIH, NCRR, BMTP. The simulations presented in this article were performed on computational resources supported by the Princeton Institute for Computational Science and Engineering (PICSciE) and the Office of Information Technology's High Performance Computing Center and Visualization Laboratory at Princeton University. We also thank Scott Semproni for assistance with X-ray diffraction.
Footnotes
Supporting Information Available. Crystallographic data for (iPrCNC)CoCl, (4-CPh2CH3- iPrCNC)CoN2, [(iPrCNC)CoN2][BArF4] and [(iPrCNC)CoCH3] [BArF4] in cif format. This material can be downloaded, free of charge, via the internet at http://pubs.acs.org.
References
- 1.Hartwig J. Organotransition Metal Chemistry: From Bonding to Catalysis. University Science Books; Sausalito, California: 2010. p. 575. [Google Scholar]
- 2.(a) Chirik PJ, Wieghardt K. Science. 2010;327:794. doi: 10.1126/science.1183281. [DOI] [PubMed] [Google Scholar]; (b) Enthaler S, Junge K, Beller M. Angew Chem Int Ed. 2008;47:3317. doi: 10.1002/anie.200800012. [DOI] [PubMed] [Google Scholar]; (c) Nakazawa H, Itazaki M. Top Organomet Chem. 2011;33:27. [Google Scholar]; (d) Junge K, Schröder K, Beller M. Chem Commun. 2011;47:4849. doi: 10.1039/c0cc05733a. [DOI] [PubMed] [Google Scholar]
- 3.(a) Bolm C, Legros J, Paith JL, Zani L. Chem Rev. 2004;104:6217. doi: 10.1021/cr040664h. [DOI] [PubMed] [Google Scholar]; (b) Bauer EB. Curr Org Chem. 2008;47:1341. [Google Scholar]; (c) Gaillard S, Renaud JL. ChemSusChem. 2008;1:505. doi: 10.1002/cssc.200800071. [DOI] [PubMed] [Google Scholar]; (d) Chirik PJ. In: Catalysis Without Precious Metals. Bullock RM, editor. Chapter 4 Wiley-VCH; Weinheim: 2010. [Google Scholar]
- 4.Crabtree RH. The Organometallic Chemistry of the Transition Metals. 4th. John Wiley & Sons; New York: 2005. [Google Scholar]
- 5.(a) DeAngelis F, Jin N, Car R, Groves JT. Inorg Chem. 2006;45:4268. doi: 10.1021/ic060306s. [DOI] [PubMed] [Google Scholar]; (b) Dhuri SN, Seo MS, Lee YM, Hirao H, Wang Y, Nam W, Shaik S. Angew Chem Int Ed. 2008;47:3356. doi: 10.1002/anie.200705880. [DOI] [PubMed] [Google Scholar]; (c) Klinker EJ, Shaik S, Hirao H, Que L. Angew Chem Int Ed. 2009;48:1291. doi: 10.1002/anie.200804029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Volpe EC, Wolczanski PT, Lobkovsky EB. Organometallics. 2010;29:364. [Google Scholar]
- 7.(a) Frankel EN, Emken EA, Peters HM, Davison VK, Butterfield RO. J Org Chem. 1964;29:3292. [Google Scholar]; (b) Frankel EN, Emken EA, Davison VK. J Org Chem. 1965;30:2739. [Google Scholar]; (c) Wrighton M. Chem Rev. 1974;74:401. [Google Scholar]; (d) Schroeder MA, Wrighton MS. J Am Chem Soc. 1976;98:551. [Google Scholar]; (e) Mitchener JC, Wrighton MS. J Am Chem Soc. 1981;103:975. [Google Scholar]; (f) Wuu YM, Bentsen JG, Brinkley CG, Wrighton MS. Inorg Chem. 1987;26:530. [Google Scholar]
- 8.(a) Das S, Li YH, Junge K, Beller M. Chem Comm. 2012;48:10742. doi: 10.1039/c2cc32142d. [DOI] [PubMed] [Google Scholar]; (b) Fleisher S, Zhou SL, Junge K, Beller M. Chem Asian J. 2011;6:2240. doi: 10.1002/asia.201100462. [DOI] [PubMed] [Google Scholar]; (c) Gartner F, Boddien A, Barsch E, Fumino K, Losse S, Junge H, Hollmann D, Bruckner A, Ludwig R, Beller M. Chem Eur J. 2011;17:6425. doi: 10.1002/chem.201003564. [DOI] [PubMed] [Google Scholar]; (d) Boddien A, Loges B, Gartner F, Torborg C, Fumino K, Junge H, Ludwig R, Beller M. J Am Chem Soc. 2010;132:8294. doi: 10.1021/ja100925n. [DOI] [PubMed] [Google Scholar]; (e) Zhou SL, Addis D, Das S, Junge K, Beller M. Chem Commun. 2009;32:4883. doi: 10.1039/b910145d. [DOI] [PubMed] [Google Scholar]; (f) Shaikh NS, Enthaler S, Junge K, Beller M. Angew Chem Int Ed. 2008;47:2497. doi: 10.1002/anie.200705624. [DOI] [PubMed] [Google Scholar]
- 9.(a) Jørgensen CK. Coord Chem Rev. 1966;1:164. [Google Scholar]; (b) Pierpont CG. Coord Chem Rev. 2001;216:99. [Google Scholar]; (c) Evangelio E, Ruiz-Molina D. Eur J Inorg Chem. 2005:2957. [Google Scholar]; (d) Ray K, Petrenko T, Wieghardt K, Neese F. Dalton Trans. 2007:1552. doi: 10.1039/b700096k. [DOI] [PubMed] [Google Scholar]; (e) De Bruin B, Hetterscheid DGH, Koekkoek AJJ, Grutzmacher H. Prog Inorg Chem. 2007;55:247–354. [Google Scholar]
- 10.Chirik PJ. Inorg Chem. 2011;50:9737. doi: 10.1021/ic201881k. [DOI] [PubMed] [Google Scholar]
- 11.Luca OR, Crabtree RH. Chem Soc Rev. 2012;42:1440. doi: 10.1039/c2cs35228a. [DOI] [PubMed] [Google Scholar]
- 12.Darmon JM, Stieber SCE, Sylvester K, Fernandez I, Lobkovsky E, Semproni SP, Bill E, Wieghardt K, DeBeer S, Chirik PJ. J Am Chem Soc. 2012;134:17125. doi: 10.1021/ja306526d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Stubbe J, Van der Donk WA. Chem Rev. 1998;98:706. doi: 10.1021/cr980059c. [DOI] [PubMed] [Google Scholar]; (b) Jazdewski BA, Tolman WB. Coord Chem Rev. 2000;633:200–202. [Google Scholar]; (c) Lewis EA, Tolman WB. Chem Rev. 2004;104:1047. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]; (d) Kaim W, Schwederski B. Coord Chem Rev. 2010;254:1580. [Google Scholar]; (e) Rittle J, Green MT. Science. 2010;330:933. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 14.Lyaskovsky V, de Bruin B. ACS Catalysis. 2012;2:270. [Google Scholar]
- 15.Praneeth VKK, Ringenberg MR, Ward TR. Angew Chem Int Ed. 2012;51:10228. doi: 10.1002/anie.201204100. [DOI] [PubMed] [Google Scholar]
- 16.(a) Heyduk AF, Zarkesh RA, Nguyen AI. Inorg Chem. 2011;50:9849. doi: 10.1021/ic200911b. [DOI] [PubMed] [Google Scholar]; (b) Nguyen N, Zarkesh RA, Lacy DC, Thorson MK, Heyduk AF. Chem Sci. 2011;2:166. [Google Scholar]; (c) Zarkesh RA, Ziller JW, Heyduk AF. Angew Chem Int Ed. 2008;47:4715. doi: 10.1002/anie.200800812. [DOI] [PubMed] [Google Scholar]; (d) King ER, Hennessy ET, Betley TA. J Am Chem Soc. 2011;133:4917. doi: 10.1021/ja110066j. [DOI] [PubMed] [Google Scholar]
- 17.(a) Lippert CA, Riener K, Soper JD. Eur J Inorg Chem. 2012:554. [Google Scholar]; (b) Lippert CA, Hardcastle KI, Soper JD. Inorg Chem. 2011;50:9864. doi: 10.1021/ic200923q. [DOI] [PubMed] [Google Scholar]; (c) Smith AL, Hardcastle KI, Soper JD. J Am Chem Soc. 2010;132:14358. doi: 10.1021/ja106212w. [DOI] [PubMed] [Google Scholar]; (d) Lippert CA, Soper JD. Inorg Chem. 2010;49:3682. doi: 10.1021/ic9024684. [DOI] [PubMed] [Google Scholar]
- 18.Bart SC, Lobkovsky E, Chirik PJ. J Am Chem Soc. 2004;126:13794. doi: 10.1021/ja046753t. [DOI] [PubMed] [Google Scholar]
- 19.(a) Russell SK, Darmon JM, Lobkovsky E, Chirik PJ. Inorg Chem. 2010;49:2782. doi: 10.1021/ic902162z. [DOI] [PubMed] [Google Scholar]; (b) Archer AM, Bouwkamp MW, Cortez MP, Lobkovsky E, Chirik PJ. Organometallics. 2006;25:4269. [Google Scholar]; (c) Trovitch RJ, Lobkovsky E, Bouwkamp MW, Chirik PJ. Organometallics. 2008;27:6264. [Google Scholar]; (d) Trovitch RJ, Lobkovsky E, Bill E, Chirik PJ. Organometallics. 2008;27:1470. [Google Scholar]
- 20.(a) Tondreau AM, Atienza CCH, Weller KJ, Nye SA, Lewis KM, Delis JGP, Chirik PJ. Science. 2012;335:567. doi: 10.1126/science.1214451. [DOI] [PubMed] [Google Scholar]; (b) Atienza CCH, Tondreau AM, Turner ZR, Weller KJ, Lewis KM, Cruse RW, Nye SA, Boyer JL, Delis JGP, Chirik PJ. ACS Catal. 2012;2:2169. [Google Scholar]
- 21.For an additional examples of iron-catalyzed hydrosilylation see:; (a) Kamata K, Suzuki A, Nakai Y, Nakazawa H. Organometallics. 2012;31:3825. [Google Scholar]; (b) Tondreau AM, Atienza CCH, Darmon JM, Milsmann C, Hoyt HM, Weller KJ, Nye SA, Lewis KM, Boyer J, Delis JGP, Lobkovsky E, Chirik PJ. Organometallics. 2012;31:4886. [Google Scholar]; (c) Naumov RN, Itazaki M, Kamitani M, Nakazawa H. J Am Chem Soc. 2012;134:804. doi: 10.1021/ja209436s. [DOI] [PubMed] [Google Scholar]; (d) Wu JY, Stanzi BN, Ritter T. J Am Chem Soc. 2010;132:13214. doi: 10.1021/ja106853y. [DOI] [PubMed] [Google Scholar]
- 22.Bouwkamp MW, Bowman AC, Lobkovsky E, Chirik PJ. J Am Chem Soc. 2006;128:13340. doi: 10.1021/ja064711u. [DOI] [PubMed] [Google Scholar]
- 23.Russell SK, Lobkovsky E, Chirik PJ. J Am Chem Soc. 2011;133:8858. doi: 10.1021/ja202992p. [DOI] [PubMed] [Google Scholar]
- 24.Hoyt JM, Sylvester KT, Semproni SP, Chirik PJ. J Am Chem Soc. 2013;135:4862. doi: 10.1021/ja400895j. [DOI] [PubMed] [Google Scholar]
- 25.Obligacion JV, Chirik PJ. Org Lett. 2013;15:2680. doi: 10.1021/ol400990u. [DOI] [PubMed] [Google Scholar]
- 26.For other examples of iron-catalyzed hydroboration see:; (a) Zhang L, Peng DJ, Leng XB, Huang Z. Angew Chem Int Ed. 2013;52:3676. doi: 10.1002/anie.201210347. [DOI] [PubMed] [Google Scholar]; (b) Wu JY, Moreau B, Ritter T. J Am Chem Soc. 2009;131:12915. doi: 10.1021/ja9048493. [DOI] [PubMed] [Google Scholar]
- 27.Blanchard S, Derat E, Desage-El Murr M, Fensterbank MalacriaM, Mouriès-Mansuy V. Eur J Inorg Chem. 2012:376. [Google Scholar]
- 28.Yu RP, Darmon JM, Hoyt JM, Margulieux GW, Turner ZR, Chirik PJ. ACS Catal. 2012;2:1760. doi: 10.1021/cs300358m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Darmon JM, Turner ZR, Lobkovsky E, Chirik PJ. Organometallics. 2012;31:2275. doi: 10.1021/om201212m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Danopoulos AA, Wright JA, Motherwell WB. Chem Commun. 2005:784. doi: 10.1039/b415562a. [DOI] [PubMed] [Google Scholar]
- 31.Roseblade SJ, Pfaltz A. Acc Chem Res. 2007;40:1402. doi: 10.1021/ar700113g. [DOI] [PubMed] [Google Scholar]
- 32.(a) Leutenegger U, Madin A, Pflatz A. Angew Chem Int Ed Engl. 1989:60. [Google Scholar]; (b) Nindakova LO, Lebed FM, Shainyan BA. Russ J Org Chem. 2007;43:1332. [Google Scholar]; (c) Inagaki T, Phong LT, Furuta A, Ito J, Nishiyama H. Chem Eur J. 2010;16:3090. doi: 10.1002/chem.200903118. [DOI] [PubMed] [Google Scholar]; (d) Yu F, Zhang XC, Wu FF, Zhou JN, Fang W, Wu J, Chan ASC. Org Biomol Chem. 2011;9:5652. doi: 10.1039/c1ob05494e. [DOI] [PubMed] [Google Scholar]; (e) Corma A, Iglesias M, del Pino C, Sánchez F. J Organomet Chem. 1992;431:233. [Google Scholar]; (f) DuBois DL, Meek DW. Inorg Chim Acta. 1976;19:L29. [Google Scholar]; (g) Ohgo Y, Takeuchi S, Natori Y, Yoshimura J. Bull Chem Soc Jpn. 1981;54:2124. [Google Scholar]; (h) Takeuchi S, Ohgo Y. Bull Chem Soc Jpn. 1987;60:1449. [Google Scholar]
- 33.Kooistra TM, Knijnenburg Q, Smits JMM, Horton AD, Budzelaar PHM, Gal AW. Angew Chem Int Ed. 2001;40:4719. doi: 10.1002/1521-3773(20011217)40:24<4719::aid-anie4719>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 34.Gibson VC, Humphries MJ, Tellmann KP, Wass DF, White AJP, Williams DJ. Chem Commun. 2001:2252. doi: 10.1039/b107490c. [DOI] [PubMed] [Google Scholar]
- 35.Knijnenburg Q, Horton AD, van der Heijden H, Kooistra TM, Hetterscheid DGH, Smits JMM, de Bruin B, Budzelaar PHM, Gal AW. J Mol Cat -A. 2005;232:151. [Google Scholar]
- 36.Monfette S, Turner ZR, Semproni SP, Chirik PJ. J Am Chem Soc. 2012;134:4561. doi: 10.1021/ja300503k. [DOI] [PubMed] [Google Scholar]
- 37.Zhang G, Scott BL, Hanson SK. Angew Chem Int Ed. 2012;51:12102. doi: 10.1002/anie.201206051. [DOI] [PubMed] [Google Scholar]
- 38.Zhang G, Hanson SK. Org Lett. 2013;15:650. doi: 10.1021/ol303479f. [DOI] [PubMed] [Google Scholar]
- 39.Zhang G, Vasudevan KV, Scott BL, Hanson SK. J Am Chem Soc. 2013;135:8668. doi: 10.1021/ja402679a. [DOI] [PubMed] [Google Scholar]
- 40.Danopoulos AA, Wright JA, Motherwell WB, Ellwood S. Organometallics. 2004;23:4807. [Google Scholar]
- 41.(a) Roseblade SJ, Pfaltz A. Acc Chem Res. 2007;40:1402. doi: 10.1021/ar700113g. [DOI] [PubMed] [Google Scholar]; (b) Broene RD, Buchwald SL. J Am Chem Soc. 1993;115:12569. [Google Scholar]; (c) Troutman MV, Appella DH, Buchwald SL. J Am Chem Soc. 1999;121:4916. [Google Scholar]
- 42.Cámpora J, Perez CM, Rodriguez-Delgado A, Naz AM, Palma P, Alvarez E. Organometallics. 2007;26:1104. [Google Scholar]
- 43.Jiao Y, Evans ME, Morris J, Brennessel WW, Jones WD. J Am Chem Soc. 2013;135:6994. doi: 10.1021/ja400966y. [DOI] [PubMed] [Google Scholar]
- 44.Hounjet LJ, Bannwarth C, Garon CN, Caputo CB, Grimme S, Stephan DW. Angew Chem Int Ed. 2013;52:7492. doi: 10.1002/anie.201303166. [DOI] [PubMed] [Google Scholar]
- 45.Hartung J, Norton JR. In: Catalysis without Precious Metals. Bullock RM, editor. Chapter 1 Wiley-VCH; Weinheim: 2010. [Google Scholar]
- 46.(a) Feder HM, Halpern JJ. Am Chem Soc. 1975;97:7186. [Google Scholar]; (b) Weil TA, Friedman S, Wender I. J Org Chem. 1974;39:48. [Google Scholar]
- 47.Sweany RL, Halpern J. J Am Chem Soc. 1977;99:8335. [Google Scholar]
- 48.Tang L, Papish ET, Abramo GP, Norton JR, Baik MH, Friesner RA, Rappé A. J Am Chem Soc. 2003;125:10093. doi: 10.1021/ja034927l. [DOI] [PubMed] [Google Scholar]
- 49.Tang L, Papish ET, Abramo GP, Norton JR, Baik MH, Friesner RA, Rappé A. J Am Chem Soc. 2006;128:11314. doi: 10.1021/ja034927l. [DOI] [PubMed] [Google Scholar]
- 50.Bullock RM, Samsel EG. J Am Chem Soc. 1990;112:6886. [Google Scholar]
- 51.Choi J, Pulling ME, Smith DM, Norton JR. J Am Chem Soc. 2008;130:4250. doi: 10.1021/ja710455c. [DOI] [PubMed] [Google Scholar]
- 52.Knijnenburg Q, Hetterscheid D, Kooistra TM, Budzelaar PHM. Eur J Inorg Chem. 2004:1204. [Google Scholar]
- 53.Tondreau AM, Darmon JM, Wile BM, Floyd SK, Lobkovsky E, Chirik PJ. Organometallics. 2009;28:2928. [Google Scholar]
- 54.Darmon JM, Turner ZR, Lobkovsky E, Chirik PJ. Organometallics. 2012;31:2275. doi: 10.1021/om201212m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.For early electrochemical studies identifying the redox-activity of bis(imino)pyridines see:; (a) Kuwabara IH, Comninos FCM, Pardini VL, Viertler H, Toma HE. Electrochim Acta. 1994;39:2401. [Google Scholar]; (b) Toma HE, Chavez-Gil TE. Inorg Chim Acta. 1997;257:197. [Google Scholar]; (c) de Bruin B, Bill E, Bothe E, Weyhermüller T, Wieghardt K. Inorg Chem. 2000;39:2936. doi: 10.1021/ic000113j. [DOI] [PubMed] [Google Scholar]; (d) Budzelaar PHM, de Bruin B, Gal AW, Wieghardt KW, van Lenthe JH. Inorg Chem. 2001;40:4649. doi: 10.1021/ic001457c. [DOI] [PubMed] [Google Scholar]
- 56.Danopoulos AA, Winston S, Motherwell WB. Chem Commun. 2002:1376. [Google Scholar]
- 57.Britovsek GJP, Clentsmith GKB, Gibson VC, Goodgame DML, McTavish SJ, Pankhurst QA. Catal Commun. 2002;3:207. [Google Scholar]
- 58.Scott J, Gambarotta S, Korobkov I, Knijnenburg Q, de Bruin B, Budzelaar PHM. J Am Chem Soc. 2005;127:17204. doi: 10.1021/ja056135s. [DOI] [PubMed] [Google Scholar]
- 59.Milsmann C, Turner ZR, Semproni SP, Chirik PJ. Angew Chem Int Ed. 2012;51:5386. doi: 10.1002/anie.201201085. [DOI] [PubMed] [Google Scholar]
- 60.Bowman AC, Milsmann C, Atienza CCH, Lobkovsky E, Wieghardt K, Chirik PJJ. Am Chem Soc. 2010;132:1676. doi: 10.1021/ja908955t. [DOI] [PubMed] [Google Scholar]
- 61.Talcott CL, Myers R. J Molecular Physics. 1967;12:549. [Google Scholar]
- 62.Danopoulos AA, Tulloch AAD, Winston S, Eastham G, Hursthouse MB. Dalton Trans. 2003:1009. [Google Scholar]
- 63.Bogdanovic B. Acc Chem Res. 1988;21:261. [Google Scholar]
- 64.Bowman AC, Milsmann C, Bill E, Lobkovsky E, Weyhermüller T, Wieghardt K, Chirik PJ. Inorg Chem. 2010;49:6110. doi: 10.1021/ic100717w. [DOI] [PubMed] [Google Scholar]
- 65.Atienza CCH, Milsmann C, Lobkovsky E, Chirik PJ. Angew Chem Int Ed. 2011;50:8143. doi: 10.1002/anie.201102825. [DOI] [PubMed] [Google Scholar]
- 66.(a) Maki AH, Edelstein N, Davison A, Holm RHJ. Am Chem Soc. 1964;86:4580. [Google Scholar]; (b) Nishida Y, Hayashida K, Sumita A, Kida S. Bull Chem Soc Jpn. 1980;53:271. [Google Scholar]; (c) Nishida Y, Kida S. Bull Chem Soc Jpn. 1978;51:143. [Google Scholar]; (d) Matsuzaki K, Yasukawa T. Chem Commun. 1968:1460. [Google Scholar]; (e) Nishida Y, Sumita A, Kida S. Bull Chem Soc Jpn. 1977;50:759. [Google Scholar]; (f) Przyojski JA, Arman HD, Tonzetich ZJ. Organometallics. 2013;32:723. doi: 10.1021/om400630q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Gregson AK, Martin RL, Mitra S. Chem Phys Lett. 1970;5:310. [Google Scholar]; (h) Mo Z, Li Y, Lee HK, Deng L. Organometallics. 2011;30:4687. [Google Scholar]; (H) Hay-Motherwell RS, Wilkinson G, Hussain B, Hursthouse MB. Polyhedron. 1990;9:931. [Google Scholar]
- 67.(a) Pierpont CG, Lange CW. Prog Inorg Chem. 1994;41:331. [Google Scholar]; (b) Ray K, Petrenko T, Wieghardt K, Neese F. Dalton Trans. 2007:1552. doi: 10.1039/b700096k. [DOI] [PubMed] [Google Scholar]
- 68.Knijnenburg Q, Gambarotta S, Budzelaar PHM. Dalton Trans. 2006;46:5542. doi: 10.1039/b612251e. [DOI] [PubMed] [Google Scholar]
- 69.(a) Tomson NC, Crimmin MR, Petrenko T, Rosebrugh LE, Sproules S, Boyd WC, Bergman RG, DeBeer S, Toste FD, Wieghardt K. J Am Chem Soc. 2011;133:18785. doi: 10.1021/ja206042k. [DOI] [PubMed] [Google Scholar]; (b) Sarangi R, Cho J, Nam W, Solomon EI. Inorg Chem. 2011;50:614. doi: 10.1021/ic101730r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stieber SCE, Milsmann C, Hoyt JM, Turner ZR, Finkelstein KD, Weighardt K, Debeer S, Chirik PJ. Inorg Chem. 2012;51:3770. doi: 10.1021/ic202750n. [DOI] [PubMed] [Google Scholar]
- 71.Bowman AC, Milsmann C, Bill E, Turner ZR, Lobkovsky E, DeBeer S, Wieghardt K, Chirik PJ. J Am Chem Soc. 2011;133:17353. doi: 10.1021/ja205736m. [DOI] [PubMed] [Google Scholar]
- 72.Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1997;119:6297. [Google Scholar]
- 73.Chandrasekaran P, Stieber SCE, Collins TJ, Que L, Neese F, DeBeer S. Dalton Trans. 2011;40:11070. doi: 10.1039/c1dt11331c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu D, Thapa I, Korobkov I, Gambarotta S, Budzelaar PHM. Inorg Chem. 2011;50:9879. doi: 10.1021/ic2002145. [DOI] [PubMed] [Google Scholar]
- 75.Bart SC, Chlopek K, Bill E, Bouwkamp MW, Lobkovsky E, Neese F, Wieghardt K, Chirik PJ. J Am Chem Soc. 2006;128:13901. doi: 10.1021/ja064557b. [DOI] [PubMed] [Google Scholar]
- 76.Humphries MJ, Tellmann KP, Gibson VC, White AJP, Williams DJ. Organometallics. 2005;24:2039. [Google Scholar]
- 77.Bart SC, Lobkovsky E, Bill E, Wieghardt K, Chirik PJ. Inorg Chem. 2007;46:7055. doi: 10.1021/ic700869h. [DOI] [PubMed] [Google Scholar]
- 78.Noodleman L, Peng CY, Case DA, Mouesca JM. Coord Chem Rev. 1995;144:199. [Google Scholar]
- 79.Ginsberg AP. J Am Chem Soc. 1980;102:111. [Google Scholar]
- 80.Kirchner B, Wennmohs F, Ye S, Neese F. Curr Opin Chem Biol. 2007;11:134. doi: 10.1016/j.cbpa.2007.02.026. [DOI] [PubMed] [Google Scholar]
- 81.(a) DeBeer GeorgeS, Petrenko T, Neese F. J Phys Chem A. 2008;112:12936. doi: 10.1021/jp803174m. [DOI] [PubMed] [Google Scholar]; (b) Chandrasekaran P, Stieber SCE, Collins TJ, Que L, Neese F, DeBeer S. Dalton Trans. 2011;40:11070. doi: 10.1039/c1dt11331c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Caulton KG. Eur J Inorg Chem. 2012:435. [Google Scholar]
- 83.Only representative procedures are reported here. Full experimental details, including General Considerations, are reported in the Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.