

Abstract
Adherens junctions, which are intercellular adhesive complexes that are crucial for maintaining epithelial homeostasis, are downregulated in many cancers to promote tumour progression. However, the role of desmosomes — adhesion complexes that are related to adherens junctions — in carcinogenesis has remained elusive. Recent studies using mouse genetic approaches have uncovered a role for desmosomes in tumour suppression, demonstrating that desmosome downregulation occurs before that of adherens junctions to drive tumour development and early invasion, suggesting a two-step model of adhesion dysfunction in cancer progression.
More than 90% of human cancers are of epithelial origin1. Elaborating the factors that promote the normal architecture and function of epithelia, and the mechanisms through which these are perturbed, is therefore fundamental for understanding the genesis of most human cancers. The formation, maturation and homeostasis of epithelia require carefully choreographed programmes of cell proliferation, adhesion, polarity, migration and differentiation2. Vital for the unity of cells in epithelial sheets are adhesion junctions, such as adherens junctions and desmosomes. These structures not only facilitate intercellular adhesion to ensure tissue integrity but also serve as crucial regulators of processes such as epithelial morphogenesis, differentiation and wound healing3,4. Moreover, dysfunction of either junctional complex is associated with specific epithelial diseases. However, until recently, only adherens junctions had been linked to the suppression of cancer development. In this Progress article, we discuss the newly appreciated role of desmosomal adhesion complexes in tumour suppression, highlighting recent mouse genetic studies and addressing how the p53 and p63 pathways might intersect with desmosome-mediated adhesion in this context.
Adherens junctions and cancer
Adherens junctions are key intercellular adhesion complexes5. Three main protein families constitute traditional adherens junction complexes: transmembrane cadherins, armadillo proteins and cytoskel-etal adaptors (FIG. 1). Classical cadherins, including the family prototype E-cadherin (encoded by CDH1), mediate cell–cell interactions in a calcium-dependent, homophilic manner through their extra-cellular domains6–9. The cytoplasmic tails of cadherins bind members of the armadillo protein family, such as β-catenin (encoded by CTNNB1) and p120 catenin (encoded by CTNND1)9–11. Cadherins communicate with the actin cytoskeleton through contacts with β-catenin, which can interact with actin-binding proteins such as α-catenin (encoded by CTNNA genes)12–16. When not bound to cadherins, β-catenin can translocate to the nucleus to promote WNT signalling, by binding to LEF/TCF transcription factors and regulating the transcription of LEF/TCF-dependent target genes17–19.
Figure 1. Desmosome deficiency can promote cancer in multiple ways.

Stable adherens junctions and desmosomes facilitate adhesion between epithelial cells. The best-characterized components are shown; the position of p53 apoptosis effector related to PMP-22 (PERP) in the desmosome is speculative. Several mechanisms through which disrupted desmosomes could promote cancer are indicated. Junction plakoglobin (JUP) is the desmosome component with the best-characterized effect on the phenotypic changes that occur in cancer cells. At high levels, JUP can compete with β-catenin for inclusion in adherens junctions and/or for interaction with the adenomatous polyposis coli (APC)-mediated degradation machinery, which regulates cellular β-catenin levels (not shown). Both scenarios result in increased nuclear β-catenin, which can stimulate the transcription of LEF/TCF -dependent target genes, promoting oncogenic effects (part a). JUP itself can also shuttle between adhesion junctions at the plasma membrane and the nucleus, where it can increase expression of LEF/TCF target genes independently of β-catenin (part b). Additionally, plakophilins (PKPs) can also shuttle between the desmosome and the nucleus, and PKP2 has been demonstrated to interact with β-catenin and to enhance LEF/TCF-mediated transactivation (part b). JUP and PKPs may also have dedicated LEF/TCF-independent target genes (part c). PKPs may also function in the cytoplasm to stimulate translational initiation (part d). Other uncharacterized molecular mechanisms of cancer promotion might also exist (part e). DSC, desmocollin; DSG, desmoglein; E-cad, E-cadherin; NRCAM, neuronal cell adhesion molecule; p120, p120 cadherin.
Although constitutive and tissue-specific ablation of adherens junction protein-encoding genes in mice has underscored the importance of adherens junctions in epithelial tissue function and homeostasis, it is the dynamic regulation of such structures that promotes tissue plasticity and reorganization during processes such as developmental epithelial to mesenchymal transition (EMT), wound healing, and cancer progression and metastasis20. Indeed, it is well established that E-cadherin-based cell–cell adhesion is lost during the progression of many types of human cancers as they acquire invasive and metastatic potential. Importantly, down-regulation of both E-cadherin and p120 catenin in human tumours is commonly associated with a poor clinical outcome21–25. The importance of adherens junction dysfunction in promoting cancer progression has been definitively demonstrated using mouse genetic models. For example, in the Rip1Tag2-transgenic mouse model, in which SV40 large T antigen expression in pancreatic β-cells causes neuroendocrine pancreatic tumours, the maintenance of E-cadherin expression causes tumours to stall at the adenoma stage. By contrast, the forced disruption of adherens junction-mediated adhesion through the expression of dominant-negative E-cadherin drives the transition of adenomas to carcinomas, which is accompanied by tumour invasion and metastasis26. Similarly, Cdh1 deletion in the mammary epithelium of mice that are prone to breast cancer — because of the loss of the tumour suppressor Trp53 — is associated with accelerated tumour development and increased invasion and metastasis27. In addition, conditional inactivation of Ctnnd1 in mouse salivary gland or skin drives tumori-genesis28,29, and ablation of Ctnna1 in mouse skin induces squamous cell carcinoma development30. Collectively, the data compiled from in vitro cell culture experiments, human tumour analysis and mouse model studies support an unambiguous function for adherens junction-mediated adhesion in tumour suppression.
Desmosomes fortify cell adhesion
Although adherens junctions are fundamental both for intercellular adhesion in epithelia and for enabling the dynamic rearrangements of epithelia, desmosomes have traditionally been viewed as static protein complexes that reinforce adhesion between epithelial cells31. The strong intercellular adhesion that is provided by desmosomes is particularly important for conferring strength to tissues that must resist large amounts of mechanical stress. Especially prominent in the skin and heart, desmosomes connect cell–cell contact sites at the plasma membrane to the intermediate filament cytoskeleton to promote tissue integrity and homeostasis4,32. Compromised desmosome function can result in various human diseases, symptoms of which typically include epidermal fragility and blistering, thickened skin of the palm or soles (palmoplantar keratoderma) and/or cardiomyopathy33.
Like adherens junctions, desmosomes comprise three main protein families: cadherins, armadillo proteins and plakins, which are arranged in a similar manner to that of adherens junction complexes (FIG. 1). However, the precise molecular composition of desmosomes can be variable and can depend on the tissue-specific or differentiation-specific expression of particular isoforms of the constituent proteins4. The two types of desmosomal cadherins — the desmogleins (DSG1–4) and the desmocollins (DSC1–3) — mediate adhesion between apposing cells through interactions of their ectodomains34–40. Intracellularly, desmosomal cadherins bind to the armadillo proteins junction plakoglobin (JUP)41–44 and plakophilins (PKP1–3)45–47, which help to bridge the cadherins to the intermediate filament cytoskeleton. Additionally, JUP is highly homologous to β-catenin and can substitute for β-catenin in adherens junctions, as well as localize to the nucleus where it can regulate the transcription of LEF/TCF-target genes48–52. PKPs also exhibit dual localization at the desmosome and in the nucleus, where their ability to affect gene expression is implicated but incompletely understood46,47,53. The most important plakin family member is desmoplakin (DSP; also known as DP), which interacts with JUP and intermediate filaments, providing the final link in the chain from the plasma membrane to the cytoskeleton54–57. Another key desmosomal protein, which was identified by its dramatic loss-of-function blistering phenotype in the epidermis and in other stratified epithelia of knockout mice, is p53 apoptosis effector related to PMP-22 (PERP)58. PERP is a tetraspan membrane protein that is transcriptionally activated by the p53 tumour suppressor during DNA damage-induced apoptosis, and by the related transcription factor p63 during the development of stratified epithelia58,59. Although PERP has been unequivocally localized to desmosomes in stratified epithelia and found to be crucial for proper desmosome assembly, its interacting partners within the desmosome remain elusive. Importantly, PERP provides a key link between the p53 family of transcriptional regulators and cell–cell adhesion. Further support for this connection is the documented activation of various cell–cell adhesion components by p63 in mammary epithelial cells60. Thus, as a target of both p53, which is inactivated in at least 50% of all human cancers, and p63, which is an important tumour suppressor in specific contexts61, PERP is a potentially crucial mediator of tumour suppression downstream of these transcription factors (FIG. 2).
Figure 2. The p53–p63 pathway regulates homeostasis in epithelial tissues.

This figure represents some of the ways in which p53 and p63 family members can regulate epithelial homeostasis. a | During the development and maintenance of epithelial tissues, p63 can directly or indirectly regulate the expression of various classes of genes, including genes that encode cell–cell adhesion proteins, such as p53 apoptosis effector related to PMP-22 (PERP). These proteins can then assemble into the intercellular adhesive complexes adherens junctions and desmosomes, which promote adhesion between adjacent epithelial cells. Adhesion between cells within a tissue contributes to its integrity, organization and function. b | Cellular stressors such as DNA damage or oncogene expression activate p53. As a sensor of stress, p53 induces the expression of genes that are involved in apoptosis, including PERP and NOXA. PERP and NOXA, and other proteins, contribute to the apoptotic programme, triggering the death of cells the survival of which would be detrimental to a tissue. Both cell–cell adhesion and apoptosis are important cellular mechanisms that contribute to tumour suppression.
Genetic loss-of-function studies in mice have reinforced the importance of desmosomes for normal tissue function. For example, constitutive deletion of Dsc3, Dsg2 or Dsp causes early embryonic lethality probably owing to defective adhesion in processes essential before, at or after implantation, respectively62–64. By contrast, Jup−/− animals typically die later in embryogenesis primarily owing to severe heart abnormalities65,66, and Perp−/& minus; mice die perinatally with profound epithelial blistering58. Mice with constitutive knockout of Dsc1 or Dsg3 or with conditional deletion of Dsp or Dsc3 in the skin survive, but exhibit epidermal integrity defects67–70. Supporting a pivotal role for desmosomes in tissue function are human diseases in which desmosome components are inactivated by mutation, targeted by autoantibodies or proteolysed by bacterial toxins. These diseases are characterized by phenotypes such as severe abnormalities of the skin, the ectodermal appendages and/or the heart and provide evidence for a crucial function for desmosome-mediated adhesion in vivo71. Interestingly, although inactivation of adherens junction components can cause tissue degeneration or can instigate cancer development and metastasis, compromised desmosome function is typically thought to only result in degenerative diseases, such as palmoplantar keratoderma and ectodermal dysplasia, and has not been clearly associated with cancer predisposition31.
Desmosomes and cancer
Direct genetic loss-of-function studies querying the role of desmosomes in cancer have been impeded by the aforementioned lethality that is typically associated with the constitutive deletion of desmosome genes in mice. In addition, data correlating the expression of particular desmosome components in human tumours with tumour progression are contradictory and confusing, with upregulation, downregulation or maintenance of desmosome components observed. For example, the expression of some desmosome proteins, including DSG2, DSG3 and PKP3, is increased compared with normal tissue in certain cancers of the skin, head and neck, prostate and lung, and this increased expression is associated with enhanced tumour progression and/or reduced patient survival72–76. By contrast, the loss or reduction of one or more desmosome components, including DSG1–3, DSC2, DSC3, JUP, PKP1–3 and DSP, is observed on the development and/or the progression of various human epithelial cancers, including skin, head and neck, gastric, colorectal, bladder, breast, prostate, cervical and endometrial cancers, often correlating with advanced tumour grade, increased metastasis and/or poor prognosis76–95. Finally, in other instances, no obvious changes in the levels of desmosomal proteins have been noted during cancer progression75,77,80,96. Attempts to clarify the role of desmosomal adhesion in cancer by modulating the expression of desmosome components in cultured cells have produced confounding results. In some cases, overexpression of desmosome components in cultured cells promotes proliferation, inhibits apoptosis and increases invasion, characteristics that are advantageous to tumour cells74,97,98. Moreover, ectopic expression of DSG2 in the upper layers of mouse skin induces tumour development99. By contrast, other experiments have shown that overexpression of desmosome components in cell lines suppresses tumour-promoting behaviour, such as invasion and anchorage-independent growth93,100. Consistent with a potential role for desmosomes in tumour suppression, overexpression of JUP in SV40-transformed fibroblasts or bladder cancer cells, and overexpression of desmosomal cadherins in squamous cell carcinoma (SCC cells, suppresses tumour formation and/or invasion in mouse xenograft assays101–103. Additionally, knockdown of PKP3 in colon cancer cells promotes anchorage-independen growth and tumour growth in immunocom-promised mice104. Adding to the uncertainty regarding the role of desmosomes in cancer is the observation that although potentially oncogenic mutations that occur in or near putative JUP phosphorylation sites have been noted in prostate and gastric cancers91,105, mutations in desmosome components seem to be rather uncommon.
Overall, the fact that some experiments support a tumour-suppressive role for desmosomes in cancer and others provide evidence for an oncogenic function could reflect real context-dependent differences in the contribution of desmosomes to cancer. Alternatively, the disparate findings could result from limitations in these surrogate models for carcinogenesis — such as, the artificial conditions under which cultured cells are grown, the analysis of transformed cells with numerous genetic alterations and the failure to recapitulate normal tissue architecture or a functional immune system in mouse xenograft tumour models. Therefore, to definitively unveil the role of desmosomes in cancer, it is imperative to use physiologically relevant in vivo genetic cancer models to accurately mimic the complexities of human cancer.
An unequivocal approach to establishing the contribution of desmosomes to cancer is the use of mouse models with intact immune systems in which cancers develop in the appropriate tissue microenvironment as a result of defined genetic lesions. Two recent studies have used this approach, consequently providing direct causal evidence linking desmosome deficiency to cancer development. The first study sought to pinpoint proteins that are crucial for restricting tumour invasion in the Rip1Tag2 model of pancreatic islet cell tumorigenesis, which proceeds from non-invasive to focally invasive and to broadly invasive carcinomas106. Gene expression analysis of non-invasive and broadly invasive pancreatic lesions derived from these mice showed that the expression of genes encoding various desmosomal components, including Dsp, Dsg2, Dsc2 and Pkp2, was significantly reduced in highly invasive tumours compared with non-invasive ones, suggesting that desmosome downregulation may contribute to malignant progression106. To test the importance of the downmodulation of desmosome genes, conditional Dsp-knockout mice were analysed. Although conditional deletion of Dsp in the pancreatic β-cells did not detrimentally affect the survival of the mice or tumour growth, loss of Dsp did enhance local invasion of tumours, without affecting broad invasion or metastasis. Interestingly, expression of E-cadherin was maintained in the locally invasive Dsp-deficient tumours, highlighting the independent nature of the desmosomes and the adherens junctions in this context, despite the fact that these two junctions are thought to regulate each other's stability68,107. These results demonstrate that loss of desmosome function is an important step towards malignant conversion by facilitating local invasion and, therefore, that desmosome-mediated adhesion is a key impediment to tumour progression. Moreover, in conjunction with previous experiments in the Rip1Tag2 mouse model, these findings suggest a two-step model for cancer progression, in which desmosomal downregulation causes local invasion in an EMT-independent manner, and subsequent loss of adherens junctions promotes full cancer progression26 (FIG. 3).
Figure 3. Desmosome downregulation is one in a series of steps occurring during cancer development.
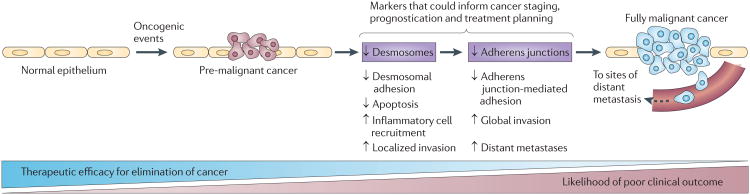
Two recent studies using mouse cancer models in which desmosome components were ablated have demonstrated key contributions of desmosome deficiency to epithelial cancer development and progression. Mutations in proto-oncogenes or tumour suppressors drive the development of nascent tumours in epithelia. In this context, desmosome deficiency, occurring before adherens junction loss, promotes several cellular phenotypes that can contribute to cancer progression: decreased desmosome-mediated intercellular adhesion, increased cell survival and inflammatory cell recruitment in ultraviolet B (UVB)-induced squamous cell carcinomas in p53 apoptosis effector related to PMP-22 (Perp)-deficient mice and increased local invasion in desmoplakin (Dsp)-deficient Rip1Tag2-driven pancreatic neuroen-docrine tumours. Subsequent dissolution of adherens junctions in tumours is associated with impaired adherens junction-mediated adhesion, enhanced global invasion and increased distant metastasis, which are features of full-blown malignancy. As desmosome downmodulation precedes that of adherens junctions, and as early diagnosis and treatment is key to achieving the optimal clinical outcome, establishing the status of desmosome and adherens junction constituents in tumours could potentially augment the current tools that are used in the staging, prognostication or treatment of cancers.
To address the role of desmosomes in a model of human skin cancer, mice with conditional deletion of Perp in the epidermis were exposed to chronic ultraviolet B (UVB) radiation to induce SCCs108. Perp loss in the skin reduced the latency of tumour development and increased the multiplicity of tumours compared with UVB-treated wild-type controls, indicating that Perp deficiency promotes tumour initiation. Moreover, tumours that developed in the Perp-deficient mice were typically less differentiated than those in control mice, suggesting that Perp loss also facilitates tumour progression. Three mechanisms were proposed to explain the propensity of Perp-deficient mice to develop skin cancer. First, as Perp-deficient keratinocytes had an impaired apoptotic response to UVB radiation, the inappropriate survival of damaged cells in Perp-deficient skin following exposure to mutagenic stimuli probably contributed to tumorigenesis. Indeed, the enhanced cell survival that is observed in UVB-treated keratinocytes, which can lack functional p53, is associated with increased carcinogenesis109. Second, Perp deficiency also compromised desmosome-mediated intercellular adhesion. Although Perp loss partially impaired desmosome function in the skin, desmosome component expression was completely lost on the development of Perp-deficient SCCs. Intriguingly, although Perp-deficient tumours showed a clear downregulation of desmosomal components, adherens junction components were maintained, suggesting that PERP and desmosome loss promote cancer by a specific mechanism rather than by a general change in differentiation status, such as EMT. The downregulation of desmosomes with the retention of adherens junctions was also observed in SCCs that formed with a longer latency in wild-type mice, indicating that although Perp depletion facilitates desmosome disassembly, it also occurs in a wild-type context. Moreover, on examining samples from different stages of human SCC development, PERP-deficient, E-cadherin-positive tumours were found to constitute a major group, suggesting that this is an important stage in human skin cancer development. Thus, as in the Rip1Tag2 model, reduced expression of desmosome proteins could be an early driver of tumour progression, and subsequent loss of adherens junctions could promote later stages, including wide-spread invasion and metastasis. Finally, gene expression profiling of the epidermis on Perp ablation revealed the induction of genes that are involved in inflammatory responses. Moreover, Perp deficiency, in conjunction with chronic UVB treatment, led to the recruitment of inflammatory cells, especially mast cells. Given the known role for inflammation in promoting cancer, this inflammatory signature and consequent infiltration of immune cells provides a clear basis for how Perp loss can enhance tumorigenesis at the cellular level. Together, these data demonstrate that Perp deficiency promotes cancer development and progression by multiple mechanisms, clearly supporting the idea that desmosomes can function as tumour suppressors (FIG. 3). Furthermore, the phenotypes induced by Perp loss may also contribute to carcinogenesis in cases of p53 or p63 inactivation (FIG. 2).
How desmosome loss promotes cancer
Various models have been proposed to provide a molecular explanation for how desmosome downregulation could promote cancer. The most extensively studied model suggests that desmosome dysfunction can provoke the release of specific desmosomal constituents that can display oncogenic activity, such as JUP. Most notably, JUP manifests β-catenin-like signalling activity, as originally shown by its ability to induce axis duplication in Xenopus laevis embryos110. Interestingly, the similar capacity of plasma membrane-anchored JUP to induce axis duplication, among other studies, suggested that the effects of JUP on WNT–β-catenin signalling were indirect and probably attributable to the ability of JUP to promote β-catenin nuclear localization and transcriptional activity111–114. Indeed, JUP can replace β-catenin in adherens junctions, freeing β-catenin to stimulate the transcription of WNT target genes (FIG. 1a), including oncogenic targets such as CCND1 (encoding cyclin D1) and neuronal cell adhesion molecule (NRCAM)52,115–119. In addition to these indirect effects on gene regulation via β-catenin, JUP can itself transit to the nucleus on release from junctions, directly activating oncogenic β-catenin–LEF/TCF target genes or potentially stimulating the expression of uncharacterized JUP-specific targets to promote proliferation or transformation51 (FIG. 1b,c). This concept was originally derived from the observation that JUP can activate β-catenin-responsive target genes in Ctnnb1-null cells or tissues52,120–122. Adding to the complexity, however, is evidence from model organisms demonstrating that JUP can antagonize WNT–β-catenin signalling. For example, cardiac-specific deletion of Dsp in mice results in the nuclear accumulation of JUP and the suppression of WNT–β-catenin signalling123, and the ablation of Jup in murine hearts or zebrafish embryos induces β-catenin transcriptional activity124,125. Although JUP-mediated inhibition of WNT–β-catenin signalling may be important in certain physiological settings, its relevance to cancer is unclear and requires further investigation.
The redistribution of PKPs from desmosomes, where they promote adhesion and differentiation, to the nucleus may also contribute to carcinogenesis. The nuclear localization of PKPs in certain settings suggests that they could modulate gene expression, and, indeed, PKP2 can interact with β-catenin and can potentiate endogenous β-catenin–TCF transcriptional activity46,126,127 (FIG. 1b). Whether PKPs regulate transcription in a β-catenin–LEF/TCF-independent manner, however, remains to be determined (FIG. 1c). In addition, PKP1 and PKP3 can localize to cytoplasmic particles where they can interact with translation-initiation factors to stimulate translation128,129 (FIG. 1d). This observation implies an oncogenic function for cytoplasmic PKPs, a concept that is supported by the observed redistribution of PKPs from the plasma membrane to the cytoplasm during tumour development75,130.
In addition to releasing components with oncogenic potential, desmosome dysfunction could also promote carcinogenesis through other means. One such mechanism is by activating signalling pathways that impinge on cancer development. For example, activation of p38MAPK is triggered by autoantibody targeting of DSG3 in the blistering disease Pemphigus vulgaris131 (FIG. 1e), and activation of ERK1, ERK2 and AKT signalling is induced by DSP knock-down in human keratinocytes132. Whatever the exact molecular alterations that occur with desmosome impairment, such changes could induce pro-tumorigenic cellular phenotypes that are associated with desmosome loss, including increased proliferation, augmented survival and enhanced inflammation. Moreover, the simple loss of the exceptional adhesive strength that is imparted by desmosomes to tissues may also contribute to cancer progression in some contexts by relieving a barrier to invasion and metastasis, perhaps in conjunction with adherens junction loss133,134.
Conclusions and future study
Although our understanding of the role of desmosomes in cancer is still evolving, genetic loss-of-function studies in vivo in physiological mouse models of cancer have revealed a causal relationship between the loss of specific desmosome proteins and the development of certain cancers. Additional studies are certainly necessary to understand the complete complexities of this relationship, but the conclusions so far support the majority of the human cancer expression data and functional studies in cultured cells, suggesting that desmosomes normally function as tumour-suppressive complexes and that loss of desmosome proteins and desmosome-mediated adhesion is associated with cancer development and/or progression.
Considerable evidence supports a tumour-suppressive function for desmosomes but this may not be the case in all circumstances, as desmosome proteins have been linked to oncogenic effects in human cancer and experimental systems. These data suggest that altered expression of desmosome proteins might promote cancer development in certain contexts. Differences in how desmosomes influence carcinogenesis could relate to differences in their composition, as well as to the expression level, subcellular localization and tissue-specific or differentiation-specific functions of their constituent proteins. Future investigation will further clarify the contribution of various desmosomal components to the development of diverse cancer types.
Interestingly, although impaired desmosome function is associated with various autoimmune, genetic and infectious human cutaneous diseases, as well as cardiomyopathy syndromes71, to our knowledge no clear cancer predisposition has been observed in individuals with these syndromes. The lack of such reports might reflect the rarity of these diseases in the general population or the often-reduced lifespan of these patients, which is perhaps not sufficient for revealing a propensity to cancer development. Alternatively, it is possible that the influence of desmosome dysfunction on cancer may be relevant only in particular contexts, such as when a specific desmosome component is targeted or in the background of particular oncogenic mutations. Future studies that assess the cancer predispositon in this population, as well as studies using patient-derived cells or tissues could help to clarify the role of impaired desmosome function in cancer and could have implications for cancer treatment.
The fact that recent genetic loss-of-function studies describe tumours that exhibit loss of desmosomes while retaining adherens junctions106,108 has considerable clinical implications. As loss of E-cadherin is a common but late event in epithelial cancer progression, identifying markers such as PERP or DSP that may be downregulated earlier could improve the diagnosis, staging and prognostication of cancers, and could also inform therapeutic decisions (FIG. 3). For example, as PERP loss seems to promote tumour progression, tumours with a PERP-deficient status might warrant more aggressive treatment approaches. Indeed, gene expression profiling identified PERP as one component of a gene signature the down-regulation of which predicts poor response to treatment in oesophageal cancers135. Additionally, reduced expression of DSP in oropharyngeal cancer was associated with poorly differentiated tumours that metastasized within a follow-up period of 3 years90. Ultimately, broadening our understanding of desmosomes and tumorigenesis, as well as context-specific distinctions in their relationship, will enhance our ability to diagnose, stage, prognosticate and treat human cancer.
Acknowledgments
R.L.D. is supported by the American Cancer Society New England-SpinOdyssey Postdoctoral Fellowship. L.D.A. is supported by the US National Cancer Institute (NCI) (R01 CA093665). The authors would like to thank K. Bieging, D. Jiang and S. Baron for thoughtful comments on the manuscript. The authors apologize to those authors whose work could not be cited owing to space limitations.
Footnotes
Competing interests statement: The authors declare no competing financial interests.
References
- 1.Cooper GM. Oncogenes. Jones and Barlett Publishers; Boston: 1995. [Google Scholar]
- 2.Schock F, Perrimon N. Molecular mechanisms of epithelial morphogenesis. Annu Rev Cell Dev Biol. 2002;18:463–493. doi: 10.1146/annurev.cellbio.18.022602.131838. [DOI] [PubMed] [Google Scholar]
- 3.Jamora C, Fuchs E. Intercellular adhesion, signalling and the cytoskeleton. Nature Cell Biol. 2002;4:e101–e108. doi: 10.1038/ncb0402-e101. [DOI] [PubMed] [Google Scholar]
- 4.Simpson CL, Green KJ. Desmosomes: new perspectives on a classic. J Invest Dermatol. 2007;127:2499–2515. doi: 10.1038/sj.jid.5701015. [DOI] [PubMed] [Google Scholar]
- 5.Vasioukhin V, Bauer C, Yin M, Fuchs E. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell. 2000;100:209–219. doi: 10.1016/s0092-8674(00)81559-7. [DOI] [PubMed] [Google Scholar]
- 6.Chen CP, Psoy S, Ben-Shaul A, Shapiro L, Honig BH. Specificity of cell-cell adhesion by classical cadherins: critical role for low-affinity dimerization through β–strand swapping. Proc Natl Acad Sci USA. 2005;102:8531–8536. doi: 10.1073/pnas.0503319102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nose A, Tsuji K, Takeichi M. Localization of specificity determining sites in cadherin cell adhesion molecules. Cell. 1990;61:147–155. doi: 10.1016/0092-8674(90)90222-z. [DOI] [PubMed] [Google Scholar]
- 8.Pertz O, et al. A new crystal structure, Ca2+ dependence and mutational analysis reveal molecular details of E-cadherin homoassociation. EMBO J. 1999;18:1738–1747. doi: 10.1093/emboj/18.7.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yap AS, Brieher WM, Gumbiner BM. Molecular and functional analysis of cadherin-based adherens junctions. Annu Rev Cell Dev Biol. 1997;13:119–146. doi: 10.1146/annurev.cellbio.13.1.119. [DOI] [PubMed] [Google Scholar]
- 10.Ozawa M, Baribault H, Kemler R. The cytoplasmic domain of the cell adhesion molecule uvomorulin associates with three independent proteins structurally related in different species. EMBO J. 1989;8:1711–1717. doi: 10.1002/j.1460-2075.1989.tb03563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reynolds AB, et al. Identification of a new catenin: the tyrosine kinase substrate p120cas associates with E-cadherin complexes. Mol Cell Biol. 1994;14:8333–8342. doi: 10.1128/mcb.14.12.8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drees F, Pokutta S, Yamada S, Nelson WJ, Weis WI. α-catenin is a molecular switch that binds E-cadherin-β-catenin and regulates actin-filament assembly. Cell. 2005;123:903–915. doi: 10.1016/j.cell.2005.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hulsken J, Birchmeier W, Behrens J. E-cadherin and APC compete for the interaction with β-catenin and the cytoskeleton. J Cell Biol. 1994;127:2061–2069. doi: 10.1083/jcb.127.6.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS. α 1(E)-catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci USA. 1995;92:8813–8817. doi: 10.1073/pnas.92.19.8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rubinfeld B, Souza B, Albert I, Munemitsu S, Polakis P. The APC protein and E-cadherin form similar but independent complexes with α-catenin, β-catenin, and plakoglobin. J Biol Chem. 1995;270:5549–5555. doi: 10.1074/jbc.270.10.5549. [DOI] [PubMed] [Google Scholar]
- 16.Yamada S, Pokutta S, Drees F, Weis WI, Nelson WJ. Deconstructing the cadherin-catenin-actin complex. Cell. 2005;123:889–901. doi: 10.1016/j.cell.2005.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Behrens J, et al. Functional interaction of β-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 18.Molenaar M, et al. XTcf-3 transcription factor mediates β-catenin-induced axis formation in Xenopus embryos. Cell. 1996;86:391–399. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- 19.van de Wetering M, et al. Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell. 1997;88:789–799. doi: 10.1016/s0092-8674(00)81925-x. [DOI] [PubMed] [Google Scholar]
- 20.D'Souza-Schorey C. Disassembling adherens junctions: breaking up is hard to do. Trends Cell Biol. 2005;15:19–26. doi: 10.1016/j.tcb.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 21.Bremnes RM, et al. High-throughput tissue microarray analysis used to evaluate biology and prognostic significance of the E-cadherin pathway in non-small-cell lung cancer. J Clin Oncol. 2002;20:2417–2428. doi: 10.1200/JCO.2002.08.159. [DOI] [PubMed] [Google Scholar]
- 22.Rakha EA, Abd El Rehim D, Pinder SE, Lewis SA, Ellis IO. E-cadherin expression in invasive non-lobular carcinoma of the breast and its prognostic significance. Histopathology. 2005;46:685–693. doi: 10.1111/j.1365-2559.2005.02156.x. [DOI] [PubMed] [Google Scholar]
- 23.Syrigos KN, et al. E-cadherin expression in bladder cancer using formalin-fixed, paraffin-embedded tissues: correlation with histopathological grade, tumour stage and survival. Int J Cancer. 1995;64:367–370. doi: 10.1002/ijc.2910640603. [DOI] [PubMed] [Google Scholar]
- 24.Wijnhoven BP, Pignatelli M, Dinjens WN, Tilanus HW. Reduced p120ctn expression correlates with poor survival in patients with adenocarcinoma of the gastroesophageal junction. J Surg Oncol. 2005;92:116–123. doi: 10.1002/jso.20344. [DOI] [PubMed] [Google Scholar]
- 25.Zheng Z, et al. Downregulation and abnormal expression of E-cadherin and β-catenin in nasopharyngeal carcinoma: close association with advanced disease stage and lymph node metastasis. Hum Pathol. 1999;30:458–466. doi: 10.1016/s0046-8177(99)90123-5. [DOI] [PubMed] [Google Scholar]
- 26.Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392:190–193. doi: 10.1038/32433. [DOI] [PubMed] [Google Scholar]
- 27.Derksen PW, et al. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell. 2006;10:437–449. doi: 10.1016/j.ccr.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 28.Davis MA, Reynolds AB. Blocked acinar development, E-cadherin reduction, and intraepithelial neoplasia upon ablation of p120-catenin in the mouse salivary gland. Dev Cell. 2006;10:21–31. doi: 10.1016/j.devcel.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 29.Perez-Moreno M, Song W, Pasolli HA, Williams SE, Fuchs E. Loss of p120 catenin and links to mitotic alterations, inflammation, and skin cancer. Proc Natl Acad Sci USA. 2008;105:15399–15404. doi: 10.1073/pnas.0807301105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kobielak A, Fuchs E. Links between α-catenin, NF-κB, and squamous cell carcinoma in skin. Proc Natl Acad Sci USA. 2006;103:2322–2327. doi: 10.1073/pnas.0510422103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kobielak A, Fuchs E. α-catenin: at the junction of intercellular adhesion and actin dynamics. Nature Rev Mol Cell Biol. 2004;5:614–625. doi: 10.1038/nrm1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Green KJ, Gaudry CA. Are desmosomes more than tethers for intermediate filaments? Nature Rev Mol Cell Biol. 2000;1:208–216. doi: 10.1038/35043032. [DOI] [PubMed] [Google Scholar]
- 33.Thomason HA, Scothern A, McHarg S, Garrod DR. Desmosomes: adhesive strength and signalling in health and disease. Biochem J. 2010;429:419–433. doi: 10.1042/BJ20100567. [DOI] [PubMed] [Google Scholar]
- 34.Chitaev NA, Troyansovsky SM. Direct Ca2+ -dependent heterophilic interaction between desmosomal cadherins, desmoglein and desmocollin, contributes to cell-cell adhesion. J Cell Biol. 1997;138:193–201. doi: 10.1083/jcb.138.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heupel WM, Zillikens D, Drenckhahn D, Waschke J. Pemphigus vulgaris IgG directly inhibit desmoglein3-mediated transinteraction. J Immunol. 2008;181:1825–1834. doi: 10.4049/jimmunol.181.3.1825. [DOI] [PubMed] [Google Scholar]
- 36.Syed SE, et al. Molecular interactions between desmosomal cadherins. Biochem J. 2002;362:317–327. doi: 10.1042/0264-6021:3620317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waschke J, Bruggeman P, Baumgartner W, Zillkens D, Drenckhahn D. Pemphigus foliaceus IgG causes dissociation of desmoglein 1 -containing junctions without blocking desmoglein 1 transinteraction. J Clin Invest. 2005;115:3157–3165. doi: 10.1172/JCI23475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koeser J, Troyanovsky SM, Grund C, Franke WW. De novo formation of desmosomes in cultured cells upon transfection of genes encoding specific desmosomal components. Exp Cell Res. 2003;285:114–130. doi: 10.1016/s0014-4827(03)00016-8. [DOI] [PubMed] [Google Scholar]
- 39.Nie Z, Merritt A, Rouhi-Parkouhi M, Tabernero L, Garrod D. Membrane-impermeable cross-linking provides evidence for homophilic, isoform-specific binding of desmosomal cadherins in epithelial cells. J Biol Chem. 2011;286:2143–2154. doi: 10.1074/jbc.M110.192245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Runswick SK, O'Hare MJ, Jones L, Streuli CH, Garrod DR. Desmosomal adhesion regulates epithelial morphogenesis and cell positioning. Nature Cell Biol. 2001;3:823–830. doi: 10.1038/ncb0901-823. [DOI] [PubMed] [Google Scholar]
- 41.Witcher LL, et al. Desmosomal cadherin binding domains of plakoglobin. J Biol Chem. 1996;271:10904–10909. doi: 10.1074/jbc.271.18.10904. [DOI] [PubMed] [Google Scholar]
- 42.Mathur M, Goodwin L, Cowin P. Interactions of the cytoplasmic domain of the desmosomal cadherin Dsg1 with plakoglobin. J Biol Chem. 1994;269:14075–14080. [PubMed] [Google Scholar]
- 43.Roh JY, Stanley JR. Plakoglobin binding by human Dsg3 (pemphigus vulgaris antigen) in keratinocytes requires the cadherin-like intracytoplasmic segment. J Invest Dermatol. 1995;104:720–724. doi: 10.1111/1523-1747.ep12606963. [DOI] [PubMed] [Google Scholar]
- 44.Troyanovsky SM, et al. Identification of the plakoglobin-binding domain in desmoglein and its role in plaque assembly and intermediate filament anchorage. J Cell Biol. 1994;127:151–160. doi: 10.1083/jcb.127.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bonne S, et al. Defining desmosomal plakophilin-3 interactions. J Cell Biol. 2003;161:403–416. doi: 10.1083/jcb.200303036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen X, Bonne S, Hatzfeld M, van Roy F, Green KJ. Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and β-catenin signaling. J Biol Chem. 2002;277:10512–10522. doi: 10.1074/jbc.M108765200. [DOI] [PubMed] [Google Scholar]
- 47.Hatzfeld M, Haffner C, Schulze K, Vinzens U. The function of plakophilin 1 in desmosome assembly and actin filament organization. J Cell Biol. 2000;149:209–222. doi: 10.1083/jcb.149.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Butz S, Stappert J, Weissig H, Kemler R. Plakoglobin and β-catenin: distinct but closely related. Science. 1992;257:1142–1144. doi: 10.1126/science.257.5073.1142-a. [DOI] [PubMed] [Google Scholar]
- 49.McCrea PD, Turck CW, Gumbiner B. A homolog of the armadillo protein in Drosophila (plakoglobin) associated with E-cadherin. Science. 1991;254:1359–1361. doi: 10.1126/science.1962194. [DOI] [PubMed] [Google Scholar]
- 50.Nathke IS, Hinck L, Swedlow JR, Papkoff J, Nelson WJ. Defining interactions and distributions of cadherin and catenin complexes in polarized epithelial cells. J Cell Biol. 1994;125:1341–1352. doi: 10.1083/jcb.125.6.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhurinsky J, Shtutman M, Ben-Ze'ev A. Plakoglobin and β-catenin: protein interaction, regulation and biological roles. J Cell Sci. 2000;113:3127–3139. doi: 10.1242/jcs.113.18.3127. [DOI] [PubMed] [Google Scholar]
- 52.Conacci-Sorrell ME, et al. Nr-CAM is a target gene of the β-catenin/LEF-1 pathway in melanoma and colon cancer and its expression enhances motility and confers tumorigenesis. Genes Dev. 2002;16:2058–2072. doi: 10.1101/gad.227502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mertens C, Kuhn C, Franke WW. Plakophilins 2a and 2b: constitutive proteins of dual location in the karyoplasm and the desmosomsal plaque. J Cell Biol. 1996;135:1009–1025. doi: 10.1083/jcb.135.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bornslaeger EA, et al. Plakophilin 1 interferes with plakoglobin binding to desmoplakin, yet together with plakoglobin promotes clustering of desmosomal plaque complexes at cell-cell borders. J Cell Sci. 2001;114:727–738. doi: 10.1242/jcs.114.4.727. [DOI] [PubMed] [Google Scholar]
- 55.Kowalczyk AP, et al. The amino-terminal domain of desmoplakin binds to plakoglobin and clusters desmosomal cadherin-plakoglobin complexes. J Cell Biol. 1997;139:773–784. doi: 10.1083/jcb.139.3.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kouklis PD, Hutton E, Fuchs E. Making a connection: direct binding between keratin intermediate filaments and desmosomal proteins. J Cell Biol. 1994;127:1049–1060. doi: 10.1083/jcb.127.4.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith EA, Fuchs E. Defining the interactions between intermediate filaments and desmosomes. J Cell Biol. 1998;141:1229–1241. doi: 10.1083/jcb.141.5.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ihrie RA, et al. Perp is a p63-regulated gene essential for epithelial integrity. Cell. 2005;120:843–856. doi: 10.1016/j.cell.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 59.Attardi LD, et al. PERP, an apoptosis-associated target of p53, is a novel member of the PMP-22/gas3 family. Genes Dev. 2000;14:704–718. [PMC free article] [PubMed] [Google Scholar]
- 60.Carroll DK, Brugge JS, Attardi LD. p63, cell adhesion, and survival. Cell Cycle. 2007;6:255–261. doi: 10.4161/cc.6.3.3799. [DOI] [PubMed] [Google Scholar]
- 61.Flores ER, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cell. 2005;7:363–373. doi: 10.1016/j.ccr.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 62.Den Z, Cheng X, Merched-Sauvage M, Koch PJ. Desmocollin 3 is required for pre-implantation development of the mouse embryo. J Cell Sci. 2006;199:482–489. doi: 10.1242/jcs.02769. [DOI] [PubMed] [Google Scholar]
- 63.Eshkind L, et al. Loss of desmoglein 2 suggests essential functions for early embryonic development and proliferation of embryonal stem cells. Eur J Cell Biol. 2002;81:592–598. doi: 10.1078/0171-9335-00278. [DOI] [PubMed] [Google Scholar]
- 64.Gallicano GI, et al. Desmoplakin is required early in development for assembly of desmosomes and cytoskeletal linkage. J Cell Biol. 1998;143:2009–2022. doi: 10.1083/jcb.143.7.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bierkamp C, Mclaughlin KJ, Schwarz H, Huber O, Kemler R. Embryonic heart and skin defects in mice lacking plakglobin. Dev Biol. 1996;180:780–785. doi: 10.1006/dbio.1996.0346. [DOI] [PubMed] [Google Scholar]
- 66.Ruiz P, et al. Targeted mutation of plakoglobin in mice reveals essential functions of desmosomes in the embryonic heart. J Cell Biol. 1996;135:215–225. doi: 10.1083/jcb.135.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koch PJ, et al. Targeted disruption of the pemphigus vulgaris antigen (desmoglein 3) gene in mice causes loss of keratinocyte cell adhesion with a phenotype similar to pemphigus vulgaris. J Cell Biol. 1997;137:1091–1102. doi: 10.1083/jcb.137.5.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vasioukhin V, Bowers E, Bauer C, Degenstein L, Fuchs E. Desmoplakin is essential in epidermal sheet formation. Nature Cell Biol. 2001;3:1076–1085. doi: 10.1038/ncb1201-1076. [DOI] [PubMed] [Google Scholar]
- 69.Chidgey M, et al. Mice lacking desmocollin 1 show epidermal fragility accompanied by barrier defects and abnormal differentiation. J Cell Biol. 2001;155:821–832. doi: 10.1083/jcb.200105009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen J, Den Z, Koch PJ. Loss of desmocollin 3 in mice leads to epidermal blistering. J Cell Sci. 2008;121:2844–2849. doi: 10.1242/jcs.031518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chidgey M. Desmosomes and disease: an update. Histol Histopathol. 2002;17:1179–1192. doi: 10.14670/HH-17.1179. [DOI] [PubMed] [Google Scholar]
- 72.Brennan D, Mahoney MG. Increased expression of Dsg2 in malignant skin carcinomas: a tissue-microarrary based study. Cell Adh Migr. 2009;3:148–154. doi: 10.4161/cam.3.2.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen YJ, et al. DSG3 is overexpressed in head neck cancer and is a potential moelcular target for inhibition of oncogenesis. Oncogene. 2007;26:467–476. doi: 10.1038/sj.onc.1209802. [DOI] [PubMed] [Google Scholar]
- 74.Furukawa C, et al. Plakophilin 3 oncogene as prognostic marker and therapeutic target for lung cancer. Cancer Res. 2005;65:7102–7110. doi: 10.1158/0008-5472.CAN-04-1877. [DOI] [PubMed] [Google Scholar]
- 75.Kurzen H, Munzing I, Hartschuh W. Expression of desmosomal proteins in squamous cell carcinomas of the skin. J Cutan Pathol. 2003;30:621–630. doi: 10.1034/j.1600-0560.2003.00122.x. [DOI] [PubMed] [Google Scholar]
- 76.Breuninger S, et al. Desmosomal plakophilins in the prostate and prostatic adenocarcinomas: implications for diagnosis and tumor progression. Am J Pathol. 2010;176:2509–2519. doi: 10.2353/ajpath.2010.090737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alazawi WO, Morris LS, Stanley MA, Garrod DR, Coleman N. Altered expression of desmosomal components in high-grade squamous intraepithelial lesions of the cervix. Virchows Arch. 2003;443:51–56. doi: 10.1007/s00428-003-0771-9. [DOI] [PubMed] [Google Scholar]
- 78.Alroy J, Pauli BU, Weinstein RS. Correlation between numbers of desmosomes and the aggressiveness of transitional cell carcinoma in human urinary bladder. Cancer. 1981;47:104–112. doi: 10.1002/1097-0142(19810101)47:1<104::aid-cncr2820470118>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 79.de Boer CJ, et al. Changing roles of cadherins and catenins during progression of squamous intraepithelial lesions in the uterine cervix. Am J Pathol. 1999;155:505–515. doi: 10.1016/S0002-9440(10)65146-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Demirag GG, Sullu Y, Gurgenyatagi D, Okumus NO, Yucel I. Expression of plakophilins (PKP1, PKP2, and PKP3) in gastric cancers. Diagn Pathol. 2011;6:1. doi: 10.1186/1746-1596-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Harada T, Shinohara M, Nakamura S, Shimada M, Oka M. Immunohistochemical detection of desmosomes in oral squamous cell carcinomas: correlation with differentiation, mode of invasion, and metastatic potential. Int J Oral Maxillofac Surg. 1992;21:346–349. doi: 10.1016/s0901-5027(05)80759-3. [DOI] [PubMed] [Google Scholar]
- 82.Hiraki A, et al. Immunohistochemical staining of desmosomal components in oral squamous cell carcinomas and its association with tumour behaviour. Br J Cancer. 1996;73:1491–1497. doi: 10.1038/bjc.1996.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kocher O, Amaudruz M, Schindler AM, Gabbiani G. Desmosomes and gap junctions in precarcinomatous and carcinomatous conditions of squamous epithelia. An electron microscopic and morphometrical study. J Submicrosc Cytol. 1981;13:267–281. [PubMed] [Google Scholar]
- 84.Nei H, et al. Expression of component desmosomal proteins in uterine endometrial carcinoma and their relation to cellular differentiation. Cancer. 1996;78:461–470. doi: 10.1002/(SICI)1097-0142(19960801)78:3<461::AID-CNCR13>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 85.Tada H, Hatoko M, Tanaka A, Kuwahara M, Muramatsu T. Expression of desmoglein I and plakoglobin in skin carcinomas. J Cutan Pathol. 2000;27:24–29. doi: 10.1034/j.1600-0560.2000.027001024.x. [DOI] [PubMed] [Google Scholar]
- 86.Biedermann K, et al. Desmoglein 2 is expressed abnormally rather than mutated in familial and sporadic gastric cancer. J Pathol. 2005;207:199–206. doi: 10.1002/path.1821. [DOI] [PubMed] [Google Scholar]
- 87.Depondt J, Shabana AH, Florescu-Zorila S, Gehanno P, Forest N. Down-regulation of desmosomal molecules in oral and pharyngeal squamous cell carcinomas as a marker for tumour growth and distant metastasis. Eur J Oral Sci. 1999;107:183–193. doi: 10.1046/j.0909-8836.1999.eos1070305.x. [DOI] [PubMed] [Google Scholar]
- 88.Kahn K, et al. Desmocollin switching in colorectal cancer. Br J Cancer. 2006;95:1367–1370. doi: 10.1038/sj.bjc.6603453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Oshiro MM, et al. Epigenetic silencing of DSC3 is a common event in human breast cancer. Breast Cancer Res. 2005;7:R669–R680. doi: 10.1186/bcr1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Papagerakis S, et al. Altered desmoplakin expression at transcriptional and protein levels provides prognostic information in human oropharyngeal cancer. Hum Pathol. 2009;40:1320–1329. doi: 10.1016/j.humpath.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 91.Shiina H, et al. Functional loss of the γ-catenin gene through epigenetic and genetic pathways in human prostate cancer. Cancer Res. 2005;65:2130–2138. doi: 10.1158/0008-5472.CAN-04-3398. [DOI] [PubMed] [Google Scholar]
- 92.Sobolik-Delmaire T, Katafiasz D, Keim SA, Mahoney MG, Wahl JK. Decreased plakophilin-1 expression promotes increased motility in head and neck squamous cell carcinoma cells. Cell Commun Adhes. 2007;14:99–109. doi: 10.1080/15419060701463082. [DOI] [PubMed] [Google Scholar]
- 93.Winn RA, et al. γ-catenin expression is reduced or absent in a subset of human lung cancers and re-expression inhibits transformed cell growth. Oncogene. 2002;21:7497–7506. doi: 10.1038/sj.onc.1205963. [DOI] [PubMed] [Google Scholar]
- 94.Yashiro M, Nishioka N, Hirakawa K. Decreased expression of the adhesion molecule desmoglein-2 is associated with diffuse-type gastric carcinoma. Eur J Cancer. 2006;42:2397–2403. doi: 10.1016/j.ejca.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 95.Wong MP, et al. Loss of desmoglein1 expression associated with worse prognosis in head and neck squamous cell carcinoma patients. Pathology. 2008;40:611–616. doi: 10.1080/00313020802320614. [DOI] [PubMed] [Google Scholar]
- 96.Collins JE, Taylor I, Garrod DR. A study of desmosomes in colorectal carcinoma. Br J Cancer. 1990;62:796–805. doi: 10.1038/bjc.1990.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hakimelahi S, et al. Plakoglobin regulates the expression of the anti-apoptotic protein BCL-2. J Biol Chem. 2000;275:10905–10911. doi: 10.1074/jbc.275.15.10905. [DOI] [PubMed] [Google Scholar]
- 98.Kolligs FT, et al. γ-catenin is regulated by the APC tumor suppressor and its oncogenic activity is distinct from that of β-catenin. Genes Dev. 2000;14:1319–1331. [PMC free article] [PubMed] [Google Scholar]
- 99.Brennan D, et al. Suprabasal Dsg2 expression in transgenic mouse skin congers a hyperproliferative and apoptosis-resistant phenotype to keratinocytes. J Cell Sci. 2007;120:758–771. doi: 10.1242/jcs.03392. [DOI] [PubMed] [Google Scholar]
- 100.Tselepis C, Chidgey M, North A, Garrod D. Desmosomal adhesion inhibits invasive behavior. Proc Natl Acad Sci USA. 1998;95:8064–8069. doi: 10.1073/pnas.95.14.8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Simcha I, Geiger B, Yehuda-Levenberg S, Salomon D, Ben-Ze'ev A. Suppression of tumorigenicity by plakoglobin: an augmenting effect of N-cadherin. J Cell Biol. 1996;133:199–209. doi: 10.1083/jcb.133.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Reiger-Christ KM, et al. Restoration of plakoglobin expression in bladder carcinoma cell lines suppresses cell migration and tumorigenic potential. Br J Cancer. 2005;92:2153–2159. doi: 10.1038/sj.bjc.6602651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.De Bruin A, et al. Loss of invasiveness in squamous cell carcinoma cells overexpressing desmosomal cadherins. Cell Adhes Commun. 1999;7:13–28. doi: 10.3109/15419069909034389. [DOI] [PubMed] [Google Scholar]
- 104.Kundu ST, et al. Plakophilin3 downregulation leads to a decrease in cell adhesion and promotes metastasis. Int J Cancer. 2008;123:2303–2314. doi: 10.1002/ijc.23797. [DOI] [PubMed] [Google Scholar]
- 105.Caca K, et al. β- and γ-catenin mutations, but not E-cadherin inactivation, underlie T-cell factor/lymphoid enhancer factor transcriptional deregulation in gastric and pancreatic cancer. Cell Growth Differ. 1999;10:369–376. [PubMed] [Google Scholar]
- 106.Chun MG, Hanahan D. Genetic deletion of the desmosomal component desmoplakin promotes tumor microinvasion in a mouse model of pancreatic neuroendocrine carcinogenesis. PLoS Genet. 2010;6:e1001120. doi: 10.1371/journal.pgen.1001120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lewis JE, Jensen PJ, Wheelock MJ. Cadherin function is required for human keratinocytes to assemble desmosomes and stratify in response to calcium. J Invest Dermatol. 1994;102:870–877. doi: 10.1111/1523-1747.ep12382690. [DOI] [PubMed] [Google Scholar]
- 108.Beaudry VG, et al. Loss of the p53/p63 regulated desmosomal protein Perp promotes tumorigenesis. PLoS Genet. 2010;6:e1001168. doi: 10.1371/journal.pgen.1001168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Melnikova VO, Ananthaswamy HN. Cellular and molecular events leading to the development of skin cancer. Mutat Res. 2005;571:91–106. doi: 10.1016/j.mrfmmm.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 110.Karnovsky A, Klymkowsky MW. Anterior axis duplication in Xenopus induced by the over-expression of the cadherin-binding protein plakoglobin. Proc Natl Acad Sci USA. 1995;92:4522–4526. doi: 10.1073/pnas.92.10.4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Klymkowsky MW, Williams BO, Barish GD, Varmus HE, Vourgourakis YE. Membrane-anchored plakoglobins have multiple mechanisms of action in Wnt signaling. Mol Biol Cell. 1999;10:3151–3169. doi: 10.1091/mbc.10.10.3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Merriam JM, Rubenstein AB, Klymkowsky MW. Cytoplasmically anchored plakoglobin induces a WNT-like phenotype in Xenopus. Dev Biol. 1997;185:67–81. doi: 10.1006/dbio.1997.8550. [DOI] [PubMed] [Google Scholar]
- 113.Miller JR, Moon RT. Analysis of the signaling activities of localization mutants of β-catenin during axis specification in Xenopus. J Cell Biol. 1997;139:229–243. doi: 10.1083/jcb.139.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Simcha I, et al. Differential nuclear translocation and transactivation potential of β-catenin and plakoglobin. J Cell Biol. 1998;141:1433–1448. doi: 10.1083/jcb.141.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li L, Chapman K, Hu X, Wong A, Pasdar M. Modulation of the oncogenic potential of β-catenin by the subcellular distribution of plakoglobin. Mol Carcinog. 2007;46:824–838. doi: 10.1002/mc.20310. [DOI] [PubMed] [Google Scholar]
- 116.Salomon D, et al. Regulation of β-catenin levels and localization by overexpression of plakoglobin and inhibition of the ubiquitin-proteasome system. J Cell Biol. 1997;139:1325–1335. doi: 10.1083/jcb.139.5.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Miravet S, et al. Tyrosine phosphorylation of plakoglobin causes contrary effects on its association with desmosomes and adherens junction components and modulates β-catenin-mediated transcription. Mol Cell Biol. 2003;23:7391–7402. doi: 10.1128/MCB.23.20.7391-7402.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhurinsky J, Shtutman M, Ben-Ze'ev A. Differential mechanisms of LEF/TCF family-dependent transcriptional activation by β-catenin and plakoglobin. Mol Cell Biol. 2000;20:4238–4252. doi: 10.1128/mcb.20.12.4238-4252.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shtutman M, et al. The cyclin D1 gene is a target of the β-catenin/LEF-1 pathway. Proc Natl Acad Sci USA. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Williamson L, et al. Pemphigus vulgaris identifies plakoglobin as key suppressor of c-Myc in the skin. EMBO J. 2006;25:3298–3309. doi: 10.1038/sj.emboj.7601224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Maeda O, et al. Plakoglobin (γ-catenin) has TCF/LEF family-dependent transcriptional activity in β-catenin-deficient cell line. Oncogene. 2004;23:964–972. doi: 10.1038/sj.onc.1207254. [DOI] [PubMed] [Google Scholar]
- 122.Teuliere J, et al. β-catenin-dependent and -independent effects of DeltaN-plakoglobin on epidermal growth and differentiation. Mol Cell Biol. 2004;24:8649–8661. doi: 10.1128/MCB.24.19.8649-8661.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Garcia-Gras E, et al. Suppression of canonical Wnt/β-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 2006;116:2012–2021. doi: 10.1172/JCI27751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li J, et al. Cardiac tissue-restricted deletion of plakoglobin results in progressive cardiomyopathy and activation of β-catenin signaling. Mol Cell Biol. 2011;31:1134–1144. doi: 10.1128/MCB.01025-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Martin ED, Moriarty MA, Byrnes L, Grealy M. Plakoglobin has both structural and signalling roles in zebrafish development. Dev Biol. 2009;327:83–96. doi: 10.1016/j.ydbio.2008.11.036. [DOI] [PubMed] [Google Scholar]
- 126.Schmidt A, et al. Plakophilins 1a and 1b: widespread nuclear proteins recruited in specific epithelial cells as desmosomal plaque components. Cell Tissue Res. 1997;290:481–499. doi: 10.1007/s004410050956. [DOI] [PubMed] [Google Scholar]
- 127.Sobolik-Delmaire T, Reddy R, Pashaj A, Roberts BJ, Wahl JK. Plakophilin-1 localizes to the nucleus and interacts with single-stranded DNA. J Invest Dermatol. 2010;130:2638–2646. doi: 10.1038/jid.2010.191. [DOI] [PubMed] [Google Scholar]
- 128.Wolf A, et al. Plakophilin 1 stimulates translation by promoting eIF4A1 activity. J Cell Biol. 2010;188:463–471. doi: 10.1083/jcb.200908135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hofmann I, et al. Identification of the junctional plaque protein plakophilin 3 in cytoplasmic particles containing RNA-binding proteins and the recruitment of plakophilins 1 and 3 to stress granules. Mol Biol Cell. 2006;17:1388–1398. doi: 10.1091/mbc.E05-08-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Papagerakis S, Shabana AH, Depondt J, Gehanno P, Forest N. Immunohistochemical localization of plakophilins (PKP1, PKP2, PKP3, and p0071) in primary oropharyngeal tumors: correlation with clinical parameters. Hum Pathol. 2003;34:565–572. doi: 10.1016/s0046-8177(03)00174-6. [DOI] [PubMed] [Google Scholar]
- 131.Berkowitz P, et al. Desmosome signaling. Inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J Biol Chem. 2005;280:23778–23784. doi: 10.1074/jbc.M501365200. [DOI] [PubMed] [Google Scholar]
- 132.Wan H, South AP, Hart IR. Increased keratinocyte proliferation initiated through downregulation of desmoplakin by RNA interference. Exp Cell Res. 2007;313:2336–2344. doi: 10.1016/j.yexcr.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 133.Garrod DR, Berika MY, Bardsley WF, Holmes D, Tabernero L. Hyper-adhesion in desmosomes: its regulation in wound healing and possible relationship to cadherin crystal structure. J Cell Sci. 2005;118:5743–5754. doi: 10.1242/jcs.02700. [DOI] [PubMed] [Google Scholar]
- 134.Kimura TE, Merritt AJ, Garrod DR. Calcium-independent desmosomes of keratinocytes are hyper-adhesive. J Invest Dermatol. 2007;127:775–781. doi: 10.1038/sj.jid.5700643. [DOI] [PubMed] [Google Scholar]
- 135.Luthra R, et al. Gene expression profiling of localized esophageal carcinomas: association with pathologic response to preoperative chemoradiation. J Clin Oncol. 2006;24:259–267. doi: 10.1200/JCO.2005.03.3688. [DOI] [PubMed] [Google Scholar]