

Abstract
Several Arenaviruses cause hemorrhagic fever (HF) disease in humans for which there are no licensed vaccines, and current therapy is limited to the use of ribavirin (Rib) that is only partially effective and associated with significant side effects. In addition, compelling evidence indicates that the prototypic arenavirus lymphocytic choriomeningitis virus (LCMV) is a neglected human pathogen of clinical significance. Therefore, it is important to develop novel and effective antiarenaviral drugs. The arenavirus Z protein is the driving force of arenavirus budding, and PPPY and PTAP late (L) domain motifs within Z are critical for Z-mediated budding, which involves the interaction of Z with a variety of host cellular factors. Compounds capable of inhibiting these virus-host cell interactions represent candidate anti-arenaviral drugs. The identification of these candidate compounds would be facilitated by the availability of a Z budding assay amenable to high throughput screens (HTS). To this end, we have developed a novel assay that allows for rapid and quantitative assessment of Z-mediated budding. We provide evidence that this novel assay is amenable to HTS to identify small molecule inhibitors of Z-mediated budding, as well as to uncover cellular genes contributing to arenavirus budding.
Introduction
Several arenaviruses, chiefly Lassa virus (LASV) in West Africa and Junin virus (JUNV) in South America, cause severe hemorrhagic fever (HF) disease in humans (Geisbert and Jahrling, 2004; McCormick and Fisher-Hoch, 2002; Peters, 2002). In addition, increasing evidence indicates that LCMV congenital infections have a significant contribution to neonatal chorioretinitis and a variety of fetal abnormalities including hydrocephalus (Barton, Mets, and Beauchamp, 2002; Jahrling and Peters, 1992; Mets et al., 2000). Morever, LCMV may pose a serious risk to immunocompromised individuals as reflected by cases of fatal LCMV disease associated with transplantation in the USA (Barton, 2006) and Australia (Palacios et al., 2008). No licensed anti-arenavirus vaccines are available, and current anti-arenavirus therapies are limited to the use of the nucleoside analogue ribavirin (Rib), which is only partially effective and often associated with severe side effects including anemia and birth defects (McKee et al., 1988). Therefore, it is important to develop novel effective antiviral strategies to combat arenaviruses. To this end, the development of arenavirus reverse genetics systems (Flatz et al., 2006; Hass et al., 2004; Lopez, Jacamo, and Franze-Fernandez, 2001; Sanchez and de la Torre, 2006) have provided investigators with novel, powerful tools to facilitate the development of assays to identify compounds that target specific steps of the arenavirus life cycle and represent novel candidate antivirals to combat pathogenic arenaviruses.
Arenaviruses are enveloped viruses with a bisegmented, single-stranded, negative sense (NS) RNA genome (Buchmeier, 2007). Each of the two segments uses an ambisense coding strategy to direct the synthesis of two polypeptides. The large segment (L, 7.2 kb) encodes a small (90–99aa, 12 kDa) RING-finger protein called Z and the RNA-dependent RNA polymerase (L protein), while the small segment (S, 3.4 kb) encodes the nucleoprotein (NP) and glycoprotein precursor (GPC). L and NP are the minimal viral trans-acting factors required for virus RNA replication and gene expression (Lee et al., 2000), whereas production of infectious particles also requires GP and Z (Lee et al., 2002). Post-translational cleavage of GPC generates the three components that form the GP complex (GPc): the stable signal peptide (SSP, 58aa), GP1 (Mr 40–46 kD), and GP2 (Mr 35 kD) (Buchmeier and Oldstone, 1979; Kunz et al., 2003; Pinschewer et al., 2003; Strecker et al., 2003; York et al., 2004). Trimers of GP1/GP2 associate via ionic interactions to form the spikes that decorate the virus surface (Eschli et al., 2006). GP1 is located at the top of the spike and mediates virus interactions with host cell surface receptors, while GP2 mediates membrane fusion. Virus replication is confined to the cytoplasm of infected cells and budding of progeny virus occurs at the plasma membrane (Borio et al., 2002; Buchmeier, de la Torre, and Peters, 2007; Dalton et al., 1968; Murphy et al., 1970).
Z has been shown to be the driving force of arenavirus budding (Perez, Craven, and de la Torre, 2003; Strecker et al., 2003; Urata et al., 2006), and is the arenavirus counterpart of the matrix (M) protein found in many enveloped NS riboviruses. These M proteins function to bridge the virus ribonucleoprotein (RNP) core with other viral components, including virus surface glycoproteins, to assemble mature infectious particles whose budding from infected cells is often directed by the interaction of M with a variety of cellular proteins (Lenard, 1996). Consistent with its M-like role, Z interacts with both the NP (Eichler et al., 2004) and the GPc of arenaviruses (Capul et al., 2007) and contains PPPY and PTAP late (L) domain motifs (Perez, Craven, and de la Torre, 2003) that have been shown to play a key role in the budding activity of several retrovirus gag proteins (Garrus et al., 2001; Martin-Serrano, Zang, and Bieniasz, 2001; VerPlank et al., 2001) and M proteins from various RNA viruses (Craven et al., 1999). Myristoylation of Z at its second position glycine also plays a critical role in membrane targeting, association with the GPc, and budding (Capul et al., 2007; Perez, Greenwald, and de la Torre, 2004; Strecker et al., 2006). Z has also been implicated in interactions, via its RING domain, with several cellular proteins including PML (Borden, Campbell Dwyer, and Salvato, 1998), PRH (Topcu et al., 1999), and eIF4E (Campbell Dwyer et al., 2000). These interactions suggest that Z might interfere with cellular processes in arenavirus infected cells, but the precise mechanism(s) underlying these Z-cellular protein interactions and their biological implications remain poorly understood. The identification of Z-interacting cellular proteins required for Z-mediated budding will provide a deeper understanding of this essential step in the arenavirus life cycle and may uncover novel targets for the design of anti-arenaviral compounds.
As with many other characterized bona fide viral budding proteins (VBP), Z-mediated budding involves its interaction with host cell proteins including members of the multivesicular body pathway such as Tsg101 (Perez, Craven, and de la Torre, 2003; Strecker et al., 2003; Urata et al., 2006). Therefore, disrupting these interactions with small molecule compounds would be predicted to reduce Z-mediated budding and consequently result in inhibition of virus propagation within the infected host. The identification of small molecule inhibitors of Z-mediated budding would benefit from using an assay to rapidly and quantitatively assess Z budding activity that would be amenable to HTS. Such an assay could also be adapted to siRNA HTS, which could uncover additional cellular proteins that are involved in Z-mediated budding.
Bona fide VBP, including many M proteins, often have the ability to form virus-like particles (VLP) when expressed independently of virus infection or other virus proteins (Harty et al., 2000; Perez, Craven, and de la Torre, 2003). Therefore, the detection and quantitation of VBP-containing VLP present in tissue culture supernatants (TCS) of VBP-transfected cells has been frequently used to assess VBP-mediated budding. Experimentally, this is done by harvesting VLP from TCS by ultracentrifugation and subsequent detection of VBP (Patch et al., 2007) by Western blotting or immunoprecipitation of VBP-containing VLP in metabolically-labeled cells (Harty et al., 2000). These methods were used to demonstrate that Z was the driving force of arenavirus budding (Perez, Craven, and de la Torre, 2003; Strecker et al., 2003); however, these assays are not suitable for HTS.
Recent work has documented the use of a firefly luciferase (FLuc)-based assay to assess the budding of Ebola virus VP40 (McCarthy, Licata, and Harty, 2006). However, a VP40-FLuc fusion protein that appeared to be incorporated into VLP was not active, and luciferase activity associated with VLP was due to incorporation of FLuc into VP40 and GP-containing VLP by mechanisms that remain uncharacterized. These results raised some concerns regarding the value of the assay to measure the specific budding activity of Ebola virus VP40. In contrast, here we present data showing that a chimeric LASV Z protein fused at its C-terminus to Gaussia luciferase retained wildtype Z budding activity that could be measured by determining levels of GLuc in TCS of Z-GLuc transfected cells. Levels of GLuc in TCS were dependent on the budding activity of Z, and the dynamic range of the assay was suitable for HTS based on the Z′ score of the assay. This novel cell-based luciferase assay to quantify Z-mediated budding performed well in proof-of-concept experiments assessing inhibition of Z-mediated budding by a myristoylation inhibitor, or siRNA mediated knockdown of Tsg101. These findings indicate that this newly developed budding assay should be instrumental for both chemical and genetic HTS aimed at identifying candidate inhibitors of arenavirus budding and additional cellular genes contributing to arenavirus budding.
Materials and methods
Plasmid construction
The plasmid pC-FLuc was generated by releasing the FLuc open reading frame from pEGSH-Luc (Stratagene, La Jolla, CA) by digestion with KpnI and SalI and cloning it into the KpnI and XhoI sites in pCAGGS (Niwa, Yamamura, and Miyazaki, 1991). Hereafter, pCAGGS is indicated by the abbreviation pC. Plasmid pC-GLuc was made by PCR-amplifying the Gaussia luciferase open reading frame from pCMV-hGLuc (New England Biolabs) using the primer pair GLuc forward2 (5′-AGGGGGTACCGGATCCTCCTCCGGGACTGTAGGG-3′) and GLuc reverse (5′-TGCATGCTCGAGCGGCCGCTTAGTC-3′) then cloning into pC using KpnI and XhoI (restriction enzyme recognition sites are underlined). Plasmid pC-LASV-Z(WT)-FLuc was made by amplifying LASV Z from pC-LVZ-HA (Perez, Greenwald, and de la Torre, 2004) using OLVZ1FEco (5′-CCTGAATTCATGGGAAACAAGCAAGCC -3′) and LASVZ1RKpn (5′-AGGGGGTACCGGATCCTCCTCCGGGACTGTAGGG-3′), then cloning the PCR product into the EcoRI and KpnI sites of pC-FLuc. Plasmid pC-LASV-Z(G2A)-FLuc was generated by amplifying LASV-Z(G2A) from pC-LVZ(G2A)-HA (Perez, Greenwald, and de la Torre, 2004) using the primers OLVZG2AFEco (5′-CCTGAATTCATGGCTAACAAGCAAGCCAAAGCC-3′) and LASVZ1RKpn then cloning the resulting PCR product into the EcoRI and KpnI sites of pC-FLuc. Plasmid pC-GLuc –SP was generated by amplifying a region of GLuc to replace the signal peptide as determined by SignalP identification of the GLuc signal peptide (Nielsen, Brunak, and von Heijne, 1999) with a start codon using the primer pair GLuc –SP ORF-F (5′-GGTCCAGGTACCAAGCCCACCGAGAACAAC-3′) and GLuc-R then cloning the resulting PCR product into the KpnI and XhoI sites of pC. To create fusions of LASV Z with GLuc, the plasmid pC-GLuc-ΔSP-intermediate was first generated by amplifying a region of GLuc lacking the signal peptide using the primer pair GLuc –SP-forward1 (5′-GGTCCAGGTACCAAGCCCACCGAGAACAAC-3′) and GLuc-reverse and cloning the resulting PCR product into the KpnI and XhoI sites of pC. The plasmid pC-LASV-Z(WT)-GLuc was generated by subcloning an EcoRI/KpnI fragment from pC-LASV-Z(WT)-FLuc into the EcoRI and KpnI sites of pC-GLuc-ΔSP-intermediate. The plasmid pC-LASV-Z(G2A)-GLuc was generated by subcloning an EcoRI/KpnI fragment from pC-LASV-Z(G2A)-FLuc into the EcoRI and KpnI sites of pC-GLuc-ΔSP-intermediate. All plasmids were prepared using Qiagen kits and were sequenced prior to transfection.
VLP assays
VLP assays were done as previously described (Perez, Craven, and de la Torre, 2003). Briefly, 293T cells were transfected with pC-LASV-Z-HA, pC-LASV-Z-G2A-HA, pC-LASV-Z(WT)-GLuc, or pC-LASV-Z(G2A)-GLuc using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to manufacturer’s instructions. Hereafter, Lipofectamine 2000 is indicated in the text with the abbreviation LF2000. At 48–72h post-transfection cell lysates (CL) were collected in lysis buffer (50mM Tris pH8.0/62.5mM EDTA/1% NP-40/0.4% DOC) and TCS centrifuged through a 20% cushion at 100,000×g, 4°C, for 1h and the resulting pellet containing VLPs was dissolved in 2X SDS sample buffer. VLPs and CL were analyzed by western blotting using antibodies to either HA or GLuc.
Western blot analysis
Aliquots of cell lysates and VLPs were loaded onto 12% polyacrylamide Tris-glycine gels and run under constant voltage in Tris-glycine buffer (Sambrook, Fritsch, and Maniatis, 1989). Gels were transferred to PVDF membranes at room temperature and blocked overnight at 4°C in TBS containing 1% blocking buffer (Roche, Indianapolis, IN). Membranes were reacted with anti-GLuc antibody (Nanolight, Pinetop, AZ) or anti-HA antibody (Santa Cruz Biotechnology, Santa Cruz, CA) followed by an anti-rabbit peroxidase conjugated antibody (Pierce, Rockford, IL). Blots were processed for ECL using Dura Stable western blotting reagent (Pierce) then exposed to X-ray film (Kodak).
Luciferase-based budding assay
293T cells were transfected with the indicated plasmids using the following conditions: 0.2 ug DNA, 0.5 μL LF2000 and 1.5 × 105 cells/96-well. 96-well plates were also coated with 0.01% poly-lysine (in PBS, Sigma, St. Louis, MO) to facilitate cell attachment. After 18h, transfection mix was changed to DMEM (Invitrogen) containing 5% FBS. At 48–72h post-transfection, TCS was collected and clarified by low-speed centrifugation to remove detached cells. 5–20% of the TCS (20 μL) was aliquoted to 96-well black plates (VWR, West Chester, PA) and either an equivalent volume of SteadyGlo luciferase reagent (Promega, Madison, WI) or 50 μL Gaussia luciferase assay reagent (New England Biolabs, Ipswich, MA) was added to each well. Firefly luciferase activity was measured after 10 minutes incubation at room temperature using a 10 sec read time per well while Gaussia luciferase activity was read for 5 sec following injection of the assay substrate and a 0.3 sec delay per well (Remy and Michnick, 2006). All measurements were done using a Berthold Centro LB 960 luminometer (Berthold Technologies, Oak Ridge, TN). To assay Gaussia luciferase activity in cells, 25 μL of luciferase lysis buffer (NEB) was added to cells in 96-well plates and lysates were transferred to 96-well black plates for measurement of GLuc activity. To measure firefly luciferase activity in cells, 50 μL of SteadyGlo luciferase reagent was added to cells and incubated for 10 minutes to achieve lysis. The cell lysate containing FLuc assay reagent was then transferred to 96-well black plates for measurement of luciferase activity.
Cell viability assay
Cells were also lysed for measurement of cell viability using CellTiter Glo (Promega, Madison, WI), which determines the quantity of ATP present and, therefore, the presence of metabolically active cells. This assay utilizes a proprietary luciferase, whose substrate (beetle luciferin) is not used by GLuc (a coelenterazine-type luciferase) and can therefore be used to measure viability in GLuc-overexpressing cells with no interference by GLuc. GLuc measurements were normalized to cell viability after setting the viability of cells transfected with empty vector to 1. This normalization was necessary to account for variations in cell number between wells following transfection.
Pharmacological inhibition of budding
When cells were treated with 2-hydoxymyristic acid, 293T cells were transfected with plasmid using the following conditions: 125ng DNA in 25 μL Opti-MEM (Invitrogen, Carlsbad, CA), 0.31 μL LF2000 in 25 μL Opti-MEM, ca. 8 × 104 cells per M96 well. Cells were transfected in the presence of 1) medium, 2) 28mM DMSO, or 3) 28mM DMSO/100 uM 2-hydroxymyristic acid (Cayman Biologicals, Ann Arbor, MI). Forty-eight hours post-transfection, TCS was collected and clarified prior to GLuc assays and cells were lysed for measurement of GLuc activity or cell viability using CellTiter Glo.
siRNA transfection
293T cells were transfected with a single dose of siRNA using reverse transfection where transfection mix is aliquoted to wells prior to the addition of cells in suspension, a common technique used to deliver siRNA to cells in high-throughput formats (Opaluch et al., 2007). Transfection was done using the following conditions: 20nM siRNA, 0.2 μL RNAiMAX (Invitrogen, Carlsbad, CA), 2×104 cells per 96 well previously coated with poly-lysine. At 8h post-transfection, medium was changed to DMEM containing 1% FBS. At 24h post-transfection cells were transfected with pC-Z(WT)-GLuc using the following conditions: 125ng DNA total, 0.32 μL LF2000, 50 μL Opti-MEM per well. Complexes were prepared in Opti-MEM and added directly to the wells. At 24h post-transfection, cells were lysed to measure cell viability using CellTiter GLO or GLuc activity and TCS was collected, clarified, and 20 μL was transferred to black 96-well plates for GLuc assays. A non-specific siRNA (control siRNA #1, Ambion, now under Invitrogen, Carlsbad, CA) was used as a negative control along with an siRNA targeted against Tsg101 (using the sequence 5′-CAG CTG AGG GCA CTA ATG CAA-3′). To assess budding efficiency, we used the following calculation: GLuc activity (TCS)/([GLuc activity (TCS)]+[ GLuc activity (CL)]). To assess siRNA transfection efficiency, cells were also transfected with a siRNA directed against the gene KIF11 (Ambion) using the same conditions as above. Cells that are positively transfected exhibit a rounded appearance due to knockdown of the cytoskeletal-related protein KIF11. We observed a strong effect in over 90% of cells transfected with this siRNA (data not shown).
Results
Budding activity of chimeric Z-luciferase proteins
The FLuc protein (550aa) has been extensively used as a reporter both in vitro (Stankunas et al., 2007) and in vivo (Gross and Piwnica-Worms, 2005). We therefore evaluated its use for the development of an assay to assess LASV Z-mediated budding. We reasoned that fusion of FLuc to the C-terminus of LASV Z would result in a chimeric protein LASV-Z(WT)-FLuc whose budding activity could be determined by measuring levels of FLuc activity in TCS of LASV-Z(WT)-FLuc transfected cells. Likewise, we predicted that fusion of FLuc to the C-terminus of the G2A mutant of LASV Z should result in a chimeric LASV-Z(G2A)-FLuc protein with severe impaired budding activity due to the lack of myristoylation required for Z-mediated budding (Perez, Greenwald, and de la Torre, 2004; Strecker et al., 2006). To examine this, we evaluated the levels of FLuc activity in cell lysates and TCS from cells transfected with either LASV-Z(WT)-FLuc or LASV-Z(G2A)-FLuc. We observed similar levels of FLuc activity in total cell lysates of cells transfected with either LASV-Z(WT)-FLuc or LASV-Z(G2A)-FLuc, which were about 2-fold lower than levels observed in FLuc-transfected cells (Figure 1A). These differences correlated well with levels of protein expression as assessed by western blotting (Figure 1C). FLuc activity in TCS correlated with the predicted budding activity of the corresponding Z fusion protein (Figure 1B), but the difference between LASV-Z(WT)-FLuc and LASV-Z(G2A)-FLuc was at best only 2-fold. We also observed that in LASV-Z(WT)-FLuc-transfected cells, levels of FLuc activity were about 1000-fold lower in TCS compared to cell lysates. These results indicated that fusion of FLuc to the C-terminus of Z resulted in a dramatic impairment of Z budding activity, thereby preventing the use of LASV-Z(WT)-FLuc for the development of a reporter for Z-mediated budding.
Figure 1. Luciferase activity in CL and TCS of cells transfected with LASV-Z-FLuc chimera.
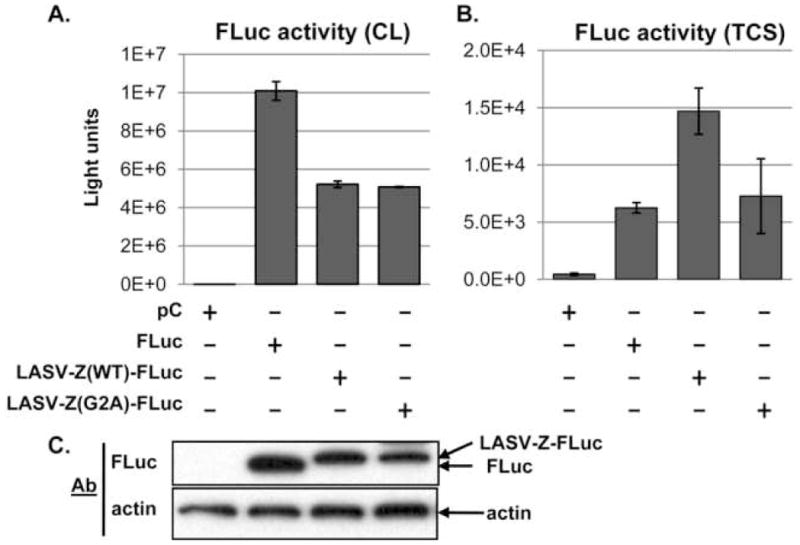
Plasmids directing the expression of the indicated LASV Z-FLuc chimeric proteins were transfected into 293T cells in a 96-well plate and either cell lysates (CL, panel A) or tissue culture supernatants (TCS, panel B) were assayed for luciferase activity. Activity in TCS was measured for 25% of total TCS volume, and the values were multiplied by four to reflect activity of 100% of TCS. Transfections were done in quadruplicate. Note the difference in the signal scale between cells and TCS. Cell lysates were also examined for FLuc protein expression levels by Western blot (panel C). A. FLuc activity in CL. B. FLuc activity in TCS. C. Detection of FLuc in CL by Western blot. Lane assignments are indicated in the figure.
We considered that the large size (550aa) of FLuc compared to Z (99aa), might have contributed to impaired Z budding activity. We therefore explored the use of the smaller Gaussia luciferase (GLuc, 185aa) (Tannous et al., 2005). To this end, we tested fusions of LASV-Z(WT) or LASV-Z(G2A) to the GLuc. The presence of a secretory signal peptide (SP) at the N-terminus of GLuc mediates its efficient secretion from cells (Knappskog et al., 2007; Tannous et al., 2005). Accordingly, TCS from GLuc-transfected cells exhibit 1000-fold higher levels of luciferase activity compared to TCS from cells transfected with a version of GLuc lacking its SP (GLuc-ΔSP). We therefore reasoned that replacing the GLuc SP with LASV-Z(WT) would result in release of the LASV-Z(WT)-GLuc protein into TCS that was dependent on the budding activity of Z. Further, we predicted that fusion of the LASV-Z G2A myristoylation mutant to GLuc-ΔSP should result in a fusion protein (LASV-Z(G2A)-GLuc) with impaired budding activity, which would be reflected in reduced levels of luciferase activity in TCS from LASV-Z(G2A)-GLuc transfected cells. We were also concerned whether overexpression of such chimeras might perturb cell viability. To this end, we measured cell viability using a firefly luciferase-based ATP assay and normalized ATP measurements of cells transfected with the GLuc constructs and Z-GLuc chimeras to cells transfected with empty vector. Since FLuc has different substrate specificity than GLuc, the viability assay is not influenced by the presence of GLuc in cells (see Materials and Methods).
TCS from cells expressing LASV-Z(WT)-GLuc showed levels of GLuc activity in TCS that were 8-fold higher compared to cells expressing LASV-Z(G2A)-GLuc (Figure 2B). Further, TCS luciferase activities of GLuc-ΔSP transfected and LASV-Z-G2A-GLuc were similar; indicating that replacement of the signal peptide with a budding-defective Z could not rescue release of GLuc from cells. GLuc activity levels correlated well with levels of Z-GLuc protein detected in VLPs recovered from TCS (Figure 2C).
Figure 2. Luciferase activity in CL and TCS of cells transfected with LASV-Z-GLuc chimera.
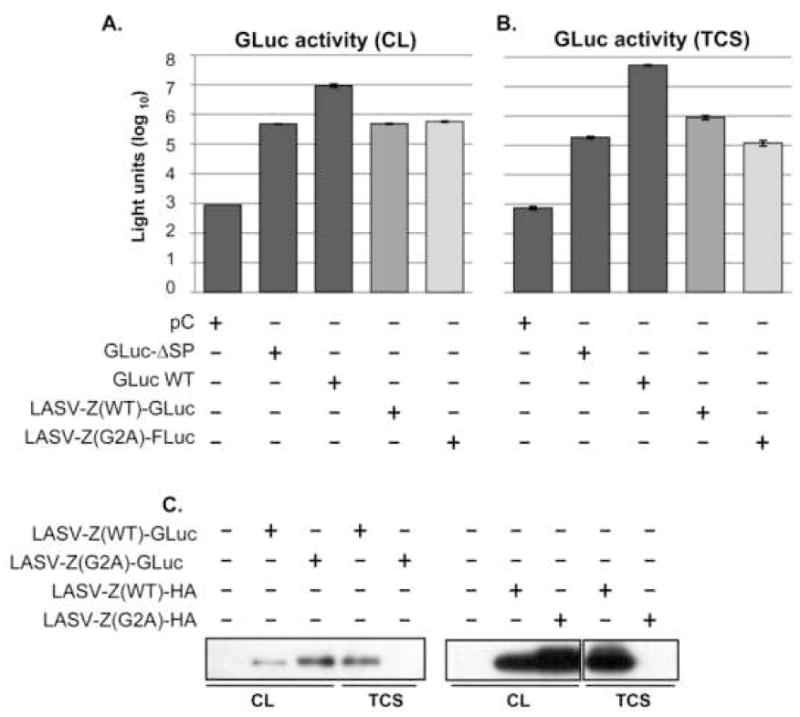
293T cells were transfected with plasmids to express the indicated proteins in quadruplicate in a 96-well plate. At 48 h post-transfection, TCS and CL were collected for measurement of GLuc activity. In a separate experiment cells were transfected in a six-well plate format and VLP were prepared from TCS as described (Perez, Craven, and de la Torre, 2003). CL and VLPs were probed for Z in western blots using antibodies to GLuc and HA. A. GLuc activity in CL. B. GLuc activity in TCS. C. Z-GLuc detected in VLPs using standard centrifugation protocol.
Together, these results validated the use of a LASV-Z-GLuc based assay to rapidly and quantitatively assess Z-mediated budding. We then asked whether LASV-Z-GLuc could be used to develop a HTS assay. For this, we calculated our assay Z′ score (Z′ = 1−[(3σc++−+ 3σc−)/|3μc+−− 3μc−|]) (Zhang, Chung, and Oldenburg, 1999), which is a measure of the quality and robustness of a HTS assay attributable to the standard deviation between multiple reads (Singh et al., 2004). Z′ values depend on the sum of the standard deviations of positive and negative controls (σc+ and σc−, respectively) as well as the difference between the mean activities of these controls (μc+ and μc−). Therefore, ideal assay conditions would show small variations within each control and a large difference between the controls. To determine the Z′ value of our assay, we used measurements of GLuc activity in TCS from cells expressing LASV-Z(WT)-GLuc or LASV-Z(G2A)-GLuc as positive or negative controls, respectively. In two independent experiments the Z′ score of our assay was >0.5 (Figure 3), a value that corresponds to an assay with excellent properties for HTS (Singh et al., 2004; Zhang, Chung, and Oldenburg, 1999).
Figure 3. Calculation of Z′ using LASV Z-GLuc fusion.
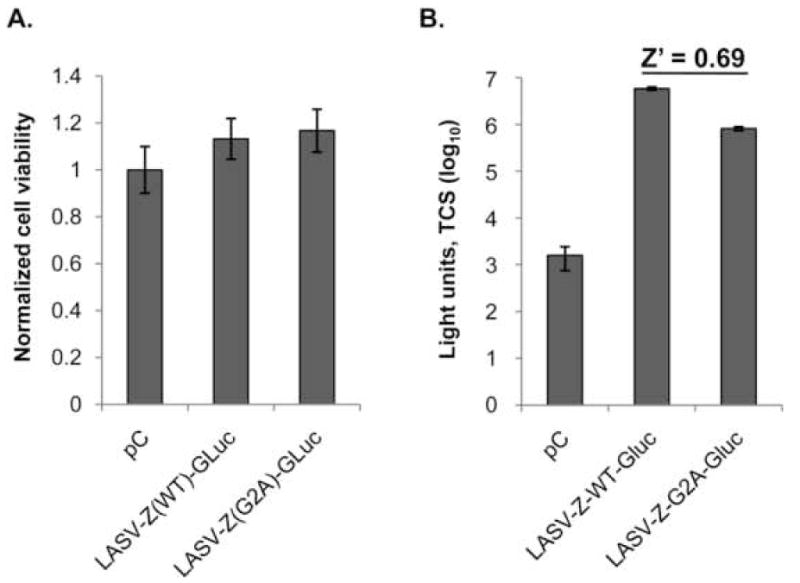
Cells were transfected overnight with the indicated plasmids in six-well plates. Transfection medium was then changed to complete cell culture medium and cells were allowed to recover for 8 hours, followed by preparation of single-cell suspension and seeding 4 ×104 cells in 96-well plates. TCS and cells were collected 48 h later for measurement of GLuc activity and cell viability, respectively. Five replicate wells were measured to calculate Z′. A. Cell viability as assessed by ATP content. B. GLuc activity from TCS, and the Z′ score is shown above the positive (LASV-Z(WT)-GLuc) and negative (LASV-Z(G2A)-GLuc) controls.
Z-GLuc budding is susceptible to myristoylation inhibitors
Based on the critical role of myristoylation in Z-mediated budding (Perez, Greenwald, and de la Torre, 2004; Strecker et al., 2006), we predicted that LASV-Z(WT)-GLuc would be susceptible to myristoylation inhibitors. To examine this we transfected 293T cells with LASV-Z(WT)-GLuc in the presence or absence of 2-hydroxymyristic acid (OHM), a myristoylation inhibitor. While GLuc activity was decreased by approximately 2-fold in cells expressing GLuc-ΔSP, LASV-Z(WT)-GLuc, or LASV-Z(G2A)-GLuc (Figure 4B), we found that the activity in TCS from LASV-Z(WT)-GLuc expressing cells was 15-fold decreased following treatment with OHM (Figure 4C), a finding consistent with published results (Perez, Greenwald, and de la Torre, 2004){Strecker, 2006 #18}. Further, when we compared LASV-Z(WT)-GLuc to LASV-Z(G2A)-GLuc directly (Figure 4D), we found that release of LASV-Z(G2A)-GLuc into TCS was 87% decreased compared to LASV-Z(WT)-GLuc, which was consistent with previous results {Perez, 2004 #14; Strecker, 2006 #18}.
Figure 4. Effect of 2-OHM on LASV Z-GLuc budding.
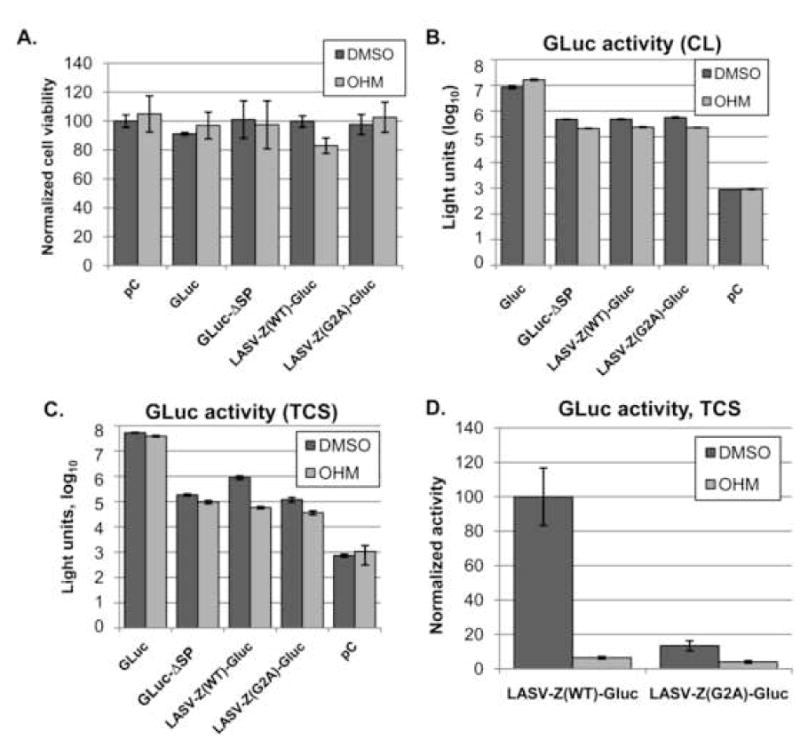
293T cells were transfected (quadruplicate) in a 96-well plate in the presence of 2-OHM (100 μM) or vehicle alone (DMSO) at the same concentration present in 2-OHM treated cells. At 48 h post-transfection, cells were measured for viability using CellTiter Glo (Promega) and TCS was collected for GLuc measurement. A. Cell viability. B. Measurement of GLuc activity in TCS. C. Measurement of GLuc activity in cell lysates. D. Comparison of LASV-Z(WT)-GLuc budding and LASV-Z(G2A)-GLuc budding, where budding activity of LASV-Z(WT)-GLuc is set at 100.
To further examine the specificity of the effect of OHM on Z-GLuc budding, we measured the effect of OHM on GLuc expression and secretion. Cells were separately transfected with WT GLuc and treated, or not, with OHM. Cell lysates were measured for either GLuc activity or ATP content (to assess cell viability), and TCS were collected for GLuc activity. After adjusting for cell viability, levels of GLuc activity were two-fold increased in lysates from OHM-treated cells compared to untreated cells (Figure 4B), whereas in TCS, GLuc activity was diminished by 26% in the presence of OHM (Figure 4C).
Z-GLuc mediated budding is susceptible to RNAi knockdown of Tsg101
Previous findings have shown that Z-mediated budding involves Z interaction with members of the MVB pathway including Tsg101 (Perez, Craven, and de la Torre, 2003) (Urata et al., 2006). Consequently, we predicted LASV-Z(WT)-GLuc budding to be sensitive to knockdown of Tsg101 by RNAi. To assess this prediction, we transfected cells with either non-specific or Tsg101-specific siRNA then subsequently with LASV-Z(WT)-GLuc and measured TCS GLuc activity. We observed a 30% reduction in TCS luciferase activity from cells transfected with the Tsg101-specific siRNA (Figure 5A), while the corresponding cell-associated luciferase activity was increased by 50% (Figure 5B). When we calculated budding efficiency (Figure 5C), we found a 24% decrease, which was consistent with previous findings (Urata et al., 2006).
Figure 5. Z-GLuc fusion is susceptible to knockdown of Tsg101.
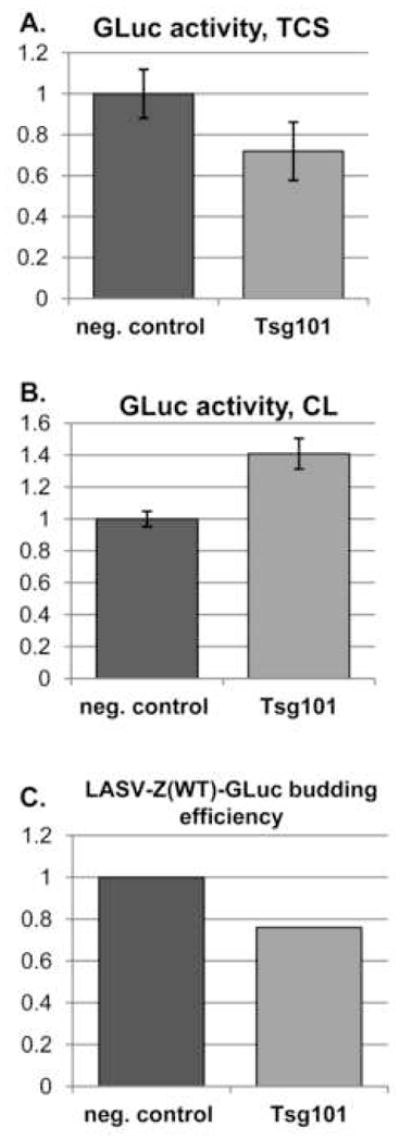
Cells were transfected in triplicate wells with siRNA directed against Tsg101, or with a control siRNA. At 24 h post-transfection, cells were transfected to express LASV-Z(WT)-GLuc. TCS was collected for GLuc assay 24 h after LASV-Z(WT)-GLuc transfection. A. Activity of GLuc in TCS when normalized for activity from LASV-Z(WT)-GLuc expressing cells in the presence of non-specific siRNA (set to 1). B. Cellular expression of GLuc when normalized for activity from LASV-Z(WT)-GLuc expressing cells in the presence of non-specific siRNA (set to1). C. Budding efficiency of LASV-Z(WT)-GLuc in the presence of Tsg101-specific siRNA where budding efficiency in the presence of non-specific siRNA was set at 1. Budding efficiency was calculated as follows: GLuc activity (TCS)/([GLuc activity (TCS)]+[ GLuc activity (CL)]).
Discussion
Budding of many enveloped NS RNA virus require interactions between cellular proteins and the VBP. Interfering with these interactions presents an opportunity to both understand the cellular biology of virus budding and to identify potential novel antiviral targets and drugs. We sought to develop a Z budding assay that would facilitate the identification of small molecule inhibitors of these interactions and therefore arenavirus budding. To this end we tested the use of LASV-Z-luciferase fusion proteins as tools to develop an assay to rapidly and quantitatively measure Z-mediated budding. A chimeric LASV-Z-FLuc protein showed 1000-fold lower activity in TCS than in cells, indicating that only small amounts of LASV-Z-FLuc could bud into TCS. In contrast, about 90% of GLuc activity in LASV-Z(WT)-GLuc expressing cells was in TCS. Moreover, levels of TCS luciferase activity were dramatically reduced (≥8-fold) in cells transfected with LASV-Z(G2A)-GLuc compared to LASV-Z(WT)-GLuc transfected cells. Previously, we showed that LASV Z tagged at its C-terminus with HA (LASV-Z(WT)-HA) has WT Z budding activity as determined by levels of Z present in VLP collected from TCS of transfected cells (Perez, Craven, and de la Torre, 2003). Using the same biochemical assay, we observed that LASV-Z(WT)-HA and LASV-Z(WT)-GLuc had similar budding activity. These results argue strongly that the LASV-Z(WT)-GLuc chimera is functionally equivalent to WT Z in budding activity and therefore levels of Z-mediated budding can be assessed based on GLuc activity in TCS from Z-GLuc transfected cells.
The reasons why LASV-Z(WT)-FLuc was severely impaired in its budding activity remain to be determined. However, a significant difference is the larger size (three times bigger) of FLuc compared to GLuc. Notably, a TAP-tagged Z (LASV-Z(WT)-TAP) retained WT Z budding activity (MP and JCT, unpublished data), and interestingly the TAP tag and GLuc are similar in size (185aa and 168aa respectively), while FLuc is 550aa. It is therefore plausible that the budding efficiency of chimeric Z proteins may depend on the size of the fusion partner added to Z.
GLuc activity from cells expressing either GLuc-ΔSP or LASV-Z(G2A)-GLuc were 1000-fold lower than GLuc WT. These results showed that consistent with previous results GLuc secretion requires its SP (Tannous et al., 2005), but also that a Z mutant defective in budding cannot functionally substitute for the SP of GLuc. We also observed that levels of GLuc in TCS from LASV-Z(WT)-GLuc transfected cells were consistently lower (50–60 fold) compared to those in TCS from GLuc WT transfected cells. Differences in the mechanism by which GLuc and LASV-Z(WT)-GLuc are released from cells likely contributed to these findings. The former involves secretion mediated by the native SP in WT GLuc (Knappskog et al., 2007), whereas the latter occurs via budding mediated by WT Z. Together, these data indicate that GLuc release from cells depends on the efficiency of SP-mediated secretion, whereas levels of released Z-GLuc chimera proteins into TCS reflect the relative efficiency of Z-mediated budding.
When LASV-Z(WT)-GLuc and LASV-Z(G2A)-GLuc were used as positive and negative controls, respectively, our assay had Z′ values significantly >0.5, thus indicating that our assay has excellent features for its use in HTS.
We showed that WT GLuc expression and secretion was somewhat affected, though modestly, by OHM (Figure 4B and 4C). These results suggest some effects of OHM that are independent of inhibiting myristoylation. Since GLuc is released from cells via secretion, OHM could have affected one or more steps in secretion by a mechanism that we do not presently understand. To address the possibility that GLuc could be myristoylated and therefore be susceptible to OHM, the amino acid sequence for GLuc was submitted to the prediction program NMT-The Predictor (http://mendel.imp.ac.at/myristate/SUPLpredictor.htm), which is available through the ExPASy (Expert Protein Analysis System) proteomics server (http://www.expasy.org/) of the Swiss Institute of Bioinformatics (SIB). We found no myristoylation sites within GLuc (data not shown), supporting the notion that OHM was somehow affecting secretion. We had also observed a decrease in intracellular GLuc-ΔSP following OHM treatment (Figure 4B). This effect was similar to the effects of OHM on intracellular expression of the Z-GLuc chimeras, suggesting that the effect of OHM on intracellular Z-GLuc was likely due to effects on expression of GLuc-ΔSP. Interestingly, these findings suggest that GLuc intracellular expression could be used to assess pleiotropic effects of small molecules to more general processes such cell viability, PolII-mediated transcription, or both, which provides this assay with an important internal control to normalize values of Z-GLuc budding.
The susceptibility of LASV-Z(WT)-GLuc to RNAi knockdown of Tsg101 indicates that Z-GLuc was also sensitive to disruption of late domain function. While the effect of Tsg101-specific siRNA on Z-GLuc budding was modest compared to previously published findings, the results presented here reflect effects following a single siRNA transfection, whereas stronger effects have been observed with two transfections of siRNA prior to transfection with the VBP of choice (Garrus et al., 2001; Urata et al., 2006). These results suggest that with refinement this assay could be adapted for use in siRNA-based HTS approaches to identify additional cellular proteins necessary for Z-mediated budding. The same screens might also identify cellular proteins involved in Z-mediated budding via interactions in regions outside the L-domains of Z. Therefore, cellular proteins identified using this method would need to be evaluated for their L-domain dependent or independent interaction with Z.
We have shown that GLuc fusion to the arenavirus LASV Z matrix protein results in a chimeric protein with similar budding properties to native Z. This would allow the use of BSL2 facilities to implement HTS aimed at identifying inhibitors of budding of LASV and HF arenaviruses, for which use of live virus requires BSL4 facilities. Importantly, because LASV Z protein contains two late domain motifs, the LASV Z-GLuc chimera can be used to identify targets necessary for VBP-mediated budding via either domain. Further, we think that similar GLuc chimeras could be done with VBP from other highly pathogenic riboviruses, making it possible to conduct assays under BSL-2 conditions to rapidly identify inhibitors of virus budding that might be developed into antiviral drugs. Likewise, the use of VBP-GLuc based assays should facilitate the use of siRNA-based HTS to globally identify host cellular proteins involved in VBP-mediated budding in different RNA viruses, including important human pathogens.
Acknowledgments
The authors would like to thank S. Kunz, S. Urata, and members of the JCT group for helpful discussion. The research herein was supported by NIH grants AI47140 and AI-065359 to JCT and T32-AI0735418 and T32-NS041219 to AAC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
H. Literature cited
- Barton LL. LCMV transmission by organ transplantation. N Engl J Med. 2006;355(16):1737. doi: 10.1056/NEJMc061706. author reply 1737–8. [DOI] [PubMed] [Google Scholar]
- Barton LL, Mets MB, Beauchamp CL. Lymphocytic choriomeningitis virus: emerging fetal teratogen. Am J Obstet Gynecol. 2002;187(6):1715–6. doi: 10.1067/mob.2002.126297. [DOI] [PubMed] [Google Scholar]
- Borden KL, Campbell Dwyer EJ, Salvato MS. An arenavirus RING (zinc-binding) protein binds the oncoprotein promyelocyte leukemia protein (PML) and relocates PML nuclear bodies to the cytoplasm. J Virol. 1998;72(1):758–66. doi: 10.1128/jvi.72.1.758-766.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borio L, Inglesby T, Peters CJ, Schmaljohn AL, Hughes JM, Jahrling PB, Ksiazek T, Johnson KM, Meyerhoff A, O’Toole T, Ascher MS, Bartlett J, Breman JG, Eitzen EM, Jr, Hamburg M, Hauer J, Henderson DA, Johnson RT, Kwik G, Layton M, Lillibridge S, Nabel GJ, Osterholm MT, Perl TM, Russell P, Tonat K. Hemorrhagic fever viruses as biological weapons: medical and public health management. Jama. 2002;287(18):2391–405. doi: 10.1001/jama.287.18.2391. [DOI] [PubMed] [Google Scholar]
- Buchmeier MJ, de la Torre J-C, Peters C. Arenaviridae: The Viruses and Their Replication. In: Knipe DM, Howley PM, editors. Fields Virology. 5. Vol. 2. Wolters Kluwer Health/Lippincott Williams & Wilkins; Philadelphia: 2007. pp. 1792–1827. [Google Scholar]
- Buchmeier MJ, Oldstone MB. Protein structure of lymphocytic choriomeningitis virus: evidence for a cell-associated precursor of the virion glycopeptides. Virology. 1979;99(1):111–20. doi: 10.1016/0042-6822(79)90042-4. [DOI] [PubMed] [Google Scholar]
- Campbell Dwyer EJ, Lai H, MacDonald RC, Salvato MS, Borden KL. The lymphocytic choriomeningitis virus RING protein Z associates with eukaryotic initiation factor 4E and selectively represses translation in a RING-dependent manner. J Virol. 2000;74(7):3293–300. doi: 10.1128/jvi.74.7.3293-3300.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capul AA, Perez M, Burke E, Kunz S, Buchmeier MJ, de la Torre JC. Arenavirus Z-glycoprotein association requires Z myristoylation but not functional RING or late domains. J Virol. 2007;81(17):9451–60. doi: 10.1128/JVI.00499-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven RC, Harty RN, Paragas J, Palese P, Wills JW. Late domain function identified in the vesicular stomatitis virus M protein by use of rhabdovirus-retrovirus chimeras. J Virol. 1999;73(4):3359–65. doi: 10.1128/jvi.73.4.3359-3365.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton AJ, Rowe WP, Smith GH, Wilsnack RE, Pugh WE. Morphological and cytochemical studies on lymphocytic choriomeningitis virus. J Virol. 1968;2(12):1465–78. doi: 10.1128/jvi.2.12.1465-1478.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler R, Strecker T, Kolesnikova L, ter Meulen J, Weissenhorn W, Becker S, Klenk HD, Garten W, Lenz O. Characterization of the Lassa virus matrix protein Z: electron microscopic study of virus-like particles and interaction with the nucleoprotein (NP) Virus Res. 2004;100(2):249–55. doi: 10.1016/j.virusres.2003.11.017. [DOI] [PubMed] [Google Scholar]
- Eschli B, Quirin K, Wepf A, Weber J, Zinkernagel R, Hengartner H. Identification of an N-terminal trimeric coiled-coil core within arenavirus glycoprotein 2 permits assignment to class I viral fusion proteins. J Virol. 2006;80(12):5897–907. doi: 10.1128/JVI.00008-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flatz L, Bergthaler A, de la Torre JC, Pinschewer DD. Recovery of an arenavirus entirely from RNA polymerase I/II-driven cDNA. Proc Natl Acad Sci U S A. 2006;103(12):4663–8. doi: 10.1073/pnas.0600652103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107(1):55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- Geisbert TW, Jahrling PB. Exotic emerging viral diseases: progress and challenges. Nat Med. 2004;10(12 Suppl):S110–21. doi: 10.1038/nm1142. [DOI] [PubMed] [Google Scholar]
- Gross S, Piwnica-Worms D. Spying on cancer: molecular imaging in vivo with genetically encoded reporters. Cancer Cell. 2005;7(1):5–15. doi: 10.1016/j.ccr.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Harty RN, Brown ME, Wang G, Huibregtse J, Hayes FP. A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc Natl Acad Sci U S A. 2000;97(25):13871–6. doi: 10.1073/pnas.250277297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hass M, Golnitz U, Muller S, Becker-Ziaja B, Gunther S. Replicon system for Lassa virus. J Virol. 2004;78(24):13793–803. doi: 10.1128/JVI.78.24.13793-13803.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahrling PB, Peters CJ. Lymphocytic choriomeningitis virus. A neglected pathogen of man. Arch Pathol Lab Med. 1992;116(5):486–8. [PubMed] [Google Scholar]
- Knappskog S, Ravneberg H, Gjerdrum C, Trosse C, Stern B, Pryme IF. The level of synthesis and secretion of Gaussia princeps luciferase in transfected CHO cells is heavily dependent on the choice of signal peptide. J Biotechnol. 2007;128(4):705–15. doi: 10.1016/j.jbiotec.2006.11.026. [DOI] [PubMed] [Google Scholar]
- Kunz S, Edelmann KH, de la Torre J-C, Gorney R, Oldstone MBA. Mechanisms for lymphocytic choriomeningitis virus glycoprotein cleavage, transport, and incorporation into virions. Virology. 2003 doi: 10.1016/s0042-6822(03)00421-5. in press. [DOI] [PubMed] [Google Scholar]
- Lee KJ, Novella IS, Teng MN, Oldstone MB, de La Torre JC. NP and L proteins of lymphocytic choriomeningitis virus (LCMV) are sufficient for efficient transcription and replication of LCMV genomic RNA analogs. J Virol. 2000;74(8):3470–7. doi: 10.1128/jvi.74.8.3470-3477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KJ, Perez M, Pinschewer DD, de la Torre JC. Identification of the lymphocytic choriomeningitis virus (LCMV) proteins required to rescue LCMV RNA analogs into LCMV-like particles. J Virol. 2002;76(12):6393–7. doi: 10.1128/JVI.76.12.6393-6397.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenard J. Negative-strand virus M and retrovirus MA proteins: all in a family? Virology. 1996;216(2):289–98. doi: 10.1006/viro.1996.0064. [DOI] [PubMed] [Google Scholar]
- Lopez N, Jacamo R, Franze-Fernandez MT. Transcription and RNA replication of tacaribe virus genome and antigenome analogs require N and L proteins: Z protein is an inhibitor of these processes. J Virol. 2001;75(24):12241–51. doi: 10.1128/JVI.75.24.12241-12251.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Serrano J, Zang T, Bieniasz PD. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat Med. 2001;7(12):1313–9. doi: 10.1038/nm1201-1313. [DOI] [PubMed] [Google Scholar]
- McCarthy SE, Licata JM, Harty RN. A luciferase-based budding assay for Ebola virus. J Virol Methods. 2006;137(1):115–9. doi: 10.1016/j.jviromet.2006.06.007. [DOI] [PubMed] [Google Scholar]
- McCormick JB, Fisher-Hoch SP. Lassa Fever. In: Oldstone MB, editor. Arenaviruses I. Vol. 262. Springer-Verlag; Berlin, Heidelberg, New York: 2002. pp. 75–110. [Google Scholar]
- McKee KT, Jr, Huggins JW, Trahan CJ, Mahlandt BG. Ribavirin prophylaxis and therapy for experimental argentine hemorrhagic fever. Antimicrob Agents Chemother. 1988;32(9):1304–9. doi: 10.1128/aac.32.9.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mets MB, Barton LL, Khan AS, Ksiazek TG. Lymphocytic choriomeningitis virus: an underdiagnosed cause of congenital chorioretinitis. Am J Ophthalmol. 2000;130(2):209–15. doi: 10.1016/s0002-9394(00)00570-5. [DOI] [PubMed] [Google Scholar]
- Murphy FA, Webb PA, Johnson KM, Whitfield SG, Chappell WA. Arenoviruses in Vero cells: ultrastructural studies. J Virol. 1970;6(4):507–18. doi: 10.1128/jvi.6.4.507-518.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen H, Brunak S, von Heijne G. Machine learning approaches for the prediction of signal peptides and other protein sorting signals. Protein Eng. 1999;12(1):3–9. doi: 10.1093/protein/12.1.3. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108(2):193–9. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Opaluch AM, Aza-Blanc P, Vang T, Williams S, Tautz L, Milan L, Mustelin T. PTPome-wide functional RNA interference screening methods. Methods. 2007;42(3):306–12. doi: 10.1016/j.ymeth.2007.02.015. [DOI] [PubMed] [Google Scholar]
- Palacios G, Druce J, Du L, Tran T, Birch C, Briese T, Conlan S, Quan PL, Hui J, Marshall J, Simons JF, Egholm M, Paddock CD, Shieh WJ, Goldsmith CS, Zaki SR, Catton M, Lipkin WI. A new arenavirus in a cluster of fatal transplant-associated diseases. N Engl J Med. 2008;358(10):991–8. doi: 10.1056/NEJMoa073785. [DOI] [PubMed] [Google Scholar]
- Patch JR, Crameri G, Wang LF, Eaton BT, Broder CC. Quantitative analysis of Nipah virus proteins released as virus-like particles reveals central role for the matrix protein. Virol J. 2007;4:1. doi: 10.1186/1743-422X-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez M, Craven RC, de la Torre JC. The small RING finger protein Z drives arenavirus budding: implications for antiviral strategies. Proc Natl Acad Sci U S A. 2003;100(22):12978–83. doi: 10.1073/pnas.2133782100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez M, Greenwald DL, de la Torre JC. Myristoylation of the RING finger Z protein is essential for arenavirus budding. J Virol. 2004;78(20):11443–8. doi: 10.1128/JVI.78.20.11443-11448.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters CJ. Human Infection with Arenaviruses in the Americas. In: Oldstone MB, editor. Arenaviruses I. Vol. 262. Springer-Verlag; Berlin Heidelberg: 2002. pp. 65–74. [DOI] [PubMed] [Google Scholar]
- Pinschewer DD, Perez M, Sanchez AB, de la Torre JC. Recombinant lymphocytic choriomeningitis virus expressing vesicular stomatitis virus glycoprotein. Proc Natl Acad Sci U S A. 2003;100(13):7895–900. doi: 10.1073/pnas.1332709100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy I, Michnick SW. A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat Methods. 2006;3(12):977–9. doi: 10.1038/nmeth979. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sanchez AB, de la Torre JC. Rescue of the prototypic Arenavirus LCMV entirely from plasmid. Virology. 2006;350(2):370–80. doi: 10.1016/j.virol.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Singh P, Harden BJ, Lillywhite BJ, Broad PM. Identification of kinase inhibitors by an ATP depletion method. Assay Drug Dev Technol. 2004;2(2):161–9. doi: 10.1089/154065804323056503. [DOI] [PubMed] [Google Scholar]
- Stankunas K, Bayle JH, Havranek JJ, Wandless TJ, Baker D, Crabtree GR, Gestwicki JE. Rescue of degradation-prone mutants of the FK506-rapamycin binding (FRB) protein with chemical ligands. Chembiochem. 2007;8(10):1162–9. doi: 10.1002/cbic.200700087. [DOI] [PubMed] [Google Scholar]
- Strecker T, Eichler R, Meulen J, Weissenhorn W, Dieter Klenk H, Garten W, Lenz O. Lassa virus Z protein is a matrix protein and sufficient for the release of virus-like particles [corrected] J Virol. 2003;77(19):10700–5. doi: 10.1128/JVI.77.19.10700-10705.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strecker T, Maisa A, Daffis S, Eichler R, Lenz O, Garten W. The role of myristoylation in the membrane association of the Lassa virus matrix protein Z. Virol J. 2006;3:93. doi: 10.1186/1743-422X-3-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannous BA, Kim DE, Fernandez JL, Weissleder R, Breakefield XO. Codon-optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo. Mol Ther. 2005;11(3):435–43. doi: 10.1016/j.ymthe.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Topcu Z, Mack DL, Hromas RA, Borden KL. The promyelocytic leukemia protein PML interacts with the proline-rich homeodomain protein PRH: a RING may link hematopoiesis and growth control. Oncogene. 1999;18(50):7091–100. doi: 10.1038/sj.onc.1203201. [DOI] [PubMed] [Google Scholar]
- Urata S, Noda T, Kawaoka Y, Yokosawa H, Yasuda J. Cellular factors required for Lassa virus budding. J Virol. 2006;80(8):4191–5. doi: 10.1128/JVI.80.8.4191-4195.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VerPlank L, Bouamr F, LaGrassa TJ, Agresta B, Kikonyogo A, Leis J, Carter CA. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag) Proc Natl Acad Sci U S A. 2001;98(14):7724–9. doi: 10.1073/pnas.131059198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- York J, Romanowski V, Lu M, Nunberg JH. The signal peptide of the Junin arenavirus envelope glycoprotein is myristoylated and forms an essential subunit of the mature G1-G2 complex. J Virol. 2004;78(19):10783–92. doi: 10.1128/JVI.78.19.10783-10792.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]