

Abstract
Glioblastoma remains one of the most challenging forms of cancer to treat. Here, we develop a computational platform that integrates the analysis of copy number variations and somatic mutations and unravels the landscape of in-frame gene fusions in glioblastoma. We find mutations with loss of heterozygosity of LZTR-1, an adaptor of Cul3-containing E3 ligase complexes. Mutations and deletions disrupt LZTR-1 function, which restrains self-renewal and growth of glioma spheres retaining stem cell features. Loss-of-function mutations of CTNND2 target a neural-specific gene and are associated with transformation of glioma cells along the very aggressive mesenchymal phenotype. We also report recurrent translocations that fuse the coding sequence of EGFR to several partners, with EGFR-SEPT14 as the most frequent functional gene fusion in human glioblastoma. EGFR-SEPT14 fusions activate Stat3 signaling and confer mitogen independency and sensitivity to EGFR inhibition. These results provide important insights into the pathogenesis of glioblastoma and highlight new targets for therapeutic intervention.
Glioblastoma (GBM) is the most common primary intrinsic malignant brain tumor affecting ∼10,000 new patients each year with a median survival rate of 12–15 months1,2. Identifying and understanding the functional significance of genetic alterations that drive initiation and progression of GBM is crucial to develop effective therapies. Previous efforts in GBM genome characterization identified somatic changes in well-known GBM genes (EGFR, PTEN, IDH1, TP53, NF1, etc.) and nominated putative cancer genes with somatic mutations, but the functional consequence of most alterations is unknown3–6. Furthermore, the abundance of passenger mutations and large regions of copy number variations (CNVs) complicates the definition of the landscape of driver mutations in glioblastoma. To address this challenge, we have employed a novel statistical approach to nominate driver genes in GBM by integrating somatic mutations identified by whole-exome sequencing with a CNVs analysis that prioritizes focality and magnitude of the genetic alterations.
Recurrent and oncogenic gene fusions are hallmarks of hematological malignancies and have also been uncovered in solid tumors7,8. Recently, we reported that a small subset of GBM harbor FGFR-TACC gene fusions and provided data to suggest that the patients with FGFR-TACC-positive tumors would benefit from targeted FGFR kinase inhibition9. It remains unknown whether gene fusions involving other RTK-coding genes exist and produce oncogene addiction in GBM. Here, we analyze a large RNA-sequencing dataset of primary GBM and Glioma Sphere Cultures (GSCs) and report the global landscape of in-frame gene fusions in human GBM.
Nomination of candidate GBM genes
We reasoned that integration of somatic point mutations and focal CNVs would uncover candidate driver GBM genes. MutComFocal is an algorithm designed to rank genes by an integrated recurrence, focality and mutation score (see Methods). We applied this strategy to 139 GBM and matched normal DNA analyzed by whole-exome sequencing to identify somatic mutations and 469 GBM analyzed by the Affymetrix SNP6.0 platform to identify CNVs.
The whole-exome analysis revealed a mean of 43 nonsynonymous somatic mutations per tumor sample (Supplementary Tables 1, 2). The distribution of substitutions shows a higher rate of transitions versus tranversions (67%), with a strong preference for C->T and G->A (55%) (Supplementary Fig. 1). As seen in other tumor types10, 19.2% of the mutations occurred in a CpG dinucleotide context (Supplementary Fig. 2). Among somatic small nucleotide variants, the most frequently mutated genes have well-established roles in cancer, including GBM (TP53, EGFR, PTEN, and IDH1, Supplementary Table 3). In addition to known cancer genes, potentially new candidate driver genes were mutated in ∼5% of tumor samples. By integrating mutational and common focal genomic lesions, MutComFocal (Supplementary Table 4) stratified somatically mutated genes into three groups: recurrently mutated genes without significant copy number alterations (Mut), in regions of focal and recurrent amplifications (Amp-Mut) and in regions of focal and recurrent deletions (Del-Mut). We generated a list of 67 genes that score at the top of each of the three categories and include nearly all the genes previously implicated in GBM (Supplementary Table 4). Among these genes, (labeled in blue in Fig. 1) are IDH1 (Mut, Fig. 1a), PIK3C2B, MDM4, MYCN, PIK3CA, PDGFRA, KIT, EGFR, and BRAF (Amp-Mut, Fig. 1b) and PIK3R1, PTEN, RB1, TP53, NF1 and ATRX (Del-Mut, Fig. 1c). The analysis also selected 52 new candidate driver genes previously unreported in GBM. Based upon their role in CNS development and homeostasis as well as their potential function in gliomagenesis, we selected 24 genes for re-sequencing in an independent dataset of 83 GBM and matched normal controls. Eighteen genes were found somatically mutated by Sanger sequencing in the independent panel (labeled in red in Fig. 1, Supplementary Table 5). Each validated new GBM gene is targeted by somatic mutations and CNVs in a cumulative fraction comprised between 2.9% and 45.7% of GBM (Supplementary Table 4). Furthermore, mutations of the 18 new GBM genes occur mostly in tumors with global mutation rates similar to the mean of 43 mutations per tumor and well within the 95% confidence interval, indicating that mutations of the 18 new genes do not cluster in hypermutated tumors (Supplementary Fig. 5).
Figure 1.
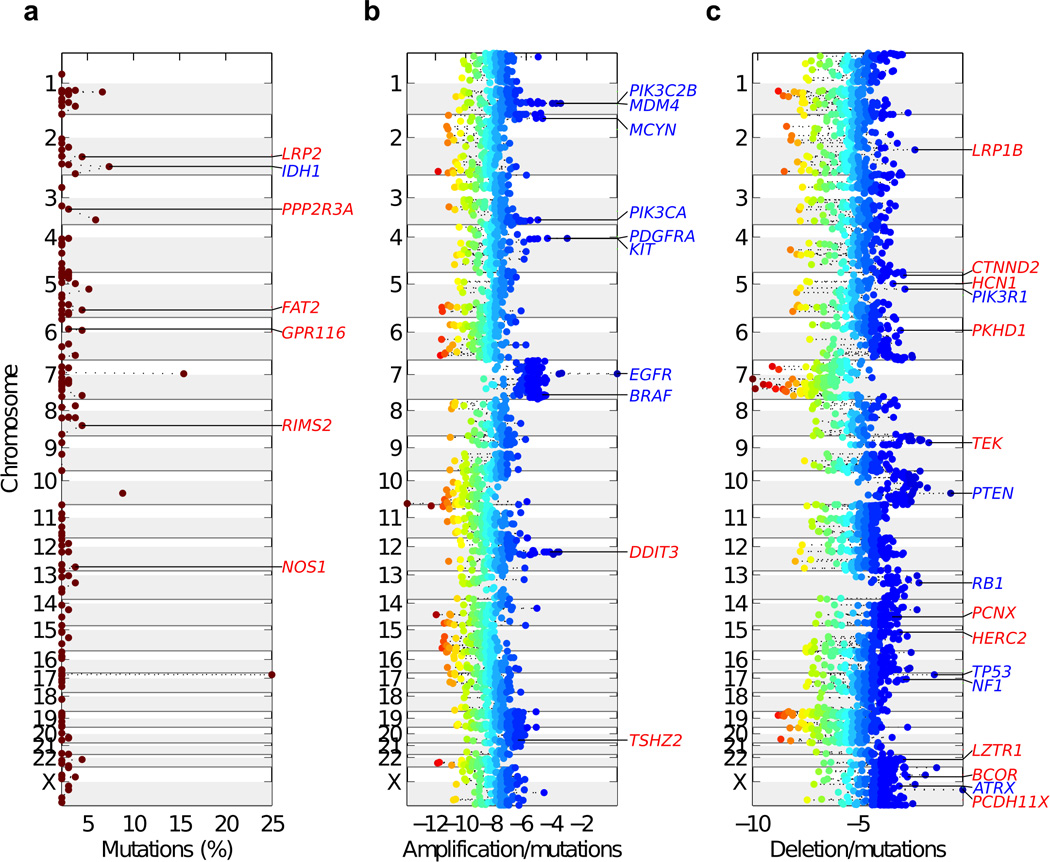
Chromosome view of validated GBM genes scoring at the top of each of the three categories by MutComFocal. a, Mutated genes without significant copy number alterations (Mut, mutation %, frequency of mutations). b, Mutated genes in regions of focal and recurrent amplifications (Amp-Mut, Amplification/mutation scores). c, Mutated genes in regions of focal and recurrent deletions (Del-Mut, Deletion/mutation scores). Previously known GBM genes are indicated in blue, new and independently validated GBM genes are indicated in red. In panels b and c, the genes scores are colored according to their corresponding tier with blue corresponding to high tiers and red to low tiers.
Among the commonly mutated and focally deleted genes exhibiting top MutComFocal scores and validated in the independent GBM dataset, we found BCOR, LRP family members, HERC2, LZTR-1 and CTNND2. BCOR, an X-linked gene, encodes for a component of the nuclear corepressor complex that is essential for normal development of neuroectoderm and stem cell functions11–13. BCOR mutations have recently been described in retinoblastoma and medulloblastoma14,15. LRP1B, a member of the LDL receptor family, is among the most frequently mutated genes in human cancer (Fig. 1c)16. Interestingly, two other LDL receptor family members (LRP2 and LRP1) are mutated in 4.4% and 2.9% of tumors, respectively (Fig. 1a, Supplementary Table 1). The LRP proteins are highly expressed in the neuroepithelium and are essential for forebrain morphogenesis in mouse and humans17,18. The tumor suppressor function of LRP proteins in GBM may relate to the ability to promote chemosensitivity and control in the Sonic hedgehog signaling pathway, which is implicated in cancer initiating cells in GBM19–21. Localized on chromosome 15q13, the Hect ubiquitin ligase Herc2 gene is deleted and mutated in 15.1% and 2.2% of GBM cases, respectively (Supplementary Table 4). Herc2 has been implicated in severe neurodevelopmental syndromes and Herc2 substrates regulate genome stability and DNA damage-repair22,23.
LZTR-1 mutations inactivate a Cullin-3 adaptor to drive self-renewal and growth of glioma spheres
A gene that received one of the highest Del-Mut score by MutComFocal is LZTR-1 (Fig. 1c, Supplementary Table 4). The LZTR-1 coding region had non-synonymous mutations in 4.4%, and the LZTR-1 locus (human chromosome 22q11) was deleted in 22.4% of GBM. Among the 18 new GBM genes, LZTR-1 had the highest co-occurrence score of mutations and deletions (Fisher’s exact test, p = 0.0007, Supplementary Table 6). It also scored at the top of the list of genes whose CNVs are statistically correlated with expression (Pearson correlation between LZTR-1 CNVs and expression is 0.36, p-value<10−6 by Student’s t-distribution, Supplementary Table 7). Finally, LZTR-1 emerged as the gene with the highest correlation for monoallelic expression of mutant alleles in tumors harboring LZTR-1 deletions (p-value = 0.0007, Supplementary Table 8). Taken together, these findings indicate that LZTR-1 is concurrently targeted in GBM by mutations and copy number loss, fulfilling the two-hits model for tumor suppressor inactivation in cancer.
LZTR-1 codes for a protein with a characteristic Kelch-BTB-BACK-BTB-BACK domain architecture (Supplementary Fig. 4, 5) and is expressed in normal brain (Supplementary Table 9). The LZTR-1 gene is highly conserved in metazoans. Although it was initially proposed that LZTR-1 functions as a transcriptional regulator, this role was not confirmed in follow-up studies24. Most proteins with BTB-BACK domains are substrate adaptors in Cullin-3 (Cul3) ubiquitin ligase complexes, in which the BTB-BACK region binds to the N-terminal domain of Cul3, while a ligand binding domain, often a Kelch 6-bladed β-propeller motif, binds to substrates targeted for ubiquitylation25. To ask whether LZTR-1 directly binds Cul3, we performed co-immunoprecipitation experiments in human glioma cells. Fig. 2a shows that Cul3 immunoprecipitates contain LZTR-1, suggesting that LZTR-1 is an adaptor in Cul3 ubiquitin ligase complexes.
Figure 2.
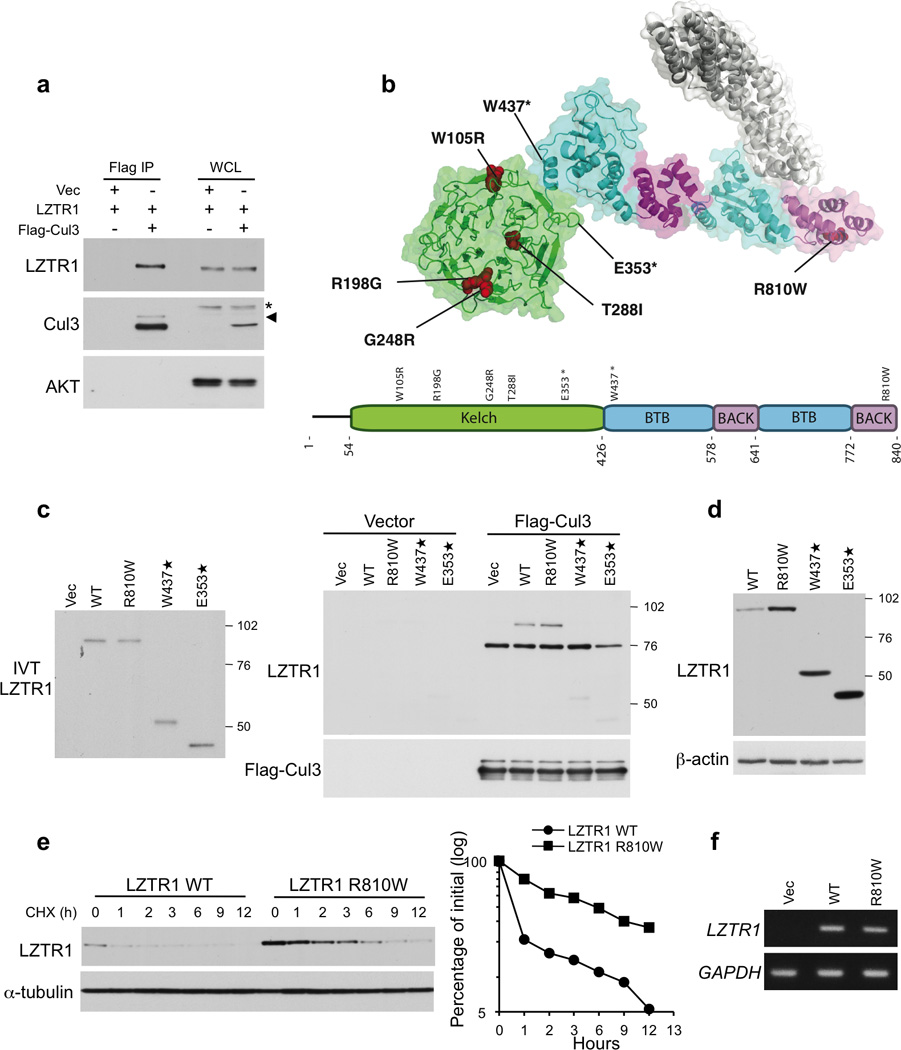
Interaction with Cul3 and protein stability of wild type and mutant LZTR-1. a, Lysates from SF188 glioma cells transfected with vectors expressing Myc-LZTR-1 and Flag-Cul3 or the empty vector were immunoprecipitated with Flag antibody and assayed by western blot with the indicated antibodies. *, non specific band; arrowhead indicates neddylated Cul3. b, Localization of altered residues in LZTR-1. Homology model of the Kelch (green), BTB (cyan) and BACK (magenta) domains of LZTR-1 with the Cul3 N-terminal domain (white) docked onto the putative binding site. GBM mutations are indicated in red. c, In vitro analysis of the interaction between Cul3 and LZTR-1 wild type and GBM related mutants. Left panel, In vitro translated Myc-LZTR-1 input. Right panel, In vitro translated Myc-LZTR-1 was mixed with Flag-Cul3 immunoprecipitated from transfected HEK-293T cells. Bound proteins were analyzed by western blot using the indicated antibodies. d, Steady state protein levels of wild type LZTR-1 and GBM-related mutants. e, Left panel, Cells transfected with LZTR-1 wild type or the R810W mutant were treated with cycloexamide for the indicated time. Right panel, Quantification of LZTR-1 wild type and LZTR-1-R810W protein from the experiment in the left panel. f, Semi-quantitative RT-PCR evaluation of LZTR-1 wild type and LZTR-1-R810W RNA expression in cells transfected as in e.
To address the function of LZTR-1 mutants, we built a homology model of LZTR-1 based partly on the crystal structures of the MATH-BTB-BACK protein SPOP26, the BTB-BACK-Kelch proteins KLHL327 and KLHL1128, and the Kelch domain of Keap129 (Fig. 2b). We predict that the second BTB-BACK region of LZTR-1 binds Cul3 because of a φ-X-E motif in this BTB domain, followed by a 3-Box/BACK region (Supplementary Fig. 5a)26. However, we cannot exclude that the preceding BTB-BACK region also participates in Cul3 binding. Five of seven LZTR-1 mutations identified in GBM are located within the Kelch domain and target highly conserved amino acids (Fig. 2b, Supplementary Fig. 4, 5b). Interestingly, the concentration of LZTR-1 mutations in the Kelch domain reflects a similar pattern of mutations in the Kelch-coding region of KLHL3, recently identified in families with hypertension and electrolytic abnormalities30,31. The R198G and G248R mutations localize to the b–c loop of the Kelch domain, in a region predicted to provide the substrate-binding surface29. The W105R mutation targets a highly conserved anchor residue in the Kelch repeats and the T288I mutation disrupts a buried residue conserved in LZTR-1 (Fig. 2b, Supplementary Fig. 4, 5b). Both mutations are expected to perturb folding of the Kelch domain. The E353STOP mutation is expected to produce a misfolded Kelch domain besides removing the C-terminal BTB-BACK regions. Located in the BTB-BACK domains, the remaining two mutations either truncate the entire BTB-BACK-BTB-BACK region (W437STOP) or are predicted to disrupt the folding of the last helical hairpin in the BTB-BACK domain (R810W, Fig. 2b).
To ask whether the mutations predicted to affect the BTB-BACK domains perturb the interaction with Cul3, we prepared in vitro translated wild type, E353STOP, W437STOP and R810W LZTR-1 Myc-tagged proteins and tested their ability to bind to Flag-Cul3 purified from mammalian cells. Wild type LZTR-1 bound Flag-Cul3, but the E353STOP and W437STOP mutants lost this property. However, the R810W mutant retained Cul3 binding in this assay (Fig. 2c). Besides promoting ubiquitin-mediated degradation of substrates, Cullin adaptors are short-lived proteins that undergo auto-ubiquitylation and destruction by the same Cullin complexes that direct substrate ubiquitylation32–34. Thus, impaired ubiquitin ligase activity of the LZTR-1-Cul3 complex should result in accumulation of mutant LZTR-1 proteins. Each of the three LZTR-1 mutants predicted to compromise integrity of the BTB-BACK domains accumulated at higher levels than wild-type LZTR-1 in transient transfection assays (Fig. 2d). The steady state and half-life of the LZTR-1 R810W mutant protein were markedly increased, in the absence of changes of the mutant mRNA (Fig. 2e, f). Thus, as for the two truncated mutants, the R810W mutation compromised protein degradation.
Next, we sought to establish the biological consequences of LZTR-1 inactivation in human GBM. Differential gene expression pattern of GBM harboring mutations and deletions of LZTR-1 or normal LZTR-1 revealed that tumors with genetic inactivation of LZTR-1 were enriched for genes associated with glioma sphere growth and proliferation35 (Fig. 3a). Introduction of LZTR-1 in three independent GBM-derived sphere cultures resulted in strong inhibition of glioma sphere formation and expression of glioma stem cell markers (Fig. 3b–e). LZTR-1 also decreased the size of tumor spheres, induced a flat and adherent phenotype and reduced proteins associated with cell cycle progression (cyclin A, PLK1, p107, Fig. 3d–e). Interestingly, both R810W and W437STOP LZTR-1 mutations abolished LZTR-1 ability to impair glioma sphere formation (Fig. 3f). The above experiments indicate that LZTR-1 inactivation in human GBM drives self-renewal and growth of glioma spheres.
Figure 3.
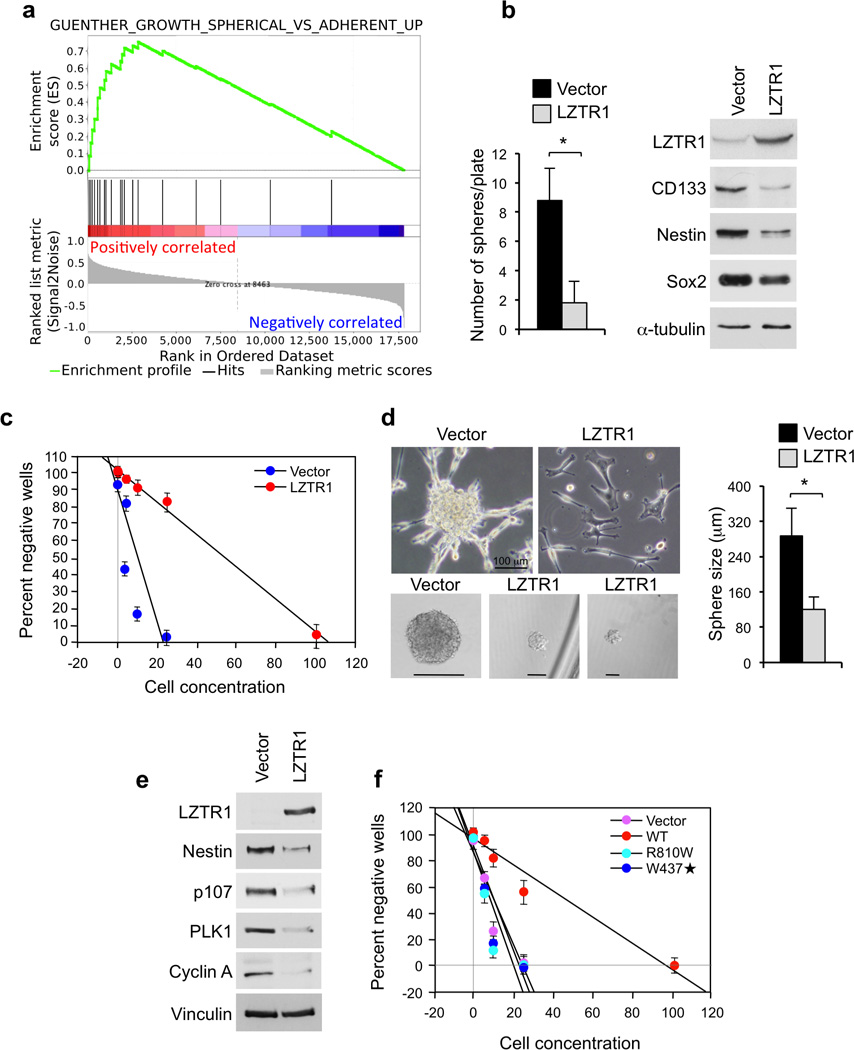
Functional analysis of LZTR-1 wild type and GBM associated mutants in GBM-derived cells. a, GSEA shows up-regulation of genes associated with the phenotype of “spherical cultures” of glioma cells in primary human GBM carrying mutations in the LZTR-1 gene [Enrichment Score (ES) = 0.754; P (family-wise error rate, FWER) = 0.000 q (false discovery rate, FDR) = 0.000]. b, Sphere forming assay (left panel) and western blot analysis (right panel) of GBM-derived glioma spheres (#48) expressing vector or LZTR-1. Data are Mean±SD of triplicate samples (t-test, p = 0.0036). c, Linear regression plot of in vitro limiting dilution assay using GBM-derived glioma spheres #46 expressing vector or LZTR-1. The frequency of sphere forming cells was 8.49±1.04 and 1.44±0.05% in vector and LZTR-1 expressing cells, respectively (t-test, p = 0.00795). Each data point represents the average of triplicates. Error bars are SD. d, Left upper panels, Bright field microphotographs of GBM-derived line 46 cells six days after transduction with vector or LZTR-1 expressing lentivirus. Left lower panels, Bright field microphotographs of spheres from GBM-derived glioma cells #46 expressing lentivirus expressing vector or LZTR-1 from experiment in c. Right panel, The size of tumor spheres from cultures in c was determined by microscopy review after 14 days of culture. n = 60 spheres from triplicates for each condition. Data are Mean±SD (t-test, p < 0.0001). e, Western blot analysis of GBM-derived cells #84 expressing vector or LZTR-1. f, Linear regression plot of in vitro limiting dilution assay using GBM-derived line 84 expressing vector, LZTR-1, LZTR-1-R810W or LZTR-1-W437STOP. The frequency of sphere forming cells was 7.2±0.92 for vector, 1.48±0.09 for LZTR-1 wild type (p = 0.0096), 7.82±0.99 for LZTR-1-R810W (p = 0.2489), and 6.74±1.07 for LZTR-1-W437STOP (p = 0.2269). Error bars are SD, p is from t-test.
Inactivation of CTNND2 induces mesenchymal transformation in glioblastoma
Among the top ranking genes in MutComFocal, CTNND2 is expressed at the highest levels in normal brain (Supplementary Table 9). CTNND2 codes for δ-catenin, a member of the p120 subfamily of catenins expressed almost exclusively in the nervous system where it is crucial for neurite elongation, dendritic morphogenesis and synaptic plasticity36–38. Germ-line hemizygous loss of CTNND2 impairs cognitive functions and underlies some forms of mental retardation39,40. CTNND2 shows pronounced clustering of mutations in GBM. The observed spectrum of mutations includes four mutations in the armadillo-coding domain and one in the region coding for the N-terminal coiled-coil domain (Supplementary Fig. 6a), the two most relevant functional domains of δ-catenin. Each mutation targets highly conserved residues with probably (K629Q, A776T, S881L, D999E) and possibly (A71T) damaging consequences41. GBM harbors focal genomic losses of CTNND2, and deletions correlate with loss of CTNND2 expression (Supplementary Fig. 6b, Supplementary Table 7).
Immunostaining experiments showed that δ-catenin is strongly expressed in normal brain, particularly in neurons, as demonstrated by co-staining with the neuronal markers β3-tubulin and MAP2 but not the astrocytic marker GFAP (Fig. 4a, b). Conversely, immunostaining of 69 GBM and western blot of 9 glioma sphere cultures revealed negligible or absent expression of δ-catenin in 21 tumors and in most glioma sphere cultures (Supplementary Fig. 6c, d). Oncogenic transformation in the CNS frequently disrupts the default proneural cell fate and induces an aberrant mesenchymal phenotype associated with aggressive clinical outcome42. Gene expression analysis of 498 GBM from ATLAS-TCGA showed that low CTNND2 expression is strongly enriched in tumors exhibiting the mesenchymal gene expression signature (t-test p-value = 2.4 10−12, Supplementary Fig. 7a). Tumors with reduced CTNND2 were characterized by poor clinical outcome and, among them, tumors with CTNND2 copy number loss displayed the worst prognosis (Supplementary Fig. 7b, c). Patients with low CTNND2 expression showed the worst clinical outcome in mesenchymal GBM, though non-mesenchymal tumors also demonstrated poor prognosis, albeit with reduced strength (Supplementary Fig. 7d). Mesenchymal transformation of GBM is associated with irreversible loss of proneural cell fate and neuronal markers42 and is detected in most established glioma cell lines. Expression of δ-catenin in the U87 human glioma cell line reduced cell proliferation (Supplementary Fig. 8a), elevated expression of neuronal proteins βIII-tubulin, PSD95 (a post-synaptic marker) and N-cadherin (Fig. 4c, Supplementary Fig. 8b) and decreased mRNA and protein levels of mesenchymal markers (Fig. 4d, e, Supplementary Fig. 8b). These effects were associated with morphologic changes characterized by neurite extension and development of branched dendritic processes (Fig. 4c, Supplementary Fig. 8c, d). Conversely, expression of the A776T, K629Q and D999E mutants of CTNND2 failed to induce neuronal features and down-regulate the mesenchymal marker fibronectin (FBN, Fig. 4e, Supplementary Fig. 8c, d). Consistent with δ-catenin inhibition of cell proliferation in glioma cells, only wild type δ-catenin decreased cyclin A, a S-phase cyclin (Fig. 4e). Next, we analyzed the effect of expressing δ-catenin in GBM-derived sphere culture #48 that lacks the endogenous δ-catenin protein (Supplementary Fig. 6d) and expresses high levels of mesenchymal markers43. Introduction of δ-catenin in sphere culture #48 strongly reduced mesenchymal proteins smooth muscle actin (SMA), collagen-5A1 (Col5A1) and FBN, as measured by quantitative immunofluorescence (Fig. 5a, b). It also induced βIII-tubulin more than eight-fold (Fig. 5c, d). Time course analysis showed the highest degree of βIII-tubulin-positive neurite extension at 4–6 days post-transduction followed by progressive depletion of neuronal-like cells from culture (Fig. 5d). Finally, we asked whether δ-catenin impacts self-renewal and growth of glioma spheres in vitro and their ability to grow as tumor masses in vivo. In a limiting dilution assay, δ-catenin inhibited glioma sphere formation more than 8-fold (Fig. 5e). To determine the effect of δ-catenin on brain tumorigenesis in vivo, we generated #48 glioma sphere cultures expressing luciferase and conducted bioluminescence imaging at different times after stereotactic transduction of control and δ-catenin-expressing cells in the mouse brain. When compared to controls, we observed a 5-fold inhibition of tumor growth by δ-catenin at each time point analyzed (Fig. 5f, Supplementary Fig. 8e). These results identify CTNND2 inactivation as a key genetic alteration driving the aggressive mesenchymal phenotype of GBM.
Figure 4.
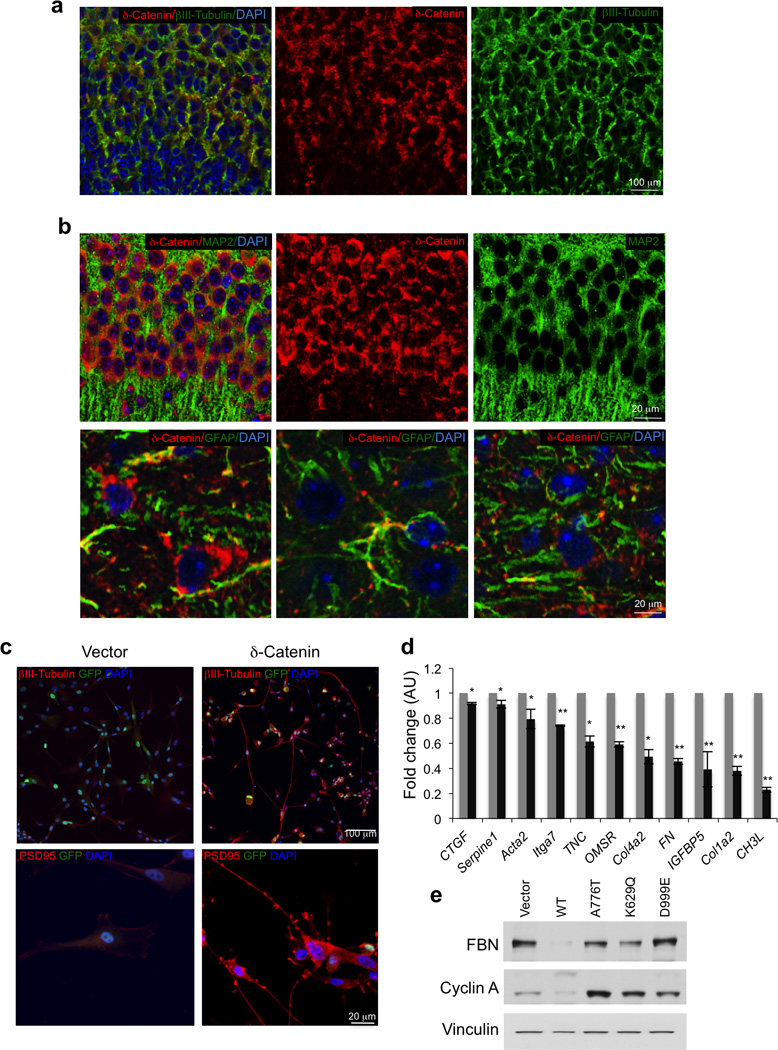
Expression of δ-catenin in neurons and δ-catenin driven loss of mesenchymal marker in GBM. a, Pattern of expression of δ-catenin in the developing brain, as determined by immunostaining. Double immunofluorescence staining of brain cortex using δ-catenin antibody (red) and βIII-tubulin (green); Nuclei are counterstained with Dapi (blue). b, Pattern of expression of δ-catenin in the adult brain, as determined by immunostaining. Upper panels, Double immunofluorescence staining of brain cortex using δ-catenin antibody (red) and MAP2 (green); Nuclei are counterstained with Dapi (blue). Lower panels, Double immunofluorescence staining of of brain cortex using δ-catenin antibody (red) and GFAP (green); Nuclei are counterstained with Dapi (blue). c, Immunofluorescence staining for βIII-tubulin (upper panels) and PSD95 (lower panels) in U87 cells expressing δ-catenin or the empty vector. d, Expression of mesenchymal genes in glioma cells expressing δ-catenin or the empty vector (averages of triplicate quantitative RT-PCR). Error bars are SD p is from t-test. *, p ≤ 0.005; **, p ≤ 0.001. e, Western blot using the indicated antibodies for U87 cells expressing δ-catenin wild type, glioma–associated δ-catenin mutants or the empty vector. FBN, fibronectin. Vinculin is shown as control for loading.
Figure 5.
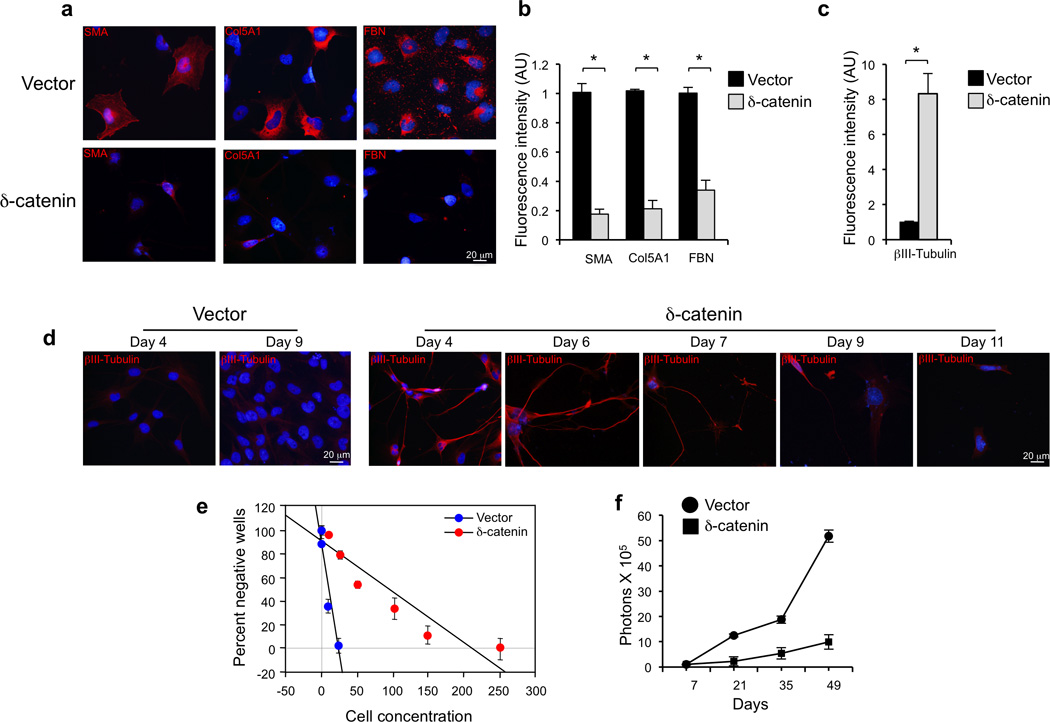
Functional analysis of CTNND2 in mesenchymal GBM. a, Immunofluorescence for fibronectin, collagen-5α1 (COL5A1) and smooth muscle actin (SMA) in glioma spheres #48 four days after infection with lentiviruses expressing δ-catenin or the empty vector. Nuclei are counterstained with Dapi. b, Quantification of fluorescence intensity for SMA, COL5A1 and FBN for cultures treated as in a. n = 3 independent experiments; data indicate mean±SD. c, Quantification of fluorescence intensity for βIII-tubulin in cells #48 infected with lentiviruses expressing CTNND2 or the empty vector. d, Time course analysis of βIII-tubulin expression in glioma spheres #48 transduced with lentiviruses expressing CTNND2 or the empty vector. Note the loss from the advanced culture of βIII-Tubulin expressing cells. e, Linear regression plot of in vitro limiting dilution assay using GBM-derived cells #48 expressing vector or δ-catenin. The frequency of sphere forming cells was 7.42±1.16 and 0.88±0.02 for vector and δ-catenin, respectively (t-test, p = 0.0098). Error bars are SD. f, Longitudinal analysis of bioluminescence imaging in mice injected intracranially with GBM-derived line 48 expressing vector or δ-catenin. n = 3 mice for vector and 5 for δ-catenin. Symbols are the mean and bars are SEM of photon counts.
Recurrent EGFR fusions in GBM
To identify gene fusions in GBM, we analyzed RNA-seq data from a total of 185 GBM samples (161 primary GBM plus 24 short-term glioma sphere cultures freshly isolated from patients carrying primary GBM). The analysis of RNA-seq led to the discovery of 92 candidate rearrangements giving rise to in-frame fusion transcripts (Supplementary Table 10). Besides previously reported FGFR3-TACC3 fusions events, the most frequent recurrent in-frame fusions involved EGFR in 7.6% of samples (14/185, 3.8%-11.3% CI). Nine of 14 EGFR fusions included recurrent partners SEPT14 (6/185, 3.2%) and PSPH (3/185, 1.6%) as the 3’ gene segment in the fusion. All EGFR-SEPT14 and two of three EGFR-PSPH gene fusions occurred within amplified regions of the fusion genes (Supplementary Fig. 9). The quantitative analysis of expressed reads spanning the fusion breakpoint versus reads spanning EGFR exons not implicated in the fusion transcripts revealed that EGFR fusion genes were expressed at higher levels in five of nine tumors (Supplementary Table 11). We also found two in-frame highly expressed fusions involving the neurotrophic tyrosine kinase receptor 1 gene (NTRK1) as the 3’ gene with two different 5’ partners (NFASC-NTRK1 and BCAN-NTRK1). Fusions involving NTRK1 are common in papillary thyroid carcinomas44. Using EXomeFuse, an algorithm that reconstructs genomic fusions from whole-exome data, we confirmed that EGFR-SEPT14 and NRTK1 fusions result from recurrent chromosomal translocations and reconstructed the corresponding genomic breakpoints (Supplementary Table 12).
The sequence of the PCR products spanning the fusion breakpoint validated all three types of recurrent in-frame fusion predictions (EGFR-SEPT14, EGFR-PSPH and NRTK1 fusions, Fig. 6, Supplementary Fig. 10 and 11). In Fig. 6a, b we show the prediction and cDNA sequence validation respectively, for one tumor harboring an EGFR-SEPT14 fusion (TCGA-27–1837). The amplified cDNA contained an open reading frame for a 1,041 amino-acid protein resulting from the fusion of EGFR residues 1–982 with SEPT14 residues 373–432 (Fig. 6c). Thus, the structure of EGFR-Septin14 fusions involve EGFR at the N-terminus, providing a receptor tyrosine kinase domain fused to a coiled-coil domain from Septin14. Exon-specific RNA-seq expression in TCGA-27–1837 demonstrated that EGFR and SEPT14 exons implicated in the fusion are highly expressed compared with mRNA sequences not included in the fusion event (Supplementary Fig. 12). Using PCR, we mapped the genomic breakpoint to chromosome 7 (#55,268,937 for EGFR and # 55,870,909 for SEPT14, genome build GRCh37/hg19) within EGFR exon 25 and SEPT14 intron 9, creating a transcript in which the 5’ EGFR exon 24 is spliced to the 3’ SEPT14 exon 10 (Fig. 6d). Interestingly, the fused EGFR-PSPH cDNA and predicted fusion protein in sample TCGA-06-5408 involves the same EGFR N-terminal region implicated in the EGFR-SEPT14 with PSPH providing a carboxy-terminal portion of 35 amino acids (Supplementary Fig. 10). An example of a fusion in which the EGFR-TK region is the 3’ partner is the CAND1-EGFR fusion in the glioma sphere culture #16 (Supplementary Fig. 13). Each fusion transcript includes the region of the EGFR mRNA coding for the TK domain (Supplementary Table 10). RT-PCR and genomic PCR followed by Sanger sequencing of GBM TCGA-06-5411 validated the NFASC-NTRK1 fusions in which the predicted fusion protein includes the TK domain of the high-affinity NGF receptor (TrkA) fused downstream to the immunoglobulin-like region of the cell adhesion and ankyrin-binding region of neurofascin (Supplementary Fig. 11).
Figure 6.
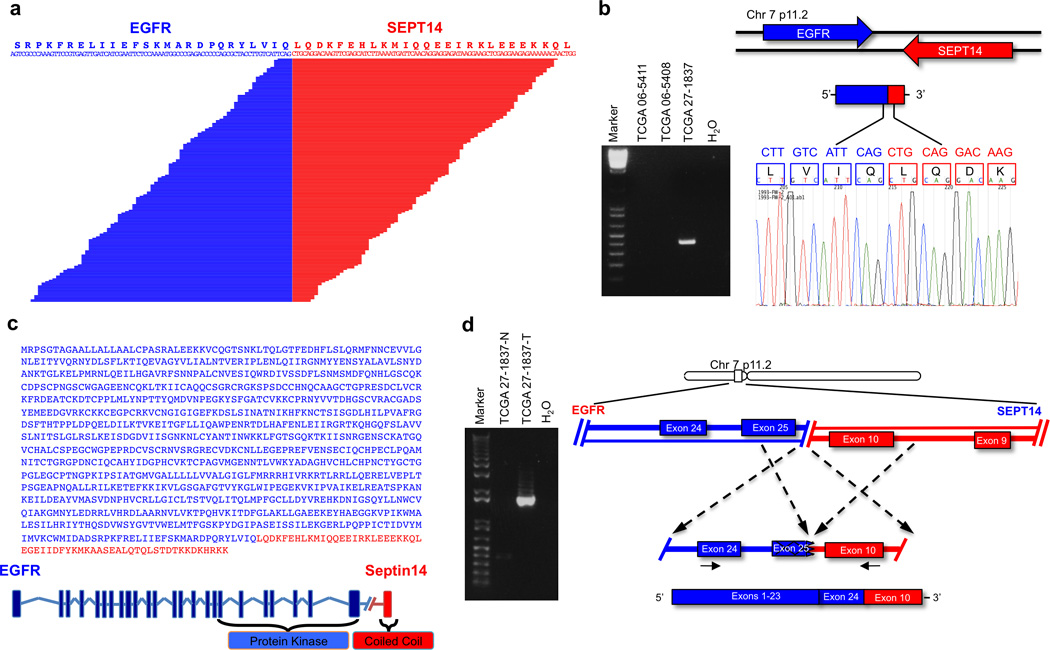
EGFR-SEPT14 gene fusion identified by whole transcriptome sequencing. a, Split reads are shown aligning on the breakpoint. The predicted reading frame at the breakpoint is shown at the top with EGFR sequences in blue and SEPT14 in red. b, (left panel), EGFR-SEPT14-specific PCR from cDNA derived from GBMs. Marker, 1kb ladder. (right panel), Sanger sequencing chromatogram showing the reading frame at the breakpoint and putative translation of the fusion protein in the positive sample. c, EGFR-Septin14 fusion protein sequence and schematics. Regions corresponding to EGFR and Septin14 are shown in blue and red, respectively. The fusion joins the tyrosine kinase domain of EGFR and the Coiled-coil domain of Septin14. d, Genomic fusion of EGFR exon 25 with intron 9 of SEPT14. In the fuse mRNA exon 24 of EGFR is spliced 5’ to exon 10 of SEPT14. Solid arrows indicate the position of the fusion genome primers that generate a fusion specific PCR product in the GBM sample TCGA-27–1837.
To confirm that GBM harbors recurrent EGFR fusions and determine the frequency in an independent dataset, we screened cDNA from a panel of 248 GBMs and discovered 10 additional cases with EGFR-SEPT14 fusions (4%). Conversely, NFASC-NTRK1 fusions were not detected in this dataset. We also determined a 2.2% (3/135) frequency of EGFR-PSPH fusions.
The discovery of recurrent EGFR fusions in GBM is of particular interest. EGFR is activated in a significant fraction of primary GBM (∼25%) by an in-frame deletion of exons 2–7 (EGFRvIII)45. However, seven of nine tumors harboring EGFR-SEPT14 and EGFR-PSPH gene fusions lacked the EGFRvIII rearrangement (Supplementary Table 13). We sought to determine whether the most frequent EGFR fusion in GBM (EGFR-SEPT14) provides an alternative mechanism of EGFR activation and confers sensitivity to EGFR inhibition. First, we asked whether EGFR gene fusions cluster into any gene expression subtype of GBM (proneural, neural, classical, mesenchymal). Although no individual subtype displayed a statistically significant enrichment of EGFR fusions, 8 of 9 GBM harboring EGFR-SEPT14 or EGFR-PSPH belonged to the classical or mesenchymal subtype (Fisher’s P value = 0.05 for classical/mesenchymal enrichment, Supplementary Table 14). Next, we compared the effects of ectopic EGFR-SEPT14, EGFRvIII or EGFR wild type on glioma cells. Lentiviral transduction of #48 human glioma sphere culture (which lacks genomic alteration of EGFR) showed that cells expressing EGFR-SEPT14 or EGFRvIII but not those expressing wild type EGFR or vector retained growth and self-renewal in the absence of EGF and bFGF (Fig. 7a). Accordingly, established glioma cell lines expressing EGFR-SEPT14 or EGFRvIII proliferated at higher rate than control cells or cells expressing wild type EGFR (Supplementary Fig. 14a, b). Furthermore, EGFR-SEPT14 and EGFRvIII markedly enhanced migration of glioma cells in a wound assay (Supplementary Fig. 14c). The above findings suggest that EGFR-SEPT14 might constitutively activate signaling events downstream of EGFR. When analyzed in the presence and absence of mitogens, the expression of EGFR-SEPT14 (or EGFRvIII) in glioma sphere cultures #48 triggered constitutive activation of phospho-STAT3 but had no effects on phospho-ERK and phospho-AKT (Fig. 7b, c). Differential gene expression analysis identified a set of 9 genes up-regulated in EGFR-SEPT14 tumors compared with EGFRvIII-positive GBM (Supplementary Fig. 15). These genes broadly relate to inflammatory/immune response, and some code for chemokines (CXCL9, 10, 11) that have been associated with aggressive glioma phenotypes46.
Figure 7.
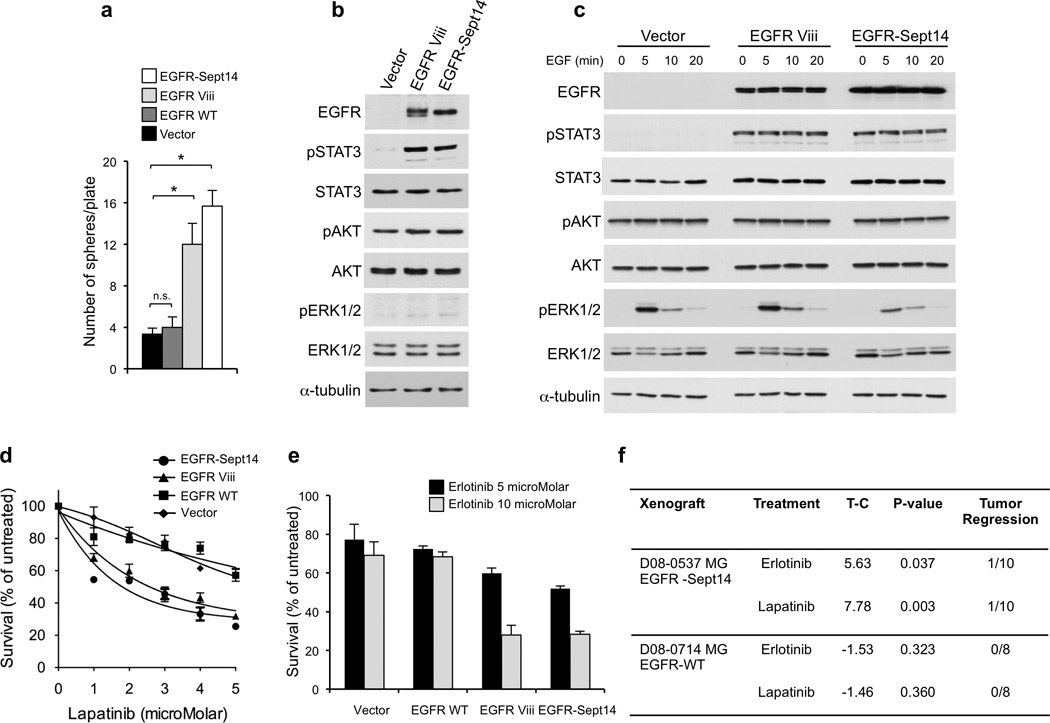
Functional analysis of EGFR-SEPT14 fusion and effect of inhibition of EGFR kinase on glioma growth. a, Sphere forming assay in the absence of EGF of GBM-derived primary cells (#48) expressing vector, EGFR wild type, EGFR Viii or EGFR-SEPT14 fusion. Data are Mean±SD of triplicate samples (t-test, p = 0.0051 and p = 0.027 for EGFR-SEPT14 fusion and EGFR Viii compared with vector, respectively). b, Western blot analysis of GBM-derived primary cells (#48) expressing vector, EGFR Viii or EGFR-SEPT14 fusion cultured in the presence of EGF. c, GBM-derived cells (#48) expressing vector, EGFR Viii or EGFR-SEPT14 fusion were cultured in the absence of EGF for 48 h and then stimulated with EGF 20ng/ml for the indicated time. Cells were assayed by western blot using the indicated antibodies. d, Survival of GBM-derived cells (#48) expressing vector, EGFR wild type, EGFR Viii or EGFR-SEPT14 fusion after treatment with lapatinib for 48 h at the indicated concentrations. Data are Mean±SD of triplicate samples. e, Survival of GBM-derived cells (#48) expressing vector, EGFR wild type, EGFR Viii or EGFR-SEPT14 fusion after treatment with erlotinib for 48 h at the indicated concentrations. Data are Mean±SD of triplicate samples. Experiments were repeated three times. f, In vivo inhibition of tumor growth by EGFR kinase inhibitors in glioma patient derived xenografts carrying EGFR-SEPT14 fusion (n = 10) but not wild type EGFR (n = 8). T-C indicates the median difference in survival between drug treated and vehicle (control) treated mice.
Finally, we sought to investigate whether EGFR-SEPT14 fusions confer sensitivity to inhibition of EGFR-TK. Treatment of #48 expressing EGFR-SEPT14, EGFRvIII, wild type EGFR or vector control with lapatinib, an irreversible EGFR inhibitor recently proposed to target EGFR alterations in GBM47, revealed that EGFR-Sept14 and EGFRvIII but not wild-type EGFR sensitized glioma cells to pharmaceutical EGFR inhibition (Fig. 7d). Similar effects were obtained following treatment of #48-derivatives with erlotinib, another inhibitor of EGFR-TK (Fig. 7e). To ask whether sensitivity to EGFR-TK inhibition is retained in human glioma cells naturally harboring EGFR-SEPT14 in vivo, we used an EGFR-SEPT14-positive GBM xenograft (D08-0537 MG) established from a heavily pretreated patient. Treatment of D08-0537 MG tumors with lapatinib or erlotinib showed that both drugs significantly delayed tumor growth, with lapatinib displaying the strongest anti-tumor effects. Conversely, EGFR inhibitors were ineffective against GBM xenograft D08-0714 MG, which lacks EGFR genomic alterations (Fig. 7f, Supplementary Fig. 14d). Taken together, these data determine that EGFR-SEPT14 fusions confer mitogen-independent growth, constitutively activate STAT3 signaling and impart sensitivity to EGFR kinase inhibition to glioma cells harboring the fusion gene.
DISCUSSION
We describe a computational pipeline that computes frequency, magnitude and focality of CNVs at any loci in the human genome with the somatic mutation rate for genes residing at that genomic location, thus integrating into a single score two genetic hallmarks of driver cancer genes (focality of CNVs and point mutations). Besides recognizing nearly all genes known to have functional relevance in GBM, our study discovered and validated somatic mutations in 18 new genes, which also harbor focal and recurrent CNVs in a significant fraction of GBM. The importance of some of these genes extends beyond GBM, as underscored by cross-tumor relevance (e.g. BCOR), and protein family recurrence (e.g. LRP family members). Also, the LZTR-1 mutations targeting highly conserved residues in the Kelch domain (W105, G248, T288) and in the second BTB-BACK domain (R810) are recurrent events in other tumor types48. Thus, understanding the nature of substrates of LZTR-1-Cul3 ubiquitin ligase activity will provide important insights into the pathogenesis of multiple cancer types. The importance of LZTR-1 genetic alterations in GBM is underscored by concurrent targeting of LZTR-1 by mutations and deletions that supports a two-hits mechanism of tumor suppressor gene inactivation as well as the impact of mutations targeting the BTB-BACK domains on Cul3 binding and/or protein stability, and their ability to release glioma cells from the restraining activity of the wild-type protein on self-renewal.
The finding that loss-of-function of CTNND2 cluster in mesenchymal GBM provides a clue to the genetic events driving this aggressive GBM subtype. The function of δ-catenin for crucial neuronal morphogenesis indicates that full-blown mesenchymal transformation in the brain requires loss of master regulators constraining cell determination along the neuronal lineage. Introduction of δ-catenin in human glioma spheres collapsed the mesenchymal phenotype and inhibited sphere formation and tumor growth. Thus, the ability of δ-catenin to reprogram glioma cells expressing mesenchymal genes towards a neuronal fate unravels an unexpected plasticity of mesenchymal GBM that might be exploited therapeutically.
In this study, we also report the landscape of gene fusions from a large dataset of GBM analyzed by RNA-Sequencing. In-frame gene fusions retaining the RTK-coding domain of EGFR emerged as the most frequent gene fusion in GBM. In this tumor, EGFR is frequently targeted by focal amplications and our finding underscores the strong recombinogenic probability of focally amplified genes, as recently reported for the myc locus in medulloblastoma49. Resembling intragenic rearrangements that generate the EGFRvIII allele, we found that EGFR-SEPT14 fusions impart to glioma cells the ability to self-renew and grow in the absence of mitogens, constitutively activate STAT3 signaling and confer sensitivity to EGFR inhibition. These findings highlight the relevance of fusions implicating RTK-coding genes in the pathogenesis of GBM9. They also provide a strong rationale for the inclusion of GBM patients harboring EGFR fusions in clinical trials based on EGFR inhibitors.
URLs
DNA and RNA sequencing and copy number variant data in The Cancer Genome Atlas (TCGA), http://cancergenome.nih.gov; glioma patient survival data from the Repository for Molecular Brain Neoplasia Data (REMBRANDT), https://caintegrator.nci.nih.gov/rembrandt/; sequence data deposition in database of Genotypes and Phenotypes (dbGaP), http://www.ncbi.nlm.nih.gov/gap; gene fusion annotation software package Pegasus, http://sourceforge.net/projects/pegasus-fus/.
Data access
RNA sequencing of twenty-four human GBM sphere cultures in this study were deposited under the dbGaP study accession phs000505.v2.p1. We also analyzed RNA and DNA sequencing of TCGA GBM samples from the dbGaP study accession phs000178.v1.p1.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by National Cancer Institute grants R01CA101644 and R01CA131126 (A.L.), R01CA085628 and R01CA127643 (A.I.), Stewart Foundation (R.R.), Partnership for Cure (R.R.), NIH 1 P50 MH094267-01 (R.R.), Lymphoma Research Foundation (R.R.), NIH 1 U54 CA121852-05 (R.R.), NIH 1R01CA164152-01 (R.R.), Leukemia and Lymphoma Society (R.R.), Canadian Cancer Society (G.G.P.), Cancer Research Society (G.G.P.), National Institute of Neurological Disorders and Stroke R01NS061776 (A.I.) and a grant from The Chemotherapy Foundation (A.I.). G.F. was supported by grants from the Associazione Italiana per la Ricerca sul Cancro and from Italian Ministry of Health. V.F., P.Z., C.D. and F.N. are supported by fellowships from the Italian Ministry of Welfare/Provincia di Benevento and F.I.A.G.O.P. (C.D.). We thank John Parkinson for helpful discussions on the phylogeny of LZTR-1 genes, Lauren Bertin for help with Western blots, Jens Kroll for the LZTR-1 plasmids and Michele Pagano for Cul3 expression plasmids.
Footnotes
AUTHOR CONTRIBUTIONS
A.L., R.R. and A.I. conceived the ideas for this study. R.R. designed and supervised the computational approach and A.L. and A.I. the experimental platform. A.L. performed or assisted in each step of the experimental platform. V.F., A.C., M.L., F.N., C.D. conducted biological experiments. V.T. performed the MutComfocal analysis. J.M.C. and F.A. performed the Gene Fusion analysis, Allele Specific Expression and most of the bioinformatics analyses. P.Z. performed bioinformatics and statistical analyses. S.T.K., H.Y., R.E.M. and D.D.B. performed the human glioma xenografts analyses to evaluated the effects of EGFR inhibitors and provided human GBM specimens. A.X.J. and G.G.P. performed the modeling analysis of LZTR-1. I.D. and A.H. conducted the targeted sequencing analysis. P.P., S.P., D.J.P., P.C., J.N.B., K.A., G.G., G.F. and T.M. provided tissue materials from study subjects. A.L., R.R. and A.I. wrote the manuscript with contributions from all other authors.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests
References
- 1.Porter KR, McCarthy BJ, Freels S, Kim Y, Davis FG. Prevalence estimates for primary brain tumors in the United States by age, gender, behavior, and histology. Neuro-oncology. 2010;12:520–527. doi: 10.1093/neuonc/nop066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl. J. Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Cancer Genome Atlas Research. N Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noushmehr H, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verhaak RG, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bass AJ, et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nat. Genet. 2011;43:964–968. doi: 10.1038/ng.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chinnaiyan AM, Palanisamy N. Chromosomal aberrations in solid tumors. Prog. Mol. Biol. Transl. Sci. 2010;95:55–94. doi: 10.1016/B978-0-12-385071-3.00004-6. [DOI] [PubMed] [Google Scholar]
- 9.Singh D, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337:1231–1235. doi: 10.1126/science.1220834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rubin AF, Green P. Mutation patterns in cancer genomes. Proc. Natl. Acad. Sci. U S A. 2009;106:21766–21770. doi: 10.1073/pnas.0912499106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan Z, et al. BCOR regulates mesenchymal stem cell function by epigenetic mechanisms. Nat. Cell Biol. 2009;11:1002–1009. doi: 10.1038/ncb1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wamstad JA, Bardwell VJ. Characterization of Bcor expression in mouse development. Gene Expr. Patterns. 2007;7:550–557. doi: 10.1016/j.modgep.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wamstad JA, Corcoran CM, Keating AM, Bardwell VJ. Role of the transcriptional corepressor Bcor in embryonic stem cell differentiation and early embryonic development. PLoS One. 2008;3:e2814. doi: 10.1371/journal.pone.0002814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pugh TJ, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–110. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J, et al. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature. 2012;481:329–334. doi: 10.1038/nature10733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beroukhim R, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kantarci S, et al. Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai-Barrow and facio-oculo-acoustico-renal syndromes. Nat. Genet. 2007;39:957–959. doi: 10.1038/ng2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Willnow TE, et al. Defective forebrain development in mice lacking gp330/megalin. Proc. Natl. Acad. Sci. U S A. 1996;93:8460–8464. doi: 10.1073/pnas.93.16.8460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christ A, et al. LRP2 is an auxiliary SHH receptor required to condition the forebrain ventral midline for inductive signals. Dev. Cell. 2012;22:268–278. doi: 10.1016/j.devcel.2011.11.023. [DOI] [PubMed] [Google Scholar]
- 20.Cowin PA, et al. LRP1B deletion in high-grade serous ovarian cancers is associated with acquired chemotherapy resistance to liposomal doxorubicin. Cancer Res. 2012;72:4060–4073. doi: 10.1158/0008-5472.CAN-12-0203. [DOI] [PubMed] [Google Scholar]
- 21.Lima FR, et al. Glioblastoma: therapeutic challenges, what lies ahead. Biochim. Biophys. Acta. 2012;1826:338–349. doi: 10.1016/j.bbcan.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 22.Bekker-Jensen S, et al. HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat. Cell Biol. 2010;12:80–86. doi: 10.1038/ncb2008. [DOI] [PubMed] [Google Scholar]
- 23.Harlalka GV, et al. Mutation of HERC2 causes developmental delay with Angelman-like features. J. Med. Genet. 2013;50:65–73. doi: 10.1136/jmedgenet-2012-101367. [DOI] [PubMed] [Google Scholar]
- 24.Nacak TG, Leptien K, Fellner D, Augustin HG, Kroll J. The BTB-kelch protein LZTR-1 is a novel Golgi protein that is degraded upon induction of apoptosis. J. Biol. Chem. 2006;281:5065–5071. doi: 10.1074/jbc.M509073200. [DOI] [PubMed] [Google Scholar]
- 25.Stogios PJ, Downs GS, Jauhal JJ, Nandra SK, Prive GG. Sequence and structural analysis of BTB domain proteins. Genome Biol. 2005;6:R82. doi: 10.1186/gb-2005-6-10-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Errington WJ, et al. Adaptor protein self-assembly drives the control of a cullin-RING ubiquitin ligase. Structure. 2012;20:1141–1153. doi: 10.1016/j.str.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 27.Ji AX, Prive GG. Crystal structure of KLHL3 in complex with Cullin3. PLoS One. 2013;8:e60445. doi: 10.1371/journal.pone.0060445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Canning P, et al. Structural basis for Cul3 assembly with the BTB-Kelch family of E3 ubiquitin ligases. J. Biol. Chem. 2013;288:7803–7814. doi: 10.1074/jbc.M112.437996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lo SC, Li X, Henzl MT, Beamer LJ, Hannink M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006;25:3605–3617. doi: 10.1038/sj.emboj.7601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boyden LM, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482:98–102. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Louis-Dit-Picard H, et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat. Genet. 2012;44:456–460. doi: 10.1038/ng.2218. S451–453. [DOI] [PubMed] [Google Scholar]
- 32.Emanuele MJ, et al. Global identification of modular cullin-RING ligase substrates. Cell. 2011;147:459–474. doi: 10.1016/j.cell.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galan JM, Peter M. Ubiquitin-dependent degradation of multiple F-box proteins by an autocatalytic mechanism. Proc. Natl. Acad. Sci. U S A. 1999;96:9124–9129. doi: 10.1073/pnas.96.16.9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang DD, et al. Ubiquitination of Keap1, a BTB-Kelch substrate adaptor protein for Cul3, targets Keap1 for degradation by a proteasome-independent pathway. J. Biol. Chem. 2005;280:30091–30099. doi: 10.1074/jbc.M501279200. [DOI] [PubMed] [Google Scholar]
- 35.Gunther HS, et al. Glioblastoma-derived stem cell-enriched cultures form distinct subgroups according to molecular and phenotypic criteria. Oncogene. 2008;27:2897–2909. doi: 10.1038/sj.onc.1210949. [DOI] [PubMed] [Google Scholar]
- 36.Abu-Elneel K, et al. A delta-catenin signaling pathway leading to dendritic protrusions. J. Biol. Chem. 2008;283:32781–32791. doi: 10.1074/jbc.M804688200. [DOI] [PubMed] [Google Scholar]
- 37.Arikkath J, et al. Delta-catenin regulates spine and synapse morphogenesis and function in hippocampal neurons during development. J. Neurosci. 2009;29:5435–5442. doi: 10.1523/JNEUROSCI.0835-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kosik KS, Donahue CP, Israely I, Liu X, Ochiishi T. Delta-catenin at the synaptic-adherens junction. Trends Cell Biol. 2005;15:172–178. doi: 10.1016/j.tcb.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 39.Israely I, et al. Deletion of the neuron-specific protein delta-catenin leads to severe cognitive and synaptic dysfunction. Curr. Biol. 2004;14:1657–1663. doi: 10.1016/j.cub.2004.08.065. [DOI] [PubMed] [Google Scholar]
- 40.Jun G, et al. delta-Catenin is genetically and biologically associated with cortical cataract and future Alzheimer-related structural and functional brain changes. PLoS One. 2012;7:e43728. doi: 10.1371/journal.pone.0043728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hicks S, Wheeler DA, Plon SE, Kimmel M. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum. Mutat. 2011;32:661–668. doi: 10.1002/humu.21490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phillips HS, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 43.Carro MS, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pierotti MA, Greco A. Oncogenic rearrangements of the NTRK1/NGF receptor. Cancer Lett. 2006;232:90–98. doi: 10.1016/j.canlet.2005.07.043. [DOI] [PubMed] [Google Scholar]
- 45.Dunn GP, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012;26:756–784. doi: 10.1101/gad.187922.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu C, et al. Chemokine receptor CXCR3 promotes growth of glioma. Carcinogenesis. 2011;32:129–137. doi: 10.1093/carcin/bgq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vivanco I, et al. Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2012;2:458–471. doi: 10.1158/2159-8290.CD-11-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Forbes SA, et al. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010;38:D652–D657. doi: 10.1093/nar/gkp995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Northcott PA, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488:49–56. doi: 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.