

Abstract
Recombinant serotype 5 adenovirus (Ad5) vectors lacking E1 expression induce robust immune responses against encoded transgenes in preclinical models, but have muted responses in human trials due to wide spread pre-existing anti-adenovirus immunity. Attempts to circumvent Ad5 specific immunity by using alternative serotypes or modifying capsid components have not yielded profound clinical improvement. To address this issue, we explored a novel alternative strategy, specifically reducing the expression of structural Ad5 genes by creating E1 and E2b deleted recombinant Ad5 vectors. Our data demonstrate that [E1−, E2b−]vectors retaining the Ad5 serotype are potent immunogens in pre-clinical models despite the presence of significant Ad5 specific immunity, in contrast to [E1−] vectors. These preclinical studies with E1 and E2b deleted recombinant Ad5 vectors suggest that anti-Ad immunity will no longer be a limiting factor and that clinical trials to evaluate their performance are warranted.
Keywords: Adenovirus, Ad5 vectors, immunotherapy
INTRODUCTION
Despite promising results in murine models, cancer vaccines have not been proven clinically effective in human clinical trials1. Many cancer vaccines target non-mutated “self” antigens expressed aberrantly by tumor cells, such as Carcinoembryonic antigen (CEA). While CEA expression in adults is normally limited to low levels in the gastrointestinal epithelium2, CEA is highly expressed in the majority of colorectal cancers and many breast, lung and pancreas cancers2, 3. Immune responses are weak to tumor antigens such as CEA in humans because of strong immunologic tolerance to these proteins. As a consequence, a minimal requirement for cancer vaccines that target non-mutated self antigens is a need to overcome immune tolerance.
Recombinant viral vector vaccines have been shown to overcome tolerance in a variety of animal models and in clinical studies. We and others have previously tested recombinant fowlpox vectors in clinical trials and observed the induction of detectable anti-CEA immunity4. Nonetheless, we have seen limited boosting effects with repeated immunizations, suggesting that anti-fowlpox immune responses were limiting the expression and subsequent immune response to CEA. Even using vaccination strategies to avoid neutralizing antibodies, such as ex vivo infection of dendritic cells (DC), did not lead to boosting4. Consequently, we sought to develop alternative viral vectors to use alone or in heterolgous vector prime-boost strategies to enhance CEA-specific immunity.
Recombinant adenovirus (Ad) vectors are the most widely used viral vector in clinical gene therapy applications including vaccines5–8. The commonly used Ad5 serotype has an extensive and accepted safety profile, as well as allowing for maximal induction of transgene-specific adaptive immune responses9–11. The prototypic Ad vectors are E1 deleted and considered to be replication-defective but have been shown to allow for limited genome replication and expression of toxic Ad gene products. Consequently, recombinant Ad5 vaccines have limited efficacy in the presence of widely prevalent neutralizing antibodies and T cell responses to Ad5. This anti-vector immunity limits the infection of host antigen presenting cells in vivo and consequently also limits immune responses to the vector encoded vaccine antigen(s).
We have a significant interest in the modification of Adenovirus based vectors to optimize them for use as cancer vaccines. We retained the Ad5 based vaccine platform and made genomic modifications to determine if we could improve its efficacy, and particularly if these modifications can evade the effects of pre-existing anti-Ad5 immunity. We constructed two new Ad5 vectors (referred to as replicating and non-replicating) expressing CEA and compared them to the prototypical Ad5[E1−] vector used in most clinical studies. Critically, we found that the non-replicating Ad5 vector deleted for the E1, E2b, E3 genes was strikingly superior to the Ad5 E1 deleted vector and was capable of generating CEA specific T cell responses in mice with pre-existing Ad5 immunity that were equal to responses in naïve mice. These results suggest that pre-existing Ad5 immunity is no longer a limiting factor to the use of Ad5-based vaccines, and that future clinical trials to evaluate their performance are warranted.
MATERIALS AND METHODS
Reagents
Flourescent-conjugated antibodies for flow cytometry assay were purchased from BD Bioscience (San Jose, CA). HLA-A*0201 restricted CMVpp65(495–503) peptide (NLVPMVATV) and MART-1(26–35) peptide (EAAGIGILTV) were purchased from Research Genetics (Huntsville, AL). 15mer peptide mix of carcinoembryonic antigen (CEA), CMVpp65 and HIV were purchased from BD Bioscience. Peptide-MHC tetramers for HLA-A*0201 restricted CMVpp65(495–503) peptides and control tetramer were purchased from Beckman Coulter (San Diego, CA).
Adenovirus Vector Preparation
Construction of a first-generation [E1−] Ad vector containing a human carcinoembryonic antigen (CEA) or human cytomegalovirus (CMV) pp65 antigen under the control of human CMV promoter/enhancer elements was performed as previously described12. Briefly, the CEA gene or CMVpp65 gene under the expression control of the 576-bp human CMV promoter/enhancer was subcloned into a shuttle plasmid, and used to generate Ad5[E1−]CEA vector or Ad5[E1−]CMV vector. The modified adenoviral vectors, Ad [E1−, E2b−], was constructed as previously described13. This vector has multiple depletion for early region 1 (E1) and E2b regions (DNA polymerase and pTP genes), and was engineered to express the identical human CMV promoter/enhancer-transgene cassette as utilized for the Ad[E1−]CEA or Ad[E1−]CMV vector. Complementing C-7 cell lines were used to support the growth and production of high titers of these vectors, and cesium chloride double banding was performed to purify the vectors, as previously reported14. The replicating Ad5[E1+] vectors, which only have the deletion of E3 gene, was also made for comparison purposes since theoretically it would produce higher levels of transgene expression.
Human peripheral blood samples
Peripheral blood samples from healthy human volunteers were obtained under Duke IRB-approved protocols.
Infection of human dendritic cells with Ad5-CEA vectors
Dendritic cells were cultured from PBMC of healthy human volunteers in AIM-V medium (Invitrogen, Carlsbad, CA) supplemented with recombinant human GM-CSF (800 IU/ml) and IL-4 (25 ng/ml, R&D Systems, Minneapolis, MN) for 7 days. DC were harvested and 3×106 cells in 1 ml of AIM-V medium was infected with Ad5-CEA or Ad5-CMV vectors at indicated multiplicity of infection (MOI of 0, 500, 5,000, 10,000, 20,000, 40,000, 80,000) numbers for 1 h in single wells of 6-well plates. Then, 4 ml of AIM-V medium was added to each well and incubation was continued for 2d. For the temporal analysis of CEA expression, DC were infected with Ad5-CEA at MOI 20,000 and incubated for 6 h to 96 h.
To analyze the efficacy of transgene expression, DC were harvested and stained with FITC conjugated anti-CD86, PE-conjugated anti-CEA, PerCP-conjugated anti-HLA-DR, APC-conjugated anti-CD14. CD14-negative HLA-DR-positive DC were analyzed for their CEA expression. To analyze the effect of Ad-vector infection on DC viability, DC were also stained labeled with Annexin-V-biotin (Beckman Coulter, Marseille, France) and then stained with APC-conjugated streptavidin together with 7-AAD (Beckman Coulter), FITC-conjugated anti-CEA, and PE-conjugated anti-HLA-DR. Large HLA-DR-positive cells (DC) were analyzed for 7-AAD/Annexin labeling.
To assess the effect of Ad-vector infection on DC maturation status, DC were stained with PerCP-conjugated anti-HLA-DR, APC-conjugated anti-CD14, and PE-conjugated antibodies against following molecules: CD40, CD54, CD80, CD83, CD86, CCR5, and CCR7. Antibodies for flow cytometry were purchased from BD Bioscience otherwise noted.
NK-mediated cell killing of Ad-vector infected DC
7-day cultured DC from a normal donor were infected with Ad5-CEA vectors at MOI 20,000 and incubated for 2 days. NK cells were harvested from the same donor’s PBMC using NK cell isolation kit (Miltenyi Biotec) and incubated overnight with IL-2 (1000 U/ml). Harvested DC were incubated with autologous NK cells for 6 h. Supernatants were used for LDH cytotoxicity assay (CytoTox 96) according to manufacturer’s instruction (Promega, Madison, MI).
Cytotoxic T lymphocyte activation by Adenovirus-infected DC
7-day cultured DC from normal donor (A2+, CMV+) were infected with Adenovirus vectors encoding CMVpp65 or CEA antigens for 2h and incubated for 2 days. 1×104 DC-transduced with the vectors (MOI 20,000) were used as stimulators and 1×104 or 5×104 of CMVpp65 CTL line was used as responder cells. CMVp65 polypeptide mix of 15mers overlapping by 11 representing the CMVpp65 protein was used as a positive control, and a HIV gag polypeptide mix as a control for the CMV mix. After 20 h incubation, IFN-γ ELISPOT assay (Mabtech Inc.) was performed according to manufacturer’s instruction. After 40 h incubation, supernatants were harvested from each well and IFN-γ ELISA was performed.
In vitro cytotoxic T lymphocytes expansion with Ad5-CMV infected DC
7-day cultured DC from normal donor (A2+, CMV+) were infected with Ad5-CMV at MOI 20,000 and incubated for 2 days. DC were harvested from plates and mixed with autologous PBLs at 10:1 (PBL:DC) ratio. On day 3 and 7, IL-2 (300IU/ml) was added to the culture and cells were harvested for tetramer assay on day 10. Cells were stained with FITC-conjugated anti-CD3, PerCP-conjugated anti-CD8, APC-conjugated anti-CD4, CD14, and CD19, and PE-conjugated CMVpp65 tetramer or control tetramer. CD3+CD4−CD14−CD19− lymphocytes were gated and the percentages of tetramer+ cells in CD8T cells were analyzed.
Mice
C57BL/6J mice were purchased from Jackson Labs (Bar Harbor, ME) and all work was conducted in accordance with Duke IACUC approved protocols.
Assesment of vector immunogenicity in vivo
In order to compare the capacity of Ad-vectors with different constructs to establish CEA-specific immunity and to induce production of anti-adenovirus antibody in vivo, mice were vaccinated once via footpad injection of Ad5-CMV vectors or Ad5-CEA vectors (2.6×1010 particles/mouse). Fourteen days later, mice were euthanized and splenocytes and sera were collected for analysis. IFN-γ ELISPOT assay was performed to examine the CEA-specific T cell responses and ELISA for CEA-specific antibodies was performed, as described below.
Modeling pre-exisiting anti-Ad immunity in vivo
To create a model of pre-existing anti-Ad immunity, mice were injected with 5×1010 particles of a Ad5[E1+]CMV vector via the footpad either on day 0, 14, and 28, or only on day 28, or received control Saline injections (Ad naïve mice). Blood was drawn on day 42 in order to quantify mouse anti-adenovirus antibody via ELISA, as described below. Mice were injected with, 2.6×1010 particles of the respective Ad5-CEA vectors on days 42, 49 and 56. Two weeks after the last vaccination, mice were euthanized and splenocytes and serum were harvested. IFN-γ ELISPOT assay was performed to examine the CEA-specific T cell responses and ELISA for CEA-specific antibodies was performed, as described below.
ELISPOT analysis to detect antigen-specific T cell responses from mouse splenocytes
Mouse IFN-γ ELISPOT assay (Mabtech Inc., Cincinnati, OH) was performed according to the manufacturer’s instruction. Splenocytes (500,000 cells/well) were added to the well, and CMVpp65 peptide mix or CEA peptide mix (2.6 μg/ml: BD Bioscience, San Jose, CA) was used as a stimulating antigen. Peptide mixes consist of 15 mers overlapping by 11 amino acids and cover the entire protein sequence. HIV peptide mix (BD Bioscience) was used as a negative control, and a mixture of PMA (50 ng/ml) and Ionomycin (1 μg/ml) as a positive control of the assay
ELISA for anti-adenovirus antibodies
Microtiter plates (Immulon 4BX, Dynex Technologies, Chantilly, VA) were coated with 1 × 109 total particles of Ad5[E1+]CEA virus in bicarbonate buffer (200 mM NaHCO3, 81 mM Na2CO3, pH 9.5) overnight. After blocking for an hour with a solution of 1% bovine serum albumin (BSA), 5% Sucrose, 0.05% NaN3 in PBS, mouse serum (diluited 1:40 to 1:320,000) were added to the wells and incubated for 2h at 37°C, followed by incubation with 100 μl of a 1:2500 dilution of sheep alkaline phosphatase-conjugated anti-mouse IgG antibody (Jackson Immunoresearch Laboratories, West Grove, PA) for 1h at 37°C. Plates were washed three times and 50 μl per well of the pNPP substrate solution (5 mg/ml, p-nitrophenyl phosphate, Sigma) were added to each well. The reaction was stopped by adding 50 μl per well of NaOH solution. Absorbance was measured at 405 nm.
ELISA for anti-transgene antibodies
Mouse sera were collected and frozen at −80°C until use. Microtiter plates (Immulon 4BX) were coated with CEA peptide mix or CMVpp65 peptide mix (BD Bioscience) at concentration of 2.6 μg/ml. Plates were incubated at 4°C overnight, washed with PBS, and blocked with 1%BSA in PBS for 1 h. Thawed sera were diluted with the titration of 1:40 to 1:320,000 with 1% BSA in PBS, added to each well and incubated for 2 h. Sera from Ad-naïve mice (saline injected mice) were used as a negative control. Incubation with anti-mouse IgG antibody and color development was done as described above.
Proliferation assay to detect anti-adenovirus T cell responses from mouse splenocytes
Mouse splenocytes (100,000 cells/well) were added to 96-well U bottomed plates and CMVpp65 peptide mix, CEA peptide mix, HIV peptide mix (2.6 μg/ml) or heat-inactivated Ad5[RC] (1 × 109 total particles/well) was used as a stimulating antigen. After 3 day-incubation, cells were pulsed with 3H-thymidine (1 μCi) for 18 hours and harvested on an automated cell harvester (TOMTEC, Turku, Finland). 3H-thymindine incorporation was determined using a MicroBeta Plus Scintillation counter (Wallac). Data are presented as stimulation index (S.I.). Stimulation Index was determined by dividing the mean cpm of wells incubated with antigen by the mean cpm of media alone.
Statistical analyses
An unpaired Student’s t test was used to analyze statistical difference.
RESULTS
Because Ad replication may significantly impact the efficacy of Ad based vaccines we developed Ad5 based vectors expressing CEA under control of an identical CMV enhancer/promoter element, but differing only in the deletion of critical Ad genes.
Specifically, we generated a prototypic CEA expressing Ad vector which had deletions of the E1 and E3 region Ad genes (Ad5[E1−, E3−]CEA; henceforth referred to as Ad5[E1−]CEA), but also two novel Ad vaccines. The first was a non-replicating vector deleted not only for the Ad E1 and E3 genes, but also the Ad E2b genes, (Ad5[E1−, E2b−, E3−]CEA; henceforth referred to as Ad5[E1−, E2b−]CEA). This gene transfer platform has been previously shown to allow for decreased toxicity and improved persistence of transgene expression in immune competent animals15. The second was an Ad vector that retains the E1 genes but is deleted for the E3 gene (Ad5[E1+, E3−]CEA; henceforth referred to as Ad5[E1+]CEA). This design allows for Ad genome replication which we hypothesized might provide improved transgene expression and possibly enhanced vaccine responses.
Prior to testing the performance of these vectors in the setting of Ad specific immunity, we set out to characterize these vectors using critical in vitro and in vivo assays of vaccine efficacy.
Infection of human DC and assessment of vector toxicity
Adenoviral vector vaccines achieve much of their effects either by the infection of antigen presenting cells (APC), or the production of antigen that is subsequently processed and presented by APC. Therefore we studied the antigen expression of CEA in human DC in vitro following co-culture with the 3 vectors.
Human monocyte derived DC were infected and their CEA expression was analyzed (Fig. 1a). At low multiplicities of infection (MOI) the replication-competent Ad5[E1+]CEA vector resulted in the highest number of Ad transduced (CEA expressing) DC. However, similar transduction efficiencies were seen with all three vectors at MOI ≥20,000. The mean fluorescence intensity (MFI) of CEA expression is a measure of the average quantity of CEA protein being expressed by the Ad5-infected DC. The MFI of CEA expression by DC tended to be highest in Ad5[E1+]CEA-infected DC, followed by Ad5[E1−]CEA and Ad5[E1−, E2b−]CEA vectors (Fig. 1b). This is perhaps not surprising given each vectors’ inherent replicative activity. When the toxicity of Ad5 vector infection was analyzed using Annexin-V labeling as a measure of apoptotic cell death of DC (Fig. 1c), the non-replicative Ad5[E1−, E2b−]CEA vector always yielded the best viability of DC, while particularly at higher MOI the Ad5[E1+]CEA led to significant apoptotic cell death.
Figure 1. Transduction efficiency, toxicity, and temporal kinetics of CEA expression in Ad5-CEA infected human dendritic cells.
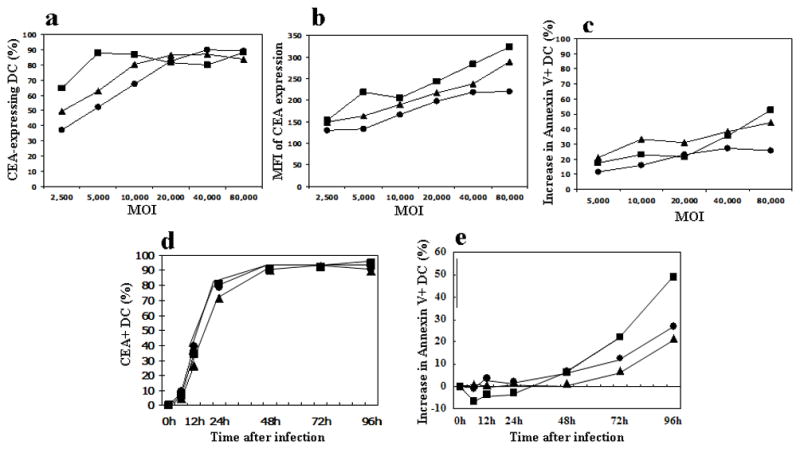
In vitro cultured human DCwere infected with either the Ad5[E1+]CEA, Ad5[E1−]CEA, or Ad5[E1−, E2b−]CEA vectors at the multiplicity of infection (MOI) shown. (a) The percentage of CEA-expressing DC is shown for each MOI. (b) Mean Fluorescent Intensity (MFI) of CEA expression on DC is shown. (c) Ad vector infection resulted in toxicity/apoptosis of DC. The increase in Annexin-V positive cells above background (mock infected DC) are plotted in the graph. To assess temporal kinetics of CEA expression and toxicity to DC, in vitro cultured human DC were infected with either the Ad[E1+]CEA, Ad[E1−]CEA, or Ad[E1−, E2b−]CEA vectors at MOI of 20,000. After 0, 6, 12, 24, 48, 72, 96 h of incubation, DCs were harvested and stained. (d) The percentage of CEA-expressing DC is plotted over time. (e) The increase of Annexin-V positive DC above background (mock infected DC) is plotted over time. Triangle: Ad5[E1−], Circle: Ad5[E1−, E2b−], Square: Ad5[E1+]. These data are representative of 3 separate experiments.
Temporal kinetics of CEA expression by human dendritic cells following Ad5-vector infection
The kinetics of CEA expression and apoptosis in the Ad-infected human DC was analyzed at MOI 20,000, because this MOI produced a similar infection rate with each vector (Fig. 1a). Interestingly, these three vectors demonstrated very similar levels of CEA expression over time (Fig. 1d), suggesting that while the total amount of CEA protein being made within the DC differed (Fig. 1b) the expression kinetics of CEA did not differ. However, the Ad5[E1+]CEA vector induced a higher rate of apoptotic cell death in DC after the 48 h time point (Fig. 1e).
Does Ad-vector infection induce maturation of human DC?
The maturation status of DC is an important determinant of the quality of the resulting immune response, whether ex vivo Ad5-modified DC are used as a vaccine, or an Ad vaccine is injected and infects endogenous DC in vivo. Consequently, expression of maturation markers on infected DC was analyzed by flow cytometry (Fig. 2). The CEA expression level and MFI demonstrated comparable levels of infection for each vector in this experiment. CD40, CD86, and CCR7 expression was moderately enhanced by Ad-vector infection, with all three Ad vectors yielding similar increases in MFI compared to mock infected DC. CD83 expression was not altered by Ad5-CEA infection.
Figure 2. Ad5 infection of human dendritic cells mediates phenotypic maturation.
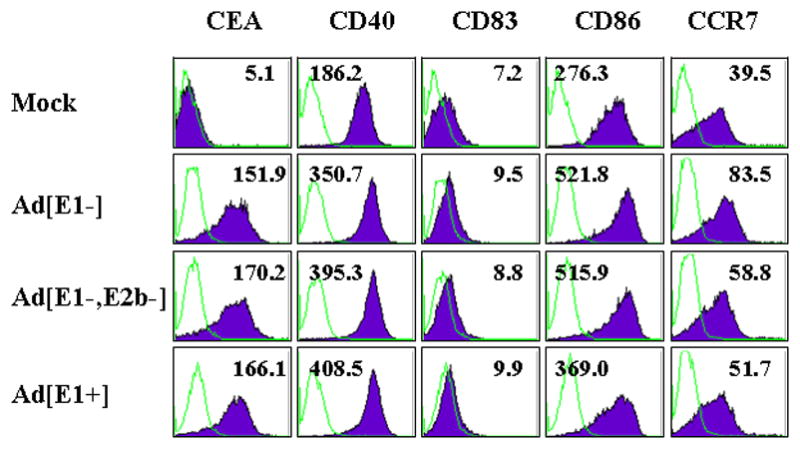
In vitro cultured human DC were infected with Ad5[E1+]CEA, Ad5[E1−]CEA, or Ad5[E1−, E2b−]CEA at MOI 20,000, or mock infected. After 2 days incubation, DC were harvested and stained for expression of the DC maturation markers CD40, CD80, CD83, CD86, and CCR7. The flow cytometry histograms show the cell population for each marker and the mean fluorescent intensity is shown in each plot. Representative data are shown from 3 independent experiments.
Are Ad-vector modified DC targets for NK cell mediated lysis?
The design of the three Ad vectors suggested that they would lead to different levels of Ad genome derived protein expression in infected APC in vivo that might render them susceptible to innate immunity mediated by NK cells. We evaluated NK cell killing of Ad5-CEA infected DC using an in vitro autologous human cell culture system consisting of Ad5-CEA infected DC and autologous NK cells. The similar levels of CEA expression on infected DC confirmed equivalent Ad infectivity for the three different Ad5-CEA vectors (data not shown). Despite this, the Ad5[E1+]CEA vector-infected DC were significantly more susceptible to NK mediated cytotoxicity (Fig. 3). Importantly, DC infected with the Ad5[E1−]CEA and Ad5[E1−, E2b−]CEA vectors were not susceptible to NK-mediated lysis, with the levels of cytotoxicity similar to that of mock infected DC.
Figure 3. Susceptibility of Ad5-infected human DC to NK-mediated cell killing.
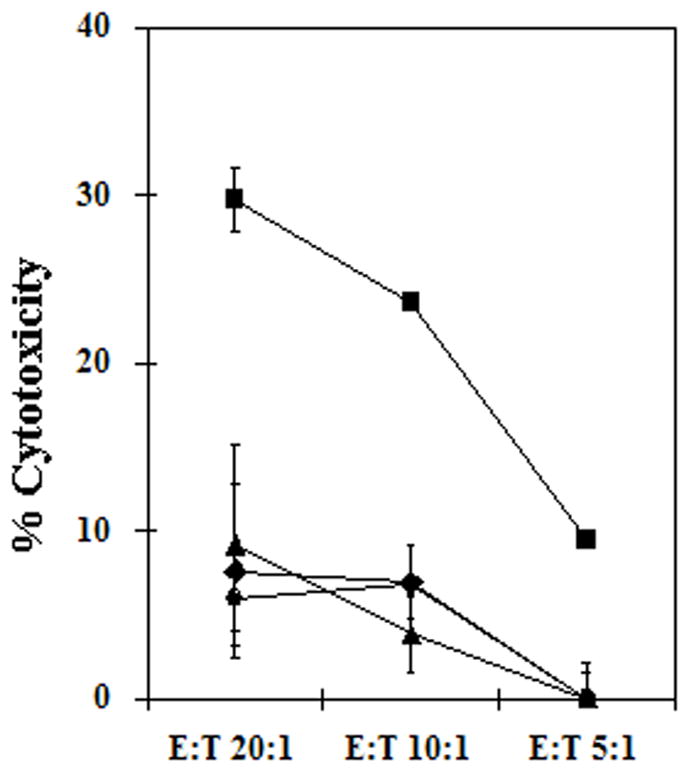
In vitro cultured human DC were infected with Ad5[E1+]CEA, Ad5[E1−]CEA, or Ad5[E1−, E2b−]CEA at MOI 20,000, or mock infected. DC were incubated with autologous NK cells and DC cytotoxicity was measured. Circle: Mock-infected, square: Ad[5E1+], triangle: Ad5[E1−], diamond: Ad5[E1−, E2b−]. Error bars represent SD. Representative data are shown from 2 independent experiments.
Ad5 transduced DC induce antigen-specific cellular responses in vitro
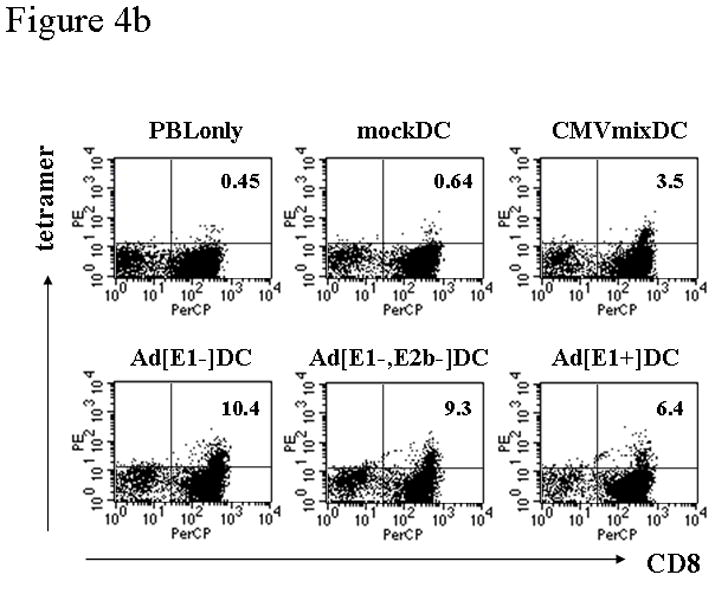
The induction of antigen-specific cytotoxic T cells will be a major component of a successful cancer vaccine strategy. Thus, we evaluated the antigen-presenting capacity of Ad5 infected DC using a CMVpp65-specific CTL line (clone 3H1) as a means of measuring T cell recognition. Ad5[E1+]CMVpp65, Ad5[E1−]CMVpp65, and Ad5[E1−, E2b−] CMVpp65 vectors were used to infect human DC, with CEA expressing vectors used as negative controls. The CMV-specific T cell clone was incubated with the infected DC and T cell recognition was measured by ELISPOT (Fig. 4a). DC infected with Ad5[E1−] and Ad5[E1−, E2b−] vectors stimulated similar high levels of T cell recognition and were superior to the Ad5[E1+] vector in this assay. Interestingly, all three Ad5-CMV vectors induced stronger antigen-specific IFN-γ production than the CMV peptide-pulsed DC.
Figure 4. Antigen presenting capacity and stimulatory capacity of Ad5-transduced human dendritic cells.
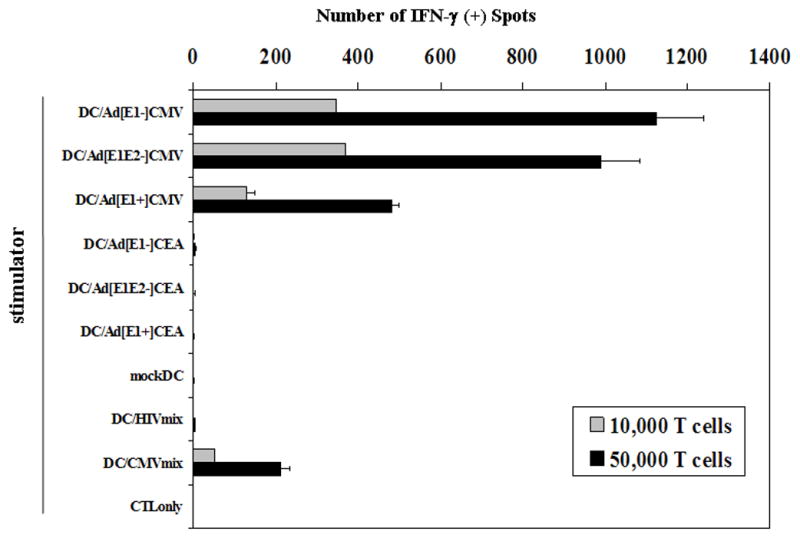
(a) The capacity of Ad-infected human DC to process and present antigen was measured by using a human T cell clone specific for HLA-A*0201/CMVpp65(495–503) peptide-MHC complexes. In vitro cultured human DC were infected with Ad5[E1+], Ad5[E1−], or Ad5[E1−, E2b−] vectors encoding CMVpp65 or CEA at MOI 20,000, or mock infected and incubated for 2 days. 1×104 DC-transduced with the vectors were used as stimulators and 1×104 or 5×104 of CMVpp65 CTL line (3H1) was used as responder cells. DC pulsed with CMVpp65 overlapping polypeptide mix and DC pulsed with a HIV gag overlapping polypeptide mix were used as positive and negative controls respectively. Interferon-gamma ELISPOT results are shown. Error bars represent standard deviation. Representative data are shown from 3 individual experiments. (b) To compare the stimulatory capacity of Ad5CMV-transduced DC, in vitro cultured human DC were infected with Ad5[E1+], Ad5[E1−], or Ad5[E1−, E2b−] vectors encoding CMVpp65 at MOI 20,000, mock infected, or pulsed with CMVpp65 or HIV overlapping polypeptide mixes as positive control negative controls respectively. DC were cultured with autologous HLA-A*0201+ peripheral blood lymphocytes (PBL) at 10:1 (PBL:DC) ratio. After 10 days of culture the percentage of CD8+ HLA-A*0201/CMVpp65(495–503) peptide-MHC tetramer positive T cells was measured by flow cytometry. Percentages of CD8+ tetramer+ cells in CD3+CD4−CD14−CD19−lymphocytes are shown in dot plots, which were calculated from duplicate samples. Representative data are shown from 3 individual experiments.
Ad5 transduced DC induce expansion of CMV-specific CTLs in vitro
While the previous experiment demonstrated the capacity of Ad-infected DC to process and present peptide to a T cell clone, we hypothesized that genome replication differences invoked by the different vaccine constructs could limit the effectiveness of the infected DC to expand antigen-specific T cells. PBL from an HLA-A2+/CMV-sero-positive donor were cultured with Ad5-CMV-transduced autologous DC, and the expansion of CMVpp65-specific CTL was analyzed (Fig. 4b). The largest expansion of CMV-pp65-specific T cells was induced by the Ad5[E1−]CMV and Ad5[E1−, E2b−]CMV-transduced DC. These results corresponded well to the results from the previous assay evaluating antigen-presenting capacity (Fig. 4a), and again indirectly suggest that Ad genome replication can negatively impact the efficacy of Ad vaccines.
Modeling anti-Ad5 immunity in mice
Because many people have been naturally infected by and recovered from wild type adenovirus, vaccination in humans commonly occurs in the setting of pre-existing Ad5 immunity, which limits the effectiveness of Ad5 vaccines clinically16–18. Once our in vitro studies demonstrated the characteristics of the three Ad5 based-vector constructs, we sought to examine each vector as a vaccine in vivo, both in naïve animals, and critically, in the context of pre-existing immunity to Ad5.
To model Ad5 immunity in mice, we exposed naïve mice to either a single dose or 3 doses of a replication competent Ad5[E1+, E3+] (Ad5[RC]) expressing a heterologous transgene encoded in the E4 region. The Ad5[RC] vector was chosen because it represented most closely the wild type Ad5 that humans are exposed to. As shown in Figure 5a, a single dose of this Ad5[WT] vector elicited anti-Ad antibody endpoint titers of 1:5,000, while 3 doses increased this to 1:20,000. The cellular immune responses to each of the vectors after three doses of the Ad[RC] vector was also determined. As we had predicted, T cell responses were greatest against the Ad5[E1+] vector. We noted an attenuated T cell response to the prototypic Ad5[E1−] vector.
Figure 5. Modeling anti-Ad5 immunity in mice.
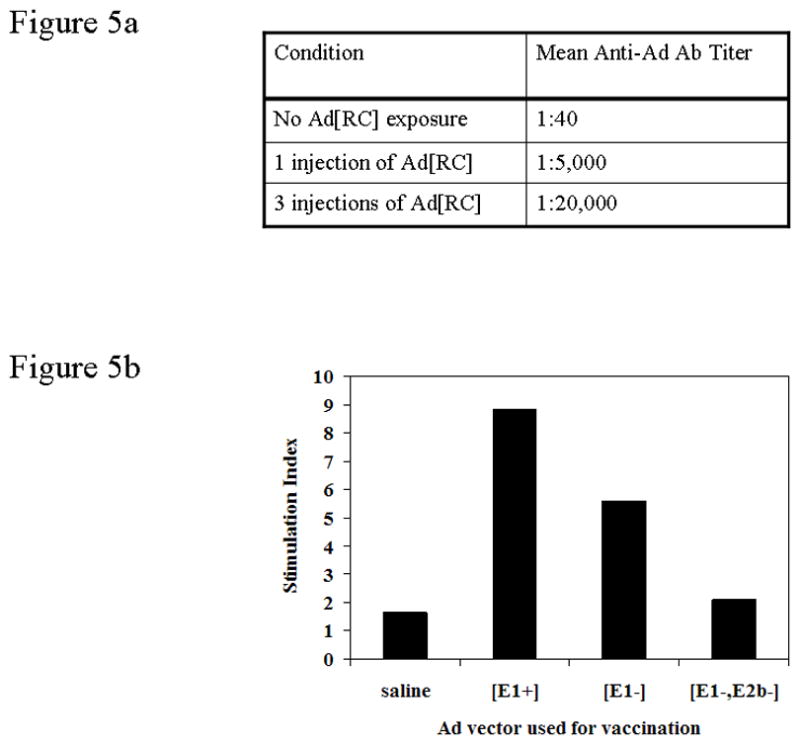
(a) Mice were immunized with 0 (saline), 1, or 3 injections of a replicating Ad5 vector (Ad5[RC]) to induce differing levels of pre-existing anti-adenovirus immunity. The anti-Ad5 antibody endpoint titer was determined by ELISA. This data is representative of 4 experiments. (b) Anti-Ad T cell proliferative responses induced by a single administration of either saline, or Ad[E1+]CEA, Ad[E1−]CEA, or Ad[E1−, E2b−]CEA vectors in mice that had been preconditioned with 3 injections of Ad5[RC] vector.
Importantly we found a dramatic and highly significant reduction in T cells response against our novel Ad5[E1−, E2−] vector. In fact, there was no proliferation of Ad5-specific T cells in response to the Ad5[E1−, E2b−]CEA (Fig. 5b).
Induction of anti-CEA T cell responses with the Ad5[E1−, E2b−]CEA vector is not diminished in mice with pre-existing anti-Ad5 immunity
We prepared either Ad5 naïve, or Ad5 immune mice, the latter receiving one or three doses of Ad5[RC]. These mice were then vaccinated with 3 doses of the Ad5[E1−, E2b−]CEA, Ad5[E1−]CEA, or Ad5[E1+]CEA vectors, and levels of CEA specific CTL responses were measured. The Ad5[E1+]CEA vector (Fig. 6a) showed the strongest CEA-specific T cell response in naïve mice, but the induction of CEA specific T cell responses was dramatically less effective when this vaccine construct was utilized in mice that had pre-existing anti-Ad5 immunity. The reduction in magnitude of the CEA specific response were 36% and 48% respectively for 1 and 3 prior Ad5[RC] injections. Similarly, the Ad5[E1−]CEA vector showed a dramatic reduction in the level of CEA-specific T cells in mice that received one and three prior doses of the Ad5[RC] vector (Fig. 6b). Responses were again diminished by 37–46% respectively in the Ad5 immune mice, as compared to that of naïve mice. In contrast, there was little effect of pre-existing anti-Ad immunity on CEA-specific T cell responses when the Ad5[E1−, E2b−]CEA vector was used. Even 3 prior doses of the Ad5[RC] vector associated with 1:20,000 anti-Ad5 antibody titers only reduced the magnitude of Ad5[E1−, E2b−]CEA induced CEA-specific T cells by 10% and there was no decrease in mice that had received a single Ad5[RC] exposure. These results were highly significant. Thus, the use of Ad5[E1−, E2b−]CEA vaccine retains the full potential of immune induction by Ad5 vaccines, but overcomes anti-Ad5 specific immune responses that significantly limit other Ad5 based vaccine constructs.
Figure 6. Induction of anti-CEA T cell responses with the Ad5[E1−, E2b−]CEA vector is not diminished in mice with pre-existing anti-Ad immunity.
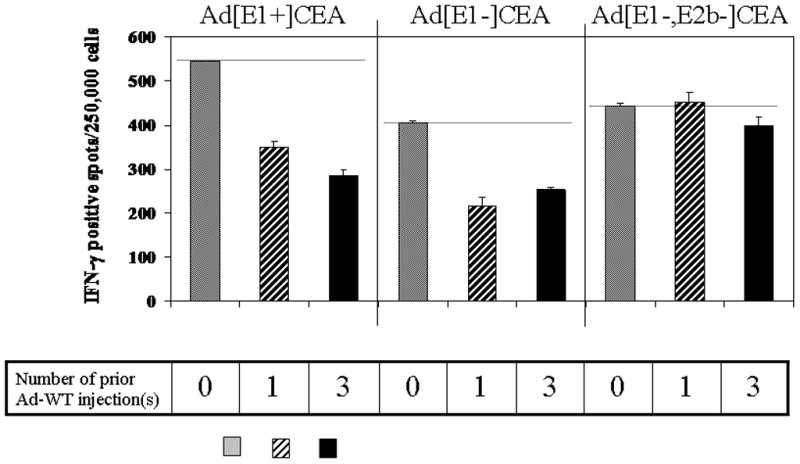
Mice were immunized with 0 (saline), 1, or 3 injections of an replicating adenovirus vector (Ad5[RC]) to induce differing levels of pre-existing anti-Ad5 immunity, reflected by the anti-adenovirus antibody titers shown in Fig. 5a and anti-Ad5 T cell responses in Fig. 5b. Subsequently, mice received 3 vaccinations with the Ad5[E1+]CEA, Ad5[E1−]CEA, or Ad5[E1−, E2b−]CEA vectors as indicated. The magnitude of anti-CEA T cell responses was measured by Interferon-gamma ELISPOT for the naïve (0 injections of Ad5[RC]) and Ad-pre-immune mice (1 or 3 injections of Ad5[RC]). SFC: spot forming cells. Error bars represent standard deviation. The data are representative of four experiments.
DISCUSSION
Unlike gene therapy applications where vector persistence requires immunologically “quiet” vectors, vaccines benefit from some degree of vector-induced inflammation that can enhance induction of transgene specific immunity by providing an adjuvant effect. Nonetheless, vaccines based on prototypic Ad5[E1−] vectors have had limited success, in part due to the significant numbers of patients with pre-existing adenoviral immunity. In contrast to previous reports which have modified capsid proteins on these vectors, we generated a novel non-replicating Ad5[E1−, E2b−] vector and compared it to the prototypic replication deficient (Ad5[E1−]) as well as a novel replicating (Ad5[E1+]) vector.
The performance of the three vectors was compared ex vivo using human DC. Notably, the Ad-infected DC were more potent than peptide loaded DC in inducing T cell responses in vitro, as has been previously reported19–21. These studies further demonstrated that all three vectors gave similar infectivity and temporal dynamics of transgene expression. There were differences however, with the replicating Ad5[E1+]CEA vector perhaps not surprisingly showing greater evidence of DC toxicity, eliciting less maturation of DC, and clearly making Ad5[E1+]CEA infected DC targets for NK-mediated innate immunity. These findings directly correlated with our findings that the Ad5[E1−] and Ad5[E1−, E2b−] vectors were superior to the Ad5[E1+] vector in their capacity to induce and expand antigen specific T cell responses. The results suggest that increased replication of an Ad vector may result in diminished efficacy of the overall platform. There were also indications that the [E1−, E2b−] performed better than the [E1−] platform, likely due to its inherent inability to replicate its genome due to the deletion of the Ad E2b genes. These indications were verified when the Ad5[E1−, E2b]CEA vaccine platform was stringently tested in vivo.
The role of anti-Ad5 immunity and neutralizing antibodies has been recognized as a major obstacle for Ad based vaccine strategies using serotype 5, as well as for some other prevalent Ad serotypes. We pre-immunized mice with single or triple doses of 1011 Ad5 replicating virus, to produce high levels of pre-existing anti-Ad5 immunity, analogous to the approach of Roberts et al.22. In order to maximize anti-Ad5 humoral and cellular immunity we used the footpad route of administration that results in greater levels of immunogenicity than sub-cutaneous or intra-muscular injection (unpublished observation). Importantly, challenge of Ad5 pre-immune mice with the Ad5[E1−, E2b−]CEA vector did not induce T cell proliferative responses, suggesting that the deletion of the E2b gene results in a loss of expression of major epitopes recognized by the anti-Ad5 T cells.
More dramatically, there was little blunting of the immune response to CEA elicited by the Ad5[E1−, E2b−]CEA vector when comparing the magnitude of the response in naïve mice to the magnitude in mice with high levels of pre-existing anti-Ad5 immunity. In contrast, the Ad5[E1+]CEA and Ad5[E1−]CEA vectors were markedly less effective when confronted with pre-existing anti-Ad5 immunity, with CEA-specific immune responses diminished by 48% and 46% respectively. Importantly, similar data has also been obtained in non-human primates (Dr. Frank Jones, personal communication; manuscript in preparation). These data not only demonstrate the improved efficacy of the Ad5[E1−, E2b−]CEA vector platform in Ad5-immune mice, but also demonstrate that antibody responses to the Ad5 capsid are not primarily responsible for the lack of efficacy of Ad5 based vaccines in the context of pre-existing Ad5 immunity. Other mechanisms targeting Ad genes may be primarily responsible. As an example, one major focus of the anti-Ad adaptive immune response is to the hexon proteins16,17,23, 24. We hypothesize that APC infected by the Ad5[E1−, E2b−]CEA vector may be less susceptible to attenuation by pre-existing anti-Ad immunity because subsequent to infection the Ad5[E1−, E2b−] vector produces significantly lower levels of multiple Ad late proteins, proteins which include highly immunogenic proteins such as hexon. We reason that DC infected by the Ad5[E1−, E2b−]CEA vector in vivo are not cleared as rapidly (possibly in a NK cell dependent mechanism) allowing more time for immune responses to the CEA transgene to develop.
Why should the additional deletion of the E2b gene have such a profound effect on Ad5 based vaccines in the face of pre-existing anti-Ad5 immunity? We have previously reported microarray studies comparing the Ad5[E1−] and Ad5[E1−, E2b−] vectors in vivo. Livers from Ad-infected mice revealed similar gene expression profiles at 6 hours post infection (hpi) for the Ad5[E1−] and Ad5[E1−, E2b−] vectors but at 72hpi there was markedly lower expression of inflammatory genes with the Ad5[E1−, E2b−] vector (published online at NCBI Gene Omnibus Express (GEO) (http://www.ncbi.nlm.nih.gov/geo/) accession numbers GSE3729 and GSE4339) 25. While the similar early gene inductions suggest that these events may be primarily capsid driven, the reduced inflammatory response at 72 hpi probably results from the significantly diminished Ad gene expression with the Ad5[E1−, E2b−] vector, since deletion of E2b prevents expression of late gene products as compared to Ad5[E1−]vectors14,26–28. The Ad5[E1−, E2b−] vectors have also been shown to have reduced hepatic toxicity when administered iv and better persistence in vivo, also suggesting a less robust immune response to the Ad5[E1−, E2b−] vector itself15. While all of these studies involved intravascular administration of vector rather than the sub-cutaneous (footpad) route used in our vaccine studies, they do suggest that the Ad5[E1−, E2b−] vector has different characteristics that mute anti-Ad immune responses, generating a more potent CEA-specific immune response in Ad5 pre-immune animals.
Alternate strategies to negate the impact of pre-existing anti-Ad5 immunity have included the vectorization of alternate serotype Ads, although several recent reports confirm that the use of alternative serotype Ads can directly lead to the altered production of important chemokines and cytokines, altered gene dysregulation, as well as significantly different biodistribution and tissue toxicities both in animal models, as well when tested in human DC, relative to Ad5 vectors29,30. The use of hexon-chimeric adenoviruses created by replacing the Ad5 hexon hypervariable regions with those from other Ad serotypes may also avoid neutralizing antibody responses to the native Ad5 capsid22,29,31,32. However, there is also evidence that hexons from other sub-groups tend to reduce vector infectivity, alter antigen specific antibody isotype responses and skew the Th1-Th2 balance11, 32. And while Ad5 mediated transduction of certain antigens can induce humoral responses, other serotypes, when vectorized, fail to do so33.
We believe our studies demonstrate that the Ad5[E1−, E2b−] vector can yield stronger immune responses in Ad5 pre-immune individuals than standard Ad5[E1−] vectors. We conclude that the Ad5[E1−, E2b−] vector platform has great potential for circumventing pre-existing anti-Ad5 immunity and warrants testing in human clinical trials.
Acknowledgments
This work was supported by NIH/NCI grant PO1-CA078673 to HKL.
References
- 1.Pardoll DM. Spinning molecular immunology into successful immunotherapy. Nat Rev Immunol. 2002;2:227–238. doi: 10.1038/nri774. [DOI] [PubMed] [Google Scholar]
- 2.Berinstein NL. Carcinoembryonic antigen as a target for therapeutic anticancer vaccines: a review. J Clin Oncol. 2002;20:2197–2207. doi: 10.1200/JCO.2002.08.017. [DOI] [PubMed] [Google Scholar]
- 3.Hammarstrom S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol. 1999;9:67–81. doi: 10.1006/scbi.1998.0119. [DOI] [PubMed] [Google Scholar]
- 4.Morse MA, Clay TM, Hobeika AC, Osada T, Khan S, Chui S, et al. Phase I study of immunization with dendritic cells modified with fowlpox encoding carcinoembryonic antigen and costimulatory molecules. Clin Cancer Res. 2005;11:3017–3024. doi: 10.1158/1078-0432.CCR-04-2172. [DOI] [PubMed] [Google Scholar]
- 5.Wilson JM. Adenoviruses as gene-delivery vehicles. N Engl J Med. 1996;334:1185–1187. doi: 10.1056/NEJM199605023341809. [DOI] [PubMed] [Google Scholar]
- 6.Barouch DH, Nabel GJ. Adenovirus vector-based vaccines for human immunodeficiency virus type 1. Hum Gene Ther. 2005;16:149–156. doi: 10.1089/hum.2005.16.149. [DOI] [PubMed] [Google Scholar]
- 7.Tatsis N, Ertl HC. Adenoviruses as vaccine vectors. Mol Ther. 2004;10:616–629. doi: 10.1016/j.ymthe.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kozarsky KF, Wilson JM. Gene therapy: adenovirus vectors. Curr Opin Genet Dev. 1993;3:499–503. doi: 10.1016/0959-437x(93)90126-a. [DOI] [PubMed] [Google Scholar]
- 9.Reyes-Sandoval A, Fitzgerald JC, Grant R, Roy S, Xiang ZQ, Li Y, et al. Human immunodeficiency virus type 1-specific immune responses in primates upon sequential immunization with adenoviral vaccine carriers of human and simian serotypes. J Virol. 2004;78:7392–7399. doi: 10.1128/JVI.78.14.7392-7399.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hensley SE, Cun AS, Giles-Davis W, Li Y, Xiang Z, Lasaro MO, et al. Type I interferon inhibits antibody responses induced by a chimpanzee adenovirus vector. Mol Ther. 2007;15:393–403. doi: 10.1038/sj.mt.6300024. [DOI] [PubMed] [Google Scholar]
- 11.Barouch DH, Pau MG, Custers JH, Kudstaal W, Kostense S, Havenga MJ, et al. Immunogenicity of recombinant adenovirus serotype 35 vaccine in the presence of pre-existing anti-Ad5 immunity. J Immunol. 2004;172:6290–6297. doi: 10.4049/jimmunol.172.10.6290. [DOI] [PubMed] [Google Scholar]
- 12.Hodges BL, Evans HK, Everett RS, Ding EY, Serra D, Amalfitano A. Adenovirus vectors with the 100K gene deleted and their potential for multiple gene therapy applications. J Virol. 2001;75:5913–5920. doi: 10.1128/JVI.75.13.5913-5920.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hodges BL, Serra D, Hu H, Begy CA, Chamberlain JS, Amalfitano A. Multiply deleted [E1, polymerase-, and pTP-] adenovirus vector persists despite deletion of the preterminal protein. J Gene Medicine. 2000;2:250–259. doi: 10.1002/1521-2254(200007/08)2:4<250::AID-JGM113>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 14.Amalfitano A, Hauser MA, Hu H, Serra D, Begy CR, Chamberlain JS. Production and characterization of improved adenovirus vectors with the E1, E2b, and E3 genes deleted. J Virol. 1998;72:926–933. doi: 10.1128/jvi.72.2.926-933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Everett RS, Hodges BL, Ding EY, Xu F, Serra D, Amalfitano A. Liver toxicities typically induced by first-generation adenoviral vectors can be reduced by use of E1, E2b-deleted adenoviral vectors. Hum Gene Ther. 2003;14:1715–1726. doi: 10.1089/104303403322611737. [DOI] [PubMed] [Google Scholar]
- 16.Sumida SM, Truitt DM, Kishko MG, Arthur JC, Jackson SS, Gorgone DA, et al. Neutralizing antibodies and CD8+ T lymphocytes both contribute to immunity to adenovirus serotype 5 vaccine vectors. J Virol. 2004;78:2666–2673. doi: 10.1128/JVI.78.6.2666-2673.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sumida SM, Truitt DM, Lemckert AA, Vogels R, Custers JH, Addo MM, et al. Neutralizing antibodies to adenovirus serotype 5 vaccine vectors are directed primarily against the adenovirus hexon protein. J Immunol. 2005;174:7179–7185. doi: 10.4049/jimmunol.174.11.7179. [DOI] [PubMed] [Google Scholar]
- 18.Shiver JW, Emini EA. Recent advances in the development of HIV-1 vaccines using replication-incompetent adenovirus vectors. Annu Rev Med. 2004;55:355–372. doi: 10.1146/annurev.med.55.091902.104344. [DOI] [PubMed] [Google Scholar]
- 19.Oh ST, Kim CH, Park MY, Won EH, Sohn HJ, Cho HI, et al. Dendritic cells transduced with recombinant adenoviruses induce more efficient anti-tumor immunity than dendritic cells pulsed with peptide. Vaccine. 2006;24:2860–2868. doi: 10.1016/j.vaccine.2005.12.056. [DOI] [PubMed] [Google Scholar]
- 20.Malowany JI, McCormick S, Santosuosso M, Zhang X, Aoki N, Ngai P, et al. Development of cell-based tuberculosis vaccines: genetically modified dendritic cell vaccine is a much more potent activator of CD4 and CD8 T cells than peptide- or protein-loaded counterparts. Mol Ther. 2006;13:766–775. doi: 10.1016/j.ymthe.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 21.Steitz J, Tormo D, Schweichel D, Tuting T. Comparison of recombinant adenovirus and synthetic peptide for DC-based melanoma vaccination. Cancer Gene Ther. 2006;13:318–325. doi: 10.1038/sj.cgt.7700894. [DOI] [PubMed] [Google Scholar]
- 22.Roberts DM, Nanda A, Havenga MJ, Abbink P, Lynch DM, Ewald BA, et al. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature. 2006;441:239–2343. doi: 10.1038/nature04721. [DOI] [PubMed] [Google Scholar]
- 23.Tang J, Olive M, Champagne K, Flomenberg N, Eisenlohr L, Hsu S, et al. Adenovirus hexon T-cell epitope is recognized by most adults and is restricted by HLA DP4, the most common class II allele. Gene Ther. 2004;11:1408–1415. doi: 10.1038/sj.gt.3302316. [DOI] [PubMed] [Google Scholar]
- 24.Molinier-Frenkel V, Lengagne R, Gaden F, Hong SS, Choppin J, Gahery-Ségard H, et al. Adenovirus hexon protein is a potent adjuvant for activation of a cellular immune response. J Virol. 2002;76:127–135. doi: 10.1128/JVI.76.1.127-135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hartman ZC, Kiang A, Everett RS, Serra D, Yang XY, Clay TM, et al. Adenovirus infection triggers a rapid, MyD88-regulated transcriptome response critical to acute-phase and adaptive immune responses in vivo. J Virol. 2007;81:1796–1812. doi: 10.1128/JVI.01936-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding EY, Hodges BL, Hu H, McVie-Wylie AJ, Serra D, Migone FK, et al. Long-term efficacy after [E1−, polymerase-] adenovirus-mediated transfer of human acid-alpha-glucosidase gene into glycogen storage disease type II knockout mice. Hum Gene Ther. 2001;12:955–965. doi: 10.1089/104303401750195917. [DOI] [PubMed] [Google Scholar]
- 27.Harris JD, Graham IR, Schepelmann S, Stannard AK, Roberts ML, Hodges BL, et al. Acute regression of advanced and retardation of early aortic atheroma in immunocompetent apolipoprotein-E (apoE) deficient mice by administration of a second generation [E1(−), E3(−), polymerase(−)] adenovirus vector expressing human apoE. Hum Mol Genet. 2002;11:43–58. doi: 10.1093/hmg/11.1.43. [DOI] [PubMed] [Google Scholar]
- 28.Ding E, Hu H, Hodges BL, Migone F, Serra D, Xu F, et al. Efficacy of gene therapy for a prototypical lysosomal storage disease (GSD-II) is critically dependent on vector dose, transgene promoter, and the tissues targeted for vector transduction. Mol Ther. 2002;5:436–446. doi: 10.1006/mthe.2002.0563. [DOI] [PubMed] [Google Scholar]
- 29.Youil R, Toner TJ, Su Q, Chen M, Tang A, Bett AJ, et al. Hexon gene switch strategy for the generation of chimeric recombinant adenovirus. Hum Gene Ther. 2002;13:311–320. doi: 10.1089/10430340252769824. [DOI] [PubMed] [Google Scholar]
- 30.Hartman ZC, Appledorn DM, Serra D, Glass O, Mendelson TB, Clay TM, et al. Replication-attenuated Human Adenoviral Type 4 vectors elicit capsid dependent enhanced innate immune responses that are partially dependent upon interactions with the complement system. Virology. 2008;374:453–467. doi: 10.1016/j.virol.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hashimoto M, Boyer JL, Hackett NR, Wilson JM, Crystal RG. Induction of protective immunity to anthrax lethal toxin with a nonhuman primate adenovirus-based vaccine in the presence of preexisting anti-human adenovirus immunity. Infect Immun. 2005;73:6885–6891. doi: 10.1128/IAI.73.10.6885-6891.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu H, Dmitriev I, Kashentseva E, Seki T, Wang M, Curiel DT. Construction and characterization of adenovirus serotype 5 packaged by serotype 3 hexon. J Virol. 2002;76:12775–12782. doi: 10.1128/JVI.76.24.12775-12782.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varnavski AN, Schlienger K, Bergelson JM, Gao GP, Wilson JM. Efficient transduction of human monocyte-derived dendritic cells by chimpanzee-derived adenoviral vector. Hum Gene Ther. 2003;14:533–544. doi: 10.1089/104303403764539323. [DOI] [PubMed] [Google Scholar]