

Abstract
5-Oxo-ETE is the most powerful eosinophil chemoattractant among lipid mediators. Eosinophil infiltration into the lungs of asthmatics may be responsible for the late phase of inflammatory asthma. We have designed and synthesized a 5-oxo-ETE receptor antagonist, the purpose of which is to prevent eosinophil migration to the lung during an asthma attack and thereby reduce asthma symptoms.
Keywords: 5-oxo-ETE, HETE, eosinophil, neutrophil, antagonist, asthma, SAR, intracellular calcium, oxovaleryl indole, design, synthesis
INTRODUCTION
5-Oxo-6E,8Z,11Z,14Z-eicosatetraenoic acid (5-oxo-ETE 5) is a metabolite of arachidonic acid (AA) that is formed as a result of the oxidation of the 5-lipoxygenase product 5S-hydroxy-6E,8Z,11Z,14Z-eicosatetraenoic acid (5-HETE 3) by 5-hydroxyeicosanoid dehydrogenase (5-HEDH). We have performed the synthesis of 5-oxo-ETE1 and a tetra deutero derivative2 and developed a mass spectrometric assay for its quantitation.3 This enzyme oxidizes eicosanoids containing a 5S-hydroxyl group followed by a 6,7-trans double bond and is highly selective for 5-HETE (Figure 1).4 It is present in neutrophils, monocytes, lymphocytes, eosinophils, platelets, and various structural cells.5,6 5-Oxo-ETE 5 acts through a specific Gi-coupled receptor7–10 that is expressed by eosinophils > neutrophils > monocytes10 as well as by certain tumor cell lines11 and has been designated as the OXE receptor (OXE-R).12 Its biological activity is limited by its metabolism to biologically inactive products, including 5-oxo-12-HETE and 5-oxo-20-HETE (Figure 1).
Figure 1.

Biosynthesis and metabolism of 5-oxo-ETE.
Although 5-oxo-ETE 5 is a chemoattractant for both neutrophils13 and monocytes,14 it is less potent than LTB4. In contrast, among lipid mediators, it is the strongest chemoattractant known for human eosinophils15 and also induces a variety of other responses in these cells,16–18 some of which are markedly enhanced by the proinflammatory cytokines GM-CSF and TNFα.19 It is active in vivo, eliciting infiltration of eosinophils and, to a lesser extent, neutrophils, into human skin.20 OXE-R is highly selective for 5-oxo-ETE 5 over a variety of its metabolites, leukotrienes (LTs) and other eicosanoids.21
Because of the above effects, 5-oxo-ETE 5 may be an important regulator of the tissue infiltration and activation of eosinophils and neutrophils in diseases such as asthma, allergic rhinitis, arthritis, and psoriasis. For this reason we set out to design a selective OXE receptor antagonist and report here the progress made.
RESULTS AND DISCUSSION
In a previous study we identified 5-oxo-12S-HETE 6 (Figure 1) as a major metabolite of 5-HETE by human platelets.22 We have performed the total synthesis of 6,23 and we found that this compound had virtually no effect on intracellular Ca++ levels in neutrophils, but blocked 5-oxo-ETE-induced Ca++ mobilization in the low µmolar range, suggesting that it has antagonist properties.22 Consistent with this, 5-oxo-12S-HETE inhibited 5-oxo-ETE-induced neutrophil migration. However, this compound was not a suitable candidate for drug development because of its instability and susceptibility to metabolism.
It has previously been shown that the conjugated triene systems of LTB424 and LTD425 are essentially flat in solution. With that in mind, we decided to synthesize a series of conformationally restricted analogs in which the carboxyl end of 5-oxo-ETE, containing the 5-oxo group, was coupled to a flat aromatic system such as benzene, naphthalene, quinoline, benzofuran, or indole. Figure 2 represents a small sample of the 45 such compounds we prepared.
Figure 2.
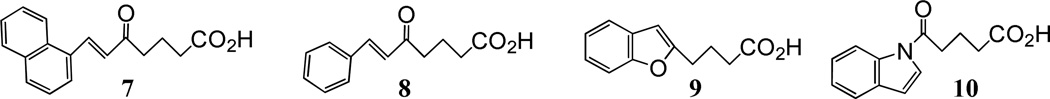
Examples of some monosubstituted aromatic compounds that have been synthesized and found to be without antagonist activity.
To screen for antagonist activity we used a calcium mobilization assay21 in which human neutrophils, loaded with the calcium-sensitive fluorescent dye indo-1, were treated first with the potential antagonist and then two min later with 5-oxo-ETE. A major advantage of this assay is that it enables real-time testing simultaneously for both agonist and antagonist activity, in contrast to a binding assay, for example, which measures only receptor affinity. Using this assay, we showed that none of these monosubstituted aromatic compounds had any agonist activity. However, they had little or no effect on 5-oxo-ETE-induced calcium mobilization and therefore showed little promise as OXE-R antagonists. A typical example is shown in Figure 3. A high concentration (30 µM) of compound 10 neither mobilized intracellular calcium when added to neutrophils, nor altered the response to 5-oxo-ETE (10 nM), which was added 2 min later. Similar results were obtained with the other three compounds shown in Figure 2.
Figure 3.

The monosubstituted indole 10 does not affect calcium mobilization in human neutrophils induced by 5-oxo-ETE. Neutrophils were loaded with the calcium-sensitive dye indo-1 and changes in fluorescence were monitored after addition of 10 (30 µM) and, 2 min later, 5-oxo-ETE (10 nM).
At this juncture, we embarked on a detailed structure-activity study to identify the major features of 5-oxo-ETE required for interaction with and activation of its receptor.21 The resulting data highlighted the importance of a free carboxyl group and a 5-oxo group conjugated with a trans-cis diene system (Figure 4A). In addition, and of particular relevance to the present study, was the requirement for the terminal hydrophobic portion of 5-oxo-ETE, as analogs with carbon chain lengths of less than 18 showed very little activity.
Figure 4.
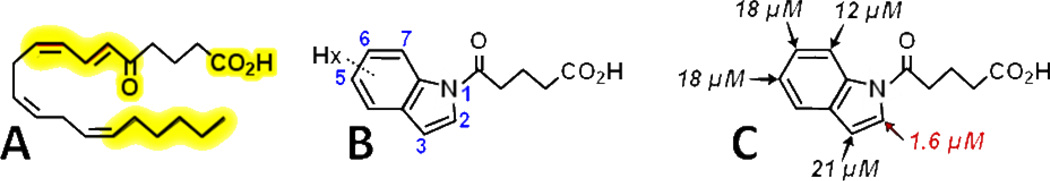
Structures of 5-oxo-ETE and indole-based conformationally restricted analogs. A: 5-Oxo-ETE with the regions required for activation of the OXE receptor in neutrophils highlighted in yellow. B: Monohexyl-substituted 1-(5-oxovalerate)indoles. C: IC50 values for 1-(5-oxovalerate)indoles containing a hexyl group in different positions.
Because of the importance of the terminal hydrophobic region of 5-oxo-ETE, we decided to investigate the antagonist effects of a series of aromatic derivatives containing two substituents: one mimicking the 5-oxo-valerate portion of 5-oxo-ETE (i.e. C1-C5) and the other mimicking the ω-end of the molecule in the form of a hexyl group. These were joined together by an aromatic group that provides the required unsaturated portion of the molecule (Figure 4B). A variety of conformations are available for 5-oxo-ETE, one of which will be preferentially bound by the OXE receptor. These different conformations can be mimicked by placing the two side chains in different positions on the aromatic scaffold.
We synthesized a series of 1-(5-oxovalerate)indoles containing a hexyl group at C-2 26, C-3 16, C-5 17, C-6 18, or C-7 19, and examined their effects on 5-oxo-ETE-induced calcium mobilization in human neutrophils. None of these compounds affected intracellular calcium levels in the absence of 5-oxo-ETE. Compounds 16–19 had only modest inhibitory effects on the response to 5-oxo-ETE, and all had IC50 values above 10 µM. In contrast, the 2-hexylindole 26, the only compound in which the two side chains are ortho to one another, strongly inhibited 5-oxo-ETE-induced calcium mobilization with an IC50 value of 1.55 µM. To ensure that the effect of 26 was selective for the OXE receptor, we determined whether it could inhibit the response to the closely related 5-lipoxygenase product leukotriene LTB4. As shown in Figure 5A, 26 (10 µM) had virtually no effect on the response of neutrophils to LTB4 while completely inhibiting their response to 5-oxo-ETE (Figure 5B). A lower concentration (1 µM) of 26 also substantially inhibited the effect of 5-oxo-ETE. The syntheses of the above molecules are shown in Schemes 1 and 2; Table 1 summarizes their IC50 values.
Figure 5.

Effects of OXE receptor antagonists on calcium mobilization in human neutrophils. Various concentrations of either 26 (A and B) or 39 (C) were added to indo-1-loaded neutrophils followed 2 min later by the addition of either LTB4 (10 nM; A) or 5-oxo-ETE (10 nM; C).
Scheme 1.

Synthesis of indole derivatives.
Scheme 2.

Synthetic approach for indole derivatives.
Table 1.
IC50 values of hexyl-substituted 1-(5-oxovalerate)indoles. A series of 1-(5-oxovalerate)indoles containing a hexyl substituent at various positions as well as 2-hexyl-1-(5-oxovalerate)indoles containing a chloro substituent at positions 5 or 6 were tested in the calcium mobilization assay as shown in Figure 5. IC50 values were determined from concentration- response curves for each individual experiment.
| Indole | IC50 (µM) |
|---|---|
| 2-Hx (26) | 1.55 ± 0.25 (11) |
| 3-Hx (16) | 20.68 ± 5.60 (4) *** |
| 5-Hx (17) | 17.67 ± 3.59 (3) ** |
| 6-Hx (18) | 18.00 ± 2.65 (3) ** |
| 7-Hx (19) | 11.77 ± 2.51 (3) * |
| 5-Cl (33) | 1.17 ± 0.32 (4) |
| 6-Cl (39) | 0.40 ± 0.03 (7) ** |
The values are means ± SE with the numbers of independent determinations shown in brackets. Statistically significant differences compared to 26 are shown as follows:
p<0.05;
p<0.005;
p<0.001
(one-way ANOVA with a Holm-Sidak post-hoc test).
We next investigated the effects of adding a chloro substituent to the benzene ring (Figure 6). The 5-chloro derivative 33 inhibited 5-oxo-ETE-induced calcium mobilization with a potency (IC50, 1.2 µM) similar to the non-chlorinated compound 26. In contrast, the addition of chlorine in the C-6 position of compound 39 (Scheme 4), increased potency by about 4-fold (IC50, 400 nM; Figure 5C). The syntheses of the two chloro analogs are shown in Schemes 3 and 4.
Figure 6.

Structures of chloro-substituted indole antagonists. The IC50 values of the 5- and 6-chloro compounds are shown on the right.
Scheme 4.
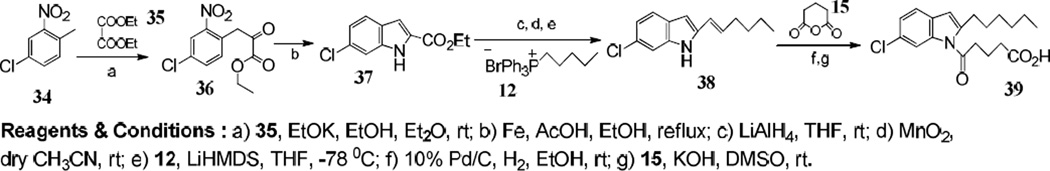
Synthesis of 5-(6-Chloro-2-hexyl-1H-indol-1-yl)-5-oxo-valeric acid 39.
Scheme 3.

Synthesis of 5-(5-Chloro-2-hexyl-1H-indol-1-yl)-5-oxo-valeric acid 33.
For the synthesis of indole-based antagonists, we started from the indole carboxaldehydes 11. Carboxaldehydes in positions 2, 6, 7 were prepared from their corresponding carboxylic acids, as illustrated for compound 20 (Scheme 2). Carboxaldehydes at C-3 and C-5 were obtained commercially. Reduction of 20 with LiAlH4 gave the alcohol which was further oxidized to aldehyde 11 by MnO2. The reaction crude was filtered through celite; however, some MnO2 filtered through. A better alternative was centrifugation followed by decanting. Next, a Wittig reaction was carried out using alkyl phosphonium salt to give the olefin which was hydrogenated to afford the alkyl indole. Glutaric anhydride was used under KOH/DMSO conditions to substitute the proton on the nitrogen of the indole ring. Schemes 1 and 2 show the synthetic approach. The synthesis of the 5-chloro analog 33 was achieved similarly as illustrated in Scheme 3.
For the synthesis of 5-(6-Chloro-2-hexyl-1H-indol-1-yl)-5-oxo-valeric acid 39 (Scheme 4), a known literature procedure was used to obtain ethyl ester 37.26 From this point onwards we used the same strategy described for the synthesis of 33 (Scheme 3). It is noteworthy that the hydrogenation of 31 and 38 with Pd/C in some instances led to a partial hydrogenolysis of the chlorine substituent.
The progression of the SAR leading to compound 39 is summarized in Figure 7, with the corresponding IC50 values. Moreover, 39 is numbered so as to show its structural similarities to 5-oxo-ETE.
Figure 7.
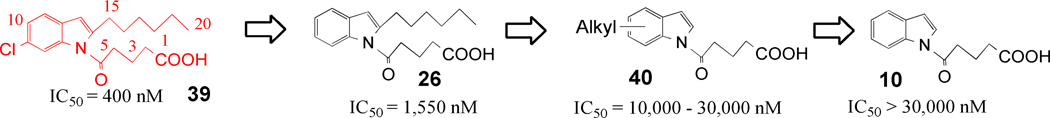
Summary of structure activity leading to 39. The numbering on 39 refers to the comparable carbons on 5-oxo-ETE.
CONCLUSION
We have described herein the design and synthesis of series of potent and selective 5-oxo-ETE antagonists with the lead compound having low micromolar activity. The initial monosubstituted aromatic oxovalerate compounds showed little to no activity. The addition of the hexyl chain proved crucial for antagonist properties to 5-oxo-ETE receptors. Moreover, optimal activity was observed with the hexyl at the C-2 position. Finally, submicromolar activity was obtained by the addition of a chlorine at C-6 of the indole ring. Figure 7 shows the progression of the structure-activity from the inactive 10 to the lead compound 39.
EXPERIMENTAL SECTION
Measurement of intracellular calcium levels
Neutrophils suspended in phosphate-buffered saline were prepared from whole blood obtained from healthy human subjects and loaded with the calcium-sensitive dye indo-1 as previously described.21 Five min before commencement of the experiment, the cells were transferred to a cuvette and CaCl2 (1.8 mM) and MgCl2 (1 mM) were added. Intracellular calcium was then measured at 37 °C in a spectrofluorimeter.21 After a stable baseline was obtained either vehicle or a synthetic indole was added, followed 2 min later by 5-oxo-ETE (10 nM). One min later digitonin was added to determine the maximal response.
Reagents and Methods
Unless stated otherwise, all reagents and chemicals were obtained from commercial sources and were used without further purification. All reactions were carried out with dry, freshly distilled solvents under anhydrous conditions. Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous materials. HRMS of positive ions were obtained on an AccuTOF mass spectrometer with a DART ion source. 1H NMR and 13C NMR spectra were recorded on a BRUKER AMX 360 MHz and BRUKER AMX 400 MHz spectrometers using CDCl3 as solvent at 25 °C. The chemical shifts (δ) for 1H NMR are expressed in ppm, followed by the multiplicity (br, broad; s, singlet; d, doublet; t, triplet; q, quadruplet; qt, quintet and m, multiplet), coupling constants (J in Hertz, Hz), and integration. All compounds were analyzed by TLC, NMR, and HRMS. All final compounds were further quantified and purified to be > 95% pure by HPLC prior to each biological assay. HPLC conditions: Waters 2695 Alliance System with Waters Novapak C18 (150 × 3.9 mm) column and photodiode array detector (Waters Model 2996), gradient mobile phase between H2O/MeCN/MeOH (56:22:22) to H2O/MeCN/MeOH (16:42:42) over 30 min, both solvents contained 0.02% acetic acid, and a flow rate of 1 mL/min.
Synthesis of monosubstituted aromatic compounds 7–10
Compounds 7 and 8 were synthesized as follows for 7:
(E)-7-(naphthalen-1-yl)-5-oxohept-6-enoic acid (7)
To a solution of methyl 5-chloro-5-oxopentanoate, 40 (3.00 g, 18.2 mmol) in dry Et2O was added 105 mL of freshly prepared CH2N2 at 0 °C over 30 min. The reaction was quenched with water; the organic layer dried with Na2SO4, and then bubbled with dry HCl. Next, it was washed with brine and the organic layer dried with Na2SO4. The crude 41 was further added to excess NaI in dry MeCN and stirred overnight. The solvent was evaporated, the crude re-suspended in CH2Cl2, washed with water and dried with Na2SO4. To this crude iodoketone (3.46 g, 12.8 mmol) in MeCN was added trimethoxyphosphine (3.23 g; 1.5 equiv). The mixture was stirred at rt for 12 h, the solvent was evaporated, and the crude was washed with brine. The organic layer was separated and dried to afford methyl 6-(dimethoxyphosphoryl)-5-oxohexanoate, 42 (3.02 g, 93.5%). 1H NMR (CDCl3): δ 3.79 (s, 3H), 3.76 (s, 3H), 3.66 (s, 3H), 3.11 (s, 1H), 3.05 (s, 1H), 2.69 (t, J = 7.0 Hz, 2H), 2.34 (t, J = 7.2 Hz, 2H), 1.90 (qt, J = 7.1 Hz, 2H). To a stirred suspension of this intermediate (242 mg, 0.960 mmol) in anhydrous THF (1 mL) was added NaHMDS (1.0M in THF, 0.800 mL, 0.800 mmol) at rt under argon. The mixture was stirred for 15 min, cooled to −78°C, and a solution of 1-naphthaldehyde, 43 (50 mg, 0.320 mmol) in 1 mL dry THF was added. The reaction mixture was stirred at −78°C for 1 h, warmed to 25°C, stirred for 3 h, quenched with saturated NH4Cl solution, and extracted with EtOAc. The organic layers were combined and dried over Na2SO4. The solvent was evaporated under reduced pressure and the crude residue was purified by silica gel chromatography using EtOAc/Hex as eluent to afford (E)-methyl 7-(naphthalen-1-yl)-5-oxohept-6-enoate (83.8 mg, 92.8%). 1H NMR (CDCl3): δ 8.44 (d, J = 15.8 Hz, 1H), 8.22 (d, J = 7.4 Hz, 1H), 7.94 (t, J = 7.6 Hz, 2H), 7.80 (d, J = 6.6 Hz, 1H), 7.48–7.67 (m, 3H), 6.85 (d, J = 15.9 Hz, 1H), 3.71 (s, 3H), 2.84 (t, J = 7.0 Hz, 2H), 2.48 (t, J = 7.0 Hz, 2H), 2.05–2.14 (m, 2H). To 10 mg of this ester was added a solution of 5% KOH in dioxane. Stirring was allowed under N2 for 2 h, then the reaction was quenched with dilute HCl, extracted with EtOAc, and dried with Na2SO4. The solvent was evaporated to afford (E)-7-(naphthalen-1-yl)-5-oxohept-6-enoic acid, 7 (9.4 mg, 99%). 1H NMR (CDCl3): δ 8.45 (d, J = 15.8 Hz, 1H), 8.22 (d, J = 8.1 Hz, 1H), 7.90 (t, J = 8.5 Hz, 2H), 7.80 (d, J = 7.1 Hz, 1H), 7.44–7.65 (m, 3H), 6.85 (d, J = 15.9 Hz, 1H), 2.87 (t, J = 6.9 Hz, 2H), 2.55 (t, J = 7.0 Hz, 2H), 2.19 (t, J = 7.0 Hz, 2H).
(E)-5-oxo-7-phenylhept-6-enoic acid (8)
1H NMR (CDCl3): δ 7.52–7.66 (m, 3H), 7.40 (br s, 3H), 6.73 (d, J = 16.0 Hz, 1H), 2.82 (t, J = 6.9 Hz, 2H), 2.50 (t, J = 7.0 Hz, 2H), 1.95–2.15 (m, 2H).
4-(benzofuran-2-yl)butanoic acid (9)
To a stirred suspension of 2-((triphenylphosphonium bromide)methyl)phenol, 45 (895 mg, 1.99 mmol) and Et3N (605 mg, 3 equiv) in toluene (8 mL) was added glutaric anhydride, 46 (250 mg, 2.19 mmol). The mixture was refluxed for 8 h, then quenched with saturated NH4Cl, extracted with EtOAc, dried and purified by silica gel chromatography to afford 9 (197 mg, 48%). 1H NMR (CDCl3): δ 7.52 (d, J = 7.4 Hz, 1H), 7.43 (d, J = 7.4 Hz, 1H), 7.18–7.28 (m, 2H), 6.42 (s, 1H), 2.82 (t, J = 6.9 Hz, 2H), 2.40 (t, J = 7.0 Hz, 2H), 1.77–1.88 (m, 2H).
5-(1H-indol-1-yl)-5-oxopentanoic acid (10)
To a stirred suspension of indole, 47 (100 mg, 0.854 mmol) in anhydrous THF (4 mL) was added EtMgBr (3M, 0.600 mL) at rt under argon. Glutaric anhydride, 46 (194 mg, 1.70 mmol) was added and stirred for 3 h. The reaction was quenched with saturated NH4Cl, extracted with EtOAc and dried to afford 10 (172 mg, 87.6%). 1H NMR (CDCl3): δ 8.46 (d, J = 8.2 Hz, 1H), 7.57 (d, J = 7.7 Hz, 1H), 7.47 (d, J = 3.5 Hz, 1H), 7.36 (t, J = 7.7 Hz, 1H), 7.31 – 7.26 (m, 1H), 6.65 (d, J = 3.8 Hz, 1H), 3.04 (td, J = 7.0, 1.6 Hz, 2H), 2.60 (t, J = 7.0 Hz, 2H), 2.18 (qt, J = 7.1 Hz, 2H). 13C NMR (CDCl3): δ 178.62 (s), 170.60 (s), 135.61 (s), 130.34 (s), 125.21 (s), 124.46 (s), 123.74 (s), 120.88 (s), 116.59 (s), 109.39 (s), 34.56 (s), 32.70 (s), 19.41 (s).
Synthesis of indole carboxaldehydes
1H-indole carboxaldehydes (at C-2, C-6 and C-7), 11, and the 5-chloro-analog were synthesized from their commercially available carboxylic acid precursors (20 and 30) as described below for 1H-indole-2-carboxaldehyde (11).
1H-Indole-2-carboxaldehyde (11)
To a stirred solution of 1H-indole-2-carboxylic acid (20) (2.0 g, 12 mmol) in THF (20 mL) at 0 °C was added LiAlH4 (1M in THF, 24.8 mmol, 25 mL) under argon. The reaction mixture was stirred at 0 °C for 30 min, then was gradually warmed to 25 °C over a period of 3 h. The reaction mixture was quenched with aqueous NH4Cl solution and then was extracted with EtOAc. The organic layers were combined and dried over Na2SO4. The solvent was evaporated under reduced pressure and the crude mixture was dried under vacuum pump. To this crude mixture in MeCN (50 mL) was added MnO2 (5.6 g, 65 mmol) and the reaction mixture was stirred overnight. Next day, the reaction mixture was diluted with ether and filtered through celite and florisil layers. The solvent was evaporated under reduced pressure and the crude residue purified by silica gel chromatography using (3:7) EtOAc/Hex solvent as eluent to afford 11 as pale brown solid (1.53 g, 81.0%). HRMS (ESI) m/z calcd for [C9H7NO+H]+: 146.0606, found 146.0613. 1H NMR (CDCl3): δ 9.85 (s, 1H), 9.01 (s, 1H), 7.76 (d, J = 8.0 Hz, 1H), 7.45 (d, J = 8.4 Hz, 1H), 7.40 (t, J = 7.5 Hz, 1H), 7.28 (s, 1H), 7.19 (t, J = 7.4 Hz, 1H). 13C NMR (CDCl3): δ 182.00 (s), 137.87 (s), 135.95 (s), 127.36 (s), 123.48 (s), 121.30 (s), 114.71 (s), 112.37 (s), 102.68 (s).
Synthesis of alk-1-en-1-yl-1H-indoles
Compounds 13, 22 ([alk-1-en-1-yl] derivatives at C-2), 31 (5-Cl analog) and 38 (6-Cl analog) were synthesized as shown for 22:
2-Hex-1-enyl-1H-indole (22)
To a stirred suspension of phosphonium salt 21 (973 mg, 2.36 mmol) in anhydrous THF (4 mL) was added LiHMDS (1.0M in THF, 2.12 mL, 2.12 mmol) at −40°C under argon. The mixture was stirred for 20 min, cooled to −78°C, and a solution of aldehyde 11 (114 mg, 0.785 mmol) in 0.5 mL dry THF was added. The reaction mixture was stirred at −78°C for 1 h, warmed to 25°C, stirred for 3 h, quenched with saturated NH4Cl solution, and extracted with EtOAc. The organic layers were combined and dried over Na2SO4. The solvent was evaporated under reduced pressure and the crude residue was purified by silica gel chromatography using (15:85) EtOAc/Hex as eluent to afford 22, as pale yellow solid (142 mg, 85.0%). HRMS (ESI) m/z calcd for [C14H17N+H]+: 200.1439, found 200.1443. 1H NMR (CDCl3): δ 7.98 (s, 1H), 7.45–7.47 (d, 1H), 7.21–7.23 (d, 1H), 7.06 (t, 1H), 6.98 (t, 1H), 6.32–6.44 (m, 2H), 5.95–6.02 (m, J = 16.04 Hz, 1H), 2.15–2.20 (q, 2H), 1.29–1.44 (m, 4H), 0.86 (t, 3H).
Synthesis of alkyl-1H-indoles
Compounds 14, 23, 32 (5-Cl analog) and 38 (6-Cl analog) were synthesized as shown for 2-hex-1-enyl-1H-indole (23):
2-Hexyl-1H-indole (23)
To a stirred solution of 22 (35 mg, 0.16 mmol) in anhydrous EtOH (1 mL) was added 10% PD/C (10 % wt) at rt under H2 atmosphere and stirred for 6 h. The reaction mixture was filtered through celite/florisil. The filtrate was concentrated under reduced pressure and the crude residue purified by silica gel chromatography using (1:9) Et2O/Hex solvent as eluent to afford 23 as white solid (32 mg, 90%). HRMS (ESI) m/z calcd for [C14H19N+H]+: 202.1596, found 202.1596. 1H NMR (CDCl3): δ 7.8 (s, 1H), 7.45 (d, 1H), 7.22 (d, 1H), 6.93–7.08 (m, 2H), 6.14 (s, 1H), 2.68 (t, 2H), 1.59–1.60 (m, 2H), 1.15–1.38 (m, 6H), 0.82 (t, 3H).
Synthesis of alkyl-(5-oxo-valeric acid)indoles
Compounds 16–19 (hexyl at C-3, 5, 6, and 7), 24–29 (alkyl at C-2 = butyl 24, pentyl 25, hexyl 26, heptyl 27, octyl 28, and undecyl 29), 33 (5-Cl analog), 39 (6-Cl analog) were synthesized using the general procedure for 24 as follows:
5-(2-Hexyl-1H-indol-1-yl)-5-oxo-valeric acid (26)
To a solution of 23 (32 mg, 0.16 mmol) in anhydrous DMSO was added crushed KOH (44.5 mg, 0.795 mmol) at rt under nitrogen. The reaction mixture was stirred for 30 min followed by addition of glutaric anhydride 15 (90.6 mg, 0.795 mmol). The stirring was continued for 3 h. The reaction mixture was quenched with saturated NH4Cl solution, extracted with EtOAc and the organic layers combined and dried over Na2SO4. The solvent was evaporated under reduced pressure and the crude residue was purified by silica gel chromatography using 50% EtOAc/Hex as eluent to afford 26 as a white solid (30.1 mg, 60%). HRMS (ESI) m/z calcd for [C19H25NO3+H]+: 316.1913, found 316.1913. 1H NMR (CDCl3): δ 7.82 (1H, d), 7.49–7.51 (1H, m), 7.21 –7.28 (2H, m), 6.43 (1H, s), 3.15 (2H, t), 3.02 (2H, t), 2.6 (2H, t), 2.18–2.25 (2H, m), 1.68–1.76 (2H, m), 1.28–1.47 (6H, m), 0.93 (3H, t). 13C NMR (CDCl3): δ 179.1, 172.7, 143.1, 136.0, 130.1, 123.4, 123.0, 120.2, 114.8, 108.3, 37.9, 32.9, 31.7, 30.7, 29.2, 28.9, 22.6, 19.8, 14.1.
5-(3-Hexyl-1H-indol-1-yl)-5-oxo-valeric acid (16)
Yield: 72%. HRMS (ESI) m/z calcd for [C19H25NO3+H]+: 316.1913, found 316.1904. 1H NMR (CDCl3): δ 8.46 (1H, s), 7.55 (1H, d), 7.37 (1H, t), 7.28–7.32 (2H, m), 7.24 (1H, s), 3.03 (2H, t), 2.67–2.72 (2H, m), 2.62 (2H, t), 2.16– 2.23 (2H, m), 1.69–1.77 (2H, m), 1.28–1.45 (6H, m), 0.92 (3H, t). 13C NMR (CDCl3): δ 178.1, 170.3, 136.1, 130.8, 125.2, 123.8, 123.4, 120.7, 119.0, 116.7, 34.6, 32.7, 31.7, 29.3, 29.1, 25.0, 22.6, 19.4, 14.1.
5-(5-Hexyl-1H-indol-1-yl)-5-oxo-valeric acid (17)
Yield: 54%. HRMS (ESI) m/z calcd for [C19H25NO3+H]+: 316.1913, found 316.1921. 1H NMR (CDCl3): δ 8.24 (s, 1H), 7.38 (d, J = 8.08 Hz, 1H), 7.32 (d, J = 3.54 Hz, 1H), 7.04 (d, J = 8.08 Hz, 1H), 6.52 (d, J = 3.54 Hz, 1H), 2.95 (t, J = 6.95 Hz, 2H), 2.65 (t, J = 7.71 Hz, 2H), 2.52 (t, J = 6.82 Hz, 2H), 2.05 – 2.16 (m, 2H), 1.59 (qt, J = 7.33 Hz, 2H), 1.16 – 1.33 (m, 6H), 0.80 (t, J = 6.44 Hz, 3H). 13C NMR (CDCl3): δ 177.9, 170.6, 140.6, 136.0, 128.2, 124.6, 123.9, 120.4, 116.3, 109.3, 36.5, 34.5, 32.6, 32.1, 31.7, 29.0, 22.6, 19.5, 14.1.
5-(6-Hexyl-1H-indol-1-yl)-5-oxo-valeric acid (18)
Yield: 66%. HRMS (ESI) m/z calcd for [C19H25NO3+H]+: 316.1913, found 316.1909. 1H NMR (CDCl3): δ 8.24 (s, 1H), 7.38 (d, J = 8.08 Hz, 1H), 7.32 (d, J = 3.54 Hz, 1H), 7.04 (d, J = 8.08 Hz, 1H), 6.52 (d, J = 3.54 Hz, 1H), 2.95 (t, J = 6.95 Hz, 2H), 2.65 (t, J = 7.71 Hz, 2H), 2.52 (t, J = 6.82 Hz, 2H), 2.05 – 2.17 (m, 2H), 1.59 (qt, J = 7.33 Hz, 2H), 1.15 – 1.33 (m, 6H), 0.80 (t, J = 6.44 Hz, 3H). 13C NMR (CDCl3): δ 177.9, 170.6 140.7, 136.0, 128.2, 124.6, 123.9, 116.3, 109.3, 36.5, 34.5, 32.6, 32.1, 31.7, 29.0, 22.6, 19.4, 14.1.
5-(7-Hexyl-1H-indol-1-yl)-5-oxo-valeric acid (19)
Yield: 74%. HRMS (ESI) m/z calcd for [C19H25NO3+H]+: 316.1913, found 316.1917. 1H NMR (CDCl3): δ 7.47 (d, J = 3.79 Hz, 1H), 7.42 (d, J = 7.33 Hz, 1H), 7.19 – 7.27 (m, 2H), 6.66 (d, J = 3.79 Hz, 1H), 3.01 – 3.12 (m, 4H), 2.61 (t, J = 6.95 Hz, 2H), 2.18 – 2.27 (m, 2H), 2.01 – 2.09 (m, 2H), 1.46 – 1.55 (m, 2H), 1.24 – 1.37 (m, 4H), 0.88 (t, 3H). 13C NMR (CDCl3): δ 177.6, 169.9, 134.3, 132.3, 131.7, 127.3, 126.0, 124.1, 118.7, 109.3, 35.7, 35.4, 31.8, 30.2, 29.7, 29.3, 22.6, 20.2, 14.1.
5-(2-Butyl-1H-indol-1-yl)-5-oxo-valeric acid (24)
Yield: 68.0%. HRMS (ESI) m/z calcd for [C17H21NO3+H]+: 288.1600, found 288.1596. 1H NMR (CDCl3): δ 7.72 (d, J = 7.83 Hz, 1H), 7.37 – 7.42 (m, 1H), 7.09 – 7.19 (m, 2H), 6.33 (s, 1H), 3.05 (t, J = 6.95 Hz, 2H), 2.92 (t, J = 7.58 Hz, 2H), 2.50 (t, J = 7.07 Hz, 2H), 2.12 (qt, J = 7.01 Hz, 2H), 1.61 (qt, J = 7.52 Hz, 2H), 1.37 (m, 2H), 0.89 (t, J = 7.20 Hz, 3H). 13C NMR (CDCl3): δ 178.5, 172.7, 143.1, 136.0, 130.1, 123.4, 123.0, 120.2, 114.7, 108.3, 37.9, 32.8, 31.1, 30.4, 22.5, 19.9, 13.9.
5-(2-Pentyl-1H-indol-1-yl)-5-oxo-valeric acid (25)
Yield: 40%. HRMS (ESI) m/z calcd for [C18H23NO3+H]+: 302.1756, found 302.1756. 1H NMR (CDCl3): δ 7.69 (d, J = 6.93 Hz, 1H), 7.32 – 7.39 (m, 1H), 6.98 – 7.12 (m, 2H), 6.27 (s, 1H), 3.18 (t, J = 7.23 Hz, 2H), 2.98 (t, J = 7.68 Hz, 2H), 2.50 (t, J = 7.16 Hz, 2H), 2.21 (m 2H), 1.58 (qt, J = 7.52 Hz, 2H), 1.04 (m, 4H), 0.92 (t, J = 6.97 Hz, 3H). 13C NMR (CDCl3): δ 179.1, 173.2, 144.7, 135.6, 129.8, 122.7, 121.9, 119.5, 113.8, 109.1, 38.2, 32.7, 31.5, 30.8, 23.1, 22.5, 19.7, 13.9.
5-(2-Heptyl-1H-indol-1-yl)-5-oxo-valeric acid (27)
Yield: 46%. HRMS (ESI) m/z calcd for [C20H27NO3+H]+: 330.2069, found 330.2058. 1H NMR (CDCl3): δ 7.72 (d, J = 7.83 Hz, 1H), 7.37 – 7.43 (m, 1H), 7.10 – 7.20 (m, 2H), 6.34 (s, 1H), 3.06 (t, J = 7.07 Hz, 2H), 2.92 (t, J = 7.58 Hz, 2H), 2.51 (t, J = 7.07 Hz, 2H), 2.12 (qt, J = 7.07 Hz, 2H), 1.62 (qt, J = 7.45 Hz, 2H), 1.15 – 1.39 (m, 8H), 0.77 – 0.85 (t, 3H). 13C NMR (CDCl3): δ 172.7, 143.1, 136.0, 130.0, 123.4, 122.9, 120.0, 114.7, 108.3, 37.9, 32.8, 31.8, 30.7, 29.4, 29.2, 29.0, 22.6, 20.0, 14.1.
5-(2-Octyl-1H-indol-1-yl)-5-oxo-valeric acid (28)
Yield: 65.5%. HRMS (ESI) m/z calcd for [C21H29NO3+H]+: 344.2226, found 344.2222. 1H NMR (CDCl3): δ 7.71 (d, J = 7.58 Hz, 1H), 7.36 – 7.43 (m, 1H), 7.10 – 7.20 (m, 2H), 6.33 (s, 1H), 3.03 – 3.12 (m, 1H), 2.87 – 2.96 (m, 3H), 2.71 (qd, J = 6.68, 13.29 Hz, 1H), 2.52 (dd, J = 6.32, 15.66 Hz, 1H), 2.34 (dd, J = 7.20, 15.54 Hz, 1H), 1.62 (qt, J = 7.52 Hz, 2H), 1.28 –1.39 (m, 3H), 1.14 – 1.28 (m, 8H), 1.08 (d, J = 6.82 Hz, 3H), 0.76 – 0.85 (t, 3H). 13C NMR (CDCl3): δ 177.6, 172.3, 143.0, 136.0, 130.0, 123.3, 122.9, 120.2, 114.7, 108.2, 45.2, 40.4, 31.8, 30.6, 29.5, 29.2, 29.0, 27.2, 22.6, 19.9, 14.1.
5-(2-Undecyl-1H-indol-1-yl)-5-oxo-valeric acid (29)
Yield: 46%. HRMS (ESI) m/z calcd for [C24H35NO3+H]+: 386.2695, found 386.2674. 1H NMR (CDCl3): δ 7.80 (d, J = 7.6 Hz, 1H), 7.48 (d, J = 6.8 Hz, 1H), 7.22 (dd, J = 12.1, 7.1 Hz, 2H), 6.41 (s, 1H), 3.13 (t, J = 6.4 Hz, 2H), 3.00 (t, J = 7.2 Hz, 2H), 2.58 (t, J = 6.5 Hz, 2H), 2.26 – 2.14 (m, 2H), 1.77 – 1.62 (m, 2H), 1.49 – 1.18 (m, 18H), 0.89 (t, J = 5.8 Hz, 3H). 13C NMR (CDCl3): δ 178.72 (s), 172.70 (s), 143.16 (s), 135.99 (s), 130.06 (s), 123.42 (s), 122.96 (s), 120.25 (s), 114.78 (s), 108.27 (s), 37.91 (s), 32.84 (s), 31.93 (s), 30.72 (s), 29.77 – 29.59 (m), 29.52 (d, J = 5.4 Hz), 29.36 (s), 28.95 (s), 22.70 (s), 19.85 (s), 14.14 (s).
5-(5-Chloro-2-hexyl-1H-indol-1-yl)-5-oxo-valeric acid (33)
Yield: 64%. HRMS (ESI) m/z calcd for [C19H24ClNO3+H]+: 350.1523 and 352.1493, found 350.1523 and 352.1567. 1H NMR (CDCl3): δ 7.72 (d, J = 7.58 Hz, 1H), 7.38 – 7.42 (m, 1H), 7.12 – 7.18 (m, 1H), 6.34 (s, 1H), 3.06 (t, J = 7.07 Hz, 2H), 2.92 (t, J = 7.71 Hz, 2H), 2.51 (t, J = 7.20 Hz, 2H), 2.13 (qt, J = 6.76 Hz, 2H), 1.62 (qt, J = 7.52 Hz, 2H), 1.14 – 1.40 (m, 6H), 0.83 (t, J = 6.95 Hz, 3H). 13C NMR (CDCl3): δ 177.63, 172.70, 143.12, 136.03, 130.07, 123.40, 122.94, 120.23, 114.74, 108.27, 37.90, 32.68, 31.70, 30.67, 29.68, 29.13, 28.93, 22.60, 19.91, 14.04.
5-(6-Chloro-2-hexyl-1H-indol-1-yl)-5-oxo-valeric acid (39)
Yield: 38.1%. HRMS (ESI) m/z calcd for [C19H24ClNO3+H]+: 350.1523 and 352.1493, found 350.1519 and 352.1568. 1H NMR (CDCl3): δ 7.83 (1H, s), 7.27–7.29 (1H, d), 7.09–7.12 (1H, dd), 6.29 (1H, s), 3.00–3.06 (1H, dd), 2.82–2.89 (2H, m), 2.64–2.73 (1H, m), 2.49–2.54 (1H, dd), 2.32–2.38 (1H, dd), 1.58– 1.65 (2H, m), 1.18–1.38 (6H, m), 0.82 (3H, t). 13C NMR (CDCl3): δ 178.0, 172.1, 143.1, 136.6, 129.3, 128.3, 123.4, 120.7, 115.3, 107.9, 44.9, 40.3, 31.7, 30.6, 29.1, 28.9, 22.6, 20.0, 14.0.
Synthesis of 3-(4-chloro-2-nitro-phenyl)-2-oxo-propionic acid ethyl ester (36)
An ethereal solution of anhydrous EtOH (15 mL EtOH + 36 mL ether) was added dropwise to a stirred mixture of potassium metal (2.26 g, 57.9 mmol) in anhydrous ether (22 mL) at rt. A large amount of effervescence was observed. After the potassium metal was completely dissolved, an ethereal solution of diethyl oxalate, 35 (9.43 mL, 69.3 mmol, 22 mL ether) was added dropwise to the reaction mixture. Then an ethereal solution of 4-chloro-1-methyl-2-nitrobenzene, 34 (10 g, 58.28 mmol) in anhydrous ether (15 mL) was added to the reaction mixture. The reaction was stirred for 15 h and then sonicated for another 7 h. The reaction mixture was neutralized by adding 1N HCl at 0°C. It was extracted with EtOAc and the organic layers washed with brine and dried over Na2SO4. The solvent was evaporated under reduced pressure to afford 36 in crude form which was used without any further purification. 1H NMR (CDCl3): δ 8.15 (d, J = 2.2 Hz, 1H), 7.58 (dd, J = 8.2, 2.2 Hz, 1H), 7.23 (d, J = 3.4 Hz, 1H), 4.49 (s, 2H), 4.36 (q, J = 7.1 Hz, 2H), 1.37 (t, J = 7.2 Hz, 3H).
Synthesis of 6-chloro-1H-indole-2-carboxylic acid ethyl ester (37)
To a solution of 36 (1 g, 3.68 mmol) in 1:1 mixture of anhydrous ethanol and glacial acetic acid (7 mL each) was added Fe powder (1.83 g, 32.76 mmol) and the reaction mixture was refluxed for 4 h. It was then cooled to 5 °C and filtered through celite and florisil layers. The filtrate was partitioned between CH2Cl2. The organic layer was washed with 1N HCl followed by saturated NaHCO3 solution and finally with brine. The crude residue was purified by crystallization to yield 37 as a pale brown solid (421 mg, 51%). HRMS (ESI) m/z calcd for [C11H10ClNO2+H]+: 224.0478 and 226.0449, found 224.0477 and 226.0451. 1H NMR (CDCl3): δ 8.84 (br s, 1H), 7.60 (d, J = 8.6 Hz, 1H), 7.42 (s, 1H), 7.19 (s, 1H), 7.13 (dd, J = 8.6, 1.1 Hz, 1H), 4.41 (q, J = 7.2 Hz, 2H), 1.42 (t, J = 7.1 Hz, 3H). 13C NMR (CDCl3): δ 161.7, 137.0, 131.3, 128.2, 126.0, 123.5, 121.8, 111.6, 108.6, 61.2, 14.3.
Synthesis of 6-chloro-2-(hex-1-en-1-yl)-1H-indole (38)
To a stirred solution of 37 (200 mg, 0.894 mmol) in THF (5 mL) at 0 °C was added a LiAlH4 (1M in THF, 1.34 mL, 1.34 mmol) under argon. The reaction mixture was stirred at 0 °C for 30 min, then was gradually warmed to rt over a period of 3 h. The reaction mixture was quenched with aqueous NH4Cl solution and then was extracted with EtOAc. The organic layers were combined and dried over Na2SO4. The solvent was evaporated under reduced pressure and the crude mixture was dried under vacuum pump. To this crude mixture in acetonitrile (5 mL) was added MnO2 (405 mg, 4.68 mmol) and the reaction mixture was stirred for 18 h. The reaction mixture was diluted with ether and filtered through celite and florisil layers. The solvent was evaporated under reduced pressure and the crude residue purified by silica gel chromatography using 30% EtOAc/Hex solvent as eluent to afford 6-chloro-1H-indole-2-carbaldehyde as a brown solid (156 mg, 92%). HRMS (ESI) m/z calcd for [C9H6ClNO+H]+: 180.0216 and 182.0187, found 180.0215 and 182.0189. 1H NMR (CDCl3): δ 9.77 (1H, s), 9.11 (1H, s), 7.58–7.60 (1H, d), 7.39 (1H, s), 7.18–7.19 (1H, m), 7.07–7.10 (1H, dd). 13C NMR (CDCl3): δ 181.96 (s), 124.41 (s), 122.48 (s), 121.43 (s), 120.66 (s), 114.68 (s), 112.23 (s), 110.88 (s), 100.13 (s). This aldehyde was reacted with the bromopentyl triphenyl phosphonium salt as described for the synthesis of 22 to afford 38: HRMS (ESI) m/z calcd for [C14H16ClN+H]+: 234.1050 and 236.1020, found 234.1053 and 236.1027. 1H NMR (CDCl3): δ 8.05 (s, 1H), 7.42 (d, J = 8.4 Hz, 1H), 7.37 – 7.26 (m, 2H), 7.02 (d, J = 8.4 Hz, 1H), 6.42 – 6.31 (m, 2H), 6.11 – 6.00 (m, 1H), 2.24 (q, J = 7.1 Hz, 2H), 1.42 (ddd, J = 31.6, 14.9, 7.4 Hz, 4H), 0.93 (t, J = 7.2 Hz, 3H). 13C NMR (CDCl3): δ 137.31 (s), 136.76 (s), 130.92 (s), 127.78 (s), 127.56 (s), 121.10 (s), 120.63 (s), 120.41 (s), 110.34 (s), 101.26 (s), 32.67 (s), 31.37 (s), 22.24 (s), 13.94 (s).
Scheme 5.

Synthesis of monosubstituted aromatic compounds 7–10.
ACKNOWLEDGEMENTS
This work was supported by grants from the American Asthma Foundation (J.R.; Award Number 12-0049), National Heart, Lung, And Blood Institute (J.R.; Award Number R01HL081873), the Canadian Institutes of Health Research (W.S.P.; MOP-6254), and the Quebec Heart and Stroke Foundation (W.S.P.). The Meakins-Christie Laboratories - MUHC-RI are supported in part by a Center grant from Le Fond de la Recherche en Santé du Québec as well as by the J. T. Costello Memorial Research Fund. J.R. also wishes to acknowledge the National Science Foundation for the AMX-360 (Grant Number CHE-90-13145) and Bruker 400 MHz (Grant Number CHE-03-42251) NMR instruments. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, And Blood Institute or the National Institutes of Health.
ABBREVIATIONS USED
- 5-HEDH
5-hydroxyeicosanoid dehydrogenase
- 5-oxo-ETE
5-Oxo-6E,8Z,11Z,14Z-eicosatetraenoic acid
- CaCl2
calcium chloride
- CDCl3
deuterated chloroform
- MeCN
acetonitrile
- CH2N2
diazomethane
- DMSO
dimethylsulfoxide
- Et2O
diethyl ether
- EtOAc
ethyl acetate
- EtOH
ethanol
- GM-CSF
granulocyte/macrophage colony stimulating factor
- HETE
hydroxy-eicosatetraenoic acid
- Hex
hexanes
- KHMDS
potassium hexamethyldisilazide
- KOH
potassium hydroxide
- LiAlH4
lithium aluminum hydride
- LiHMDS
lithium hexamethyldisilazide
- LO
lipoxygenase
- LT
leukotriene
- MgCl2
magnesium chloride
- MnO2
manganese dioxide
- NaHMDS
sodium hexamethyldisilazide
- Na2SO4
sodium sulfate
- NaI
sodium iodide
- NH4Cl
ammonium chloride
- OXE-R
oxoeicosanoid receptor
- SE
standard error
- THF
tetrahydrofuran
REFERENCES
- 1.Khanapure SP, Shi XX, Powell WS, Rokach J. Total Synthesis of a Potent Proinflammatory 5-Oxo-ETE and Its 6,7-Dihydro Biotransformation Product. J. Org. Chem. 1998;63:337–342. [Google Scholar]
- 2.Khanapure SP, Shi XX, Powell WS, Rokach J. The Total Synthesis of Tritiated and Deuterated 5-Oxo-ETE, A Novel Inflammatory Mediator. J. Org. Chem. 1998;63:4098–4102. [Google Scholar]
- 3.Powell WS, Boismenu D, Khanapure SP, Rokach J. Quantitative Analysis of 5-Oxo-6,8,11,14-Eicosatetraenoic Acid by Electrospray Mass Spectrometry Using a Deuterium-Labeled Internal Standard. Anal. Biochem. 2001;295:262–266. doi: 10.1006/abio.2001.5206. [DOI] [PubMed] [Google Scholar]
- 4.Powell WS, Gravelle F, Gravel S. Metabolism of 5(S)-Hydroxy-6,8,11,14-Eicosatetraenoic Acid and Other 5(S)-Hydroxyeicosanoids by a Specific Dehydrogenase in Human Polymorphonuclear Leukocytes. J. Biol. Chem. 1992;267:19233–19241. [PubMed] [Google Scholar]
- 5.Powell WS, Rokach J. Biochemistry, Biology and Chemistry of the 5-Lipoxygenase Product 5-Oxo-ETE. Prog. Lipid Res. 2005;44:154–183. doi: 10.1016/j.plipres.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Grant GE, Rokach J, Powell WS. 5-Oxo-ETE and the OXE Receptor. Prostag. Oth. Lipid M. 2009;89:98–104. doi: 10.1016/j.prostaglandins.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Powell WS, MacLeod R, Gravel S, Gravelle F, Bhakar A. Metabolism and Biologic Effects of 5-Oxoeicosanoids on Human Neutrophils. J. Immunol. 1996;156:336–342. [PubMed] [Google Scholar]
- 8.O'Flaherty JT, Taylor JS, Thomas MJ. Receptors for the 5-Oxo Class of Eicosanoids in Neutrophils. J. Biol. Chem. 1998;273:32535–32541. doi: 10.1074/jbc.273.49.32535. [DOI] [PubMed] [Google Scholar]
- 9.Hosoi T, Koguchi Y, Sugikawa E, Chikada A, Ogawa K, Tsuda N, Suto N, Tsunoda S, Taniguchi T, Ohnuki T. Identification of a Novel Human Eicosanoid Receptor Coupled to Gi/O. J. Biol. Chem. 2002;277:31459–31465. doi: 10.1074/jbc.M203194200. [DOI] [PubMed] [Google Scholar]
- 10.Jones CE, Holden S, Tenaillon L, Bhatia U, Seuwen K, Tranter P, Turner J, Kettle R, Bouhelal R, Charlton S, Nirmala NR, Jarai G, Finan P. Expression and Characterization of a 5-Oxo-6e,8z,11z, 14z-Eicosatetraenoic Acid Receptor Highly Expressed on Human Eosinophils and Neutrophils. Mol. Pharmacol. 2003;63:471–477. doi: 10.1124/mol.63.3.471. [DOI] [PubMed] [Google Scholar]
- 11.Sundaram S, Ghosh J. Expression of 5-Oxo-ETE Receptor in Prostate Cancer Cells: Critical Role in Survival. Biochem. Biophys. Res. Commun. 2006;339:93–98. doi: 10.1016/j.bbrc.2005.10.189. [DOI] [PubMed] [Google Scholar]
- 12.Brink C, Dahlen SE, Drazen J, Evans JF, Hay DWP, Rovati GE, Serhan CN, Shimizu T, Yokomizo T. International Union of Pharmacology XLIV. Nomenclature for the Oxoeicosanoid Receptor. Pharmacol. Rev. 2004;56:149–157. doi: 10.1124/pr.56.1.4. [DOI] [PubMed] [Google Scholar]
- 13.Powell WS, Gravel S, MacLeod RJ, Mills E, Hashefi M. Stimulation of Human Neutrophils by 5-Oxo-6,8,11,14-Eicosatetraenoic Acid by a Mechanism Independent of the Leukotriene B4 Receptor. J. Biol. Chem. 1993;268:9280–9286. [PubMed] [Google Scholar]
- 14.Sozzani S, Zhou D, Locati M, Bernasconi S, Luini W, Mantovani A, O'Flaherty JT. Stimulating Properties of 5-Oxo-Eicosanoids for Human Monocytes: Synergism with Monocyte Chemotactic Protein-1 and -3. J. Immunol. 1996;157:4664–4671. [PubMed] [Google Scholar]
- 15.Powell WS, Chung D, Gravel S. 5-Oxo-6,8,11,14-Eicosatetraenoic Acid Is a Potent Stimulator of Human Eosinophil Migration. J. Immunol. 1995;154:4123–4132. [PubMed] [Google Scholar]
- 16.Guilbert M, Ferland C, Bosse M, Flamand N, Lavigne S, Laviolette M. 5-Oxo-6,8,11,14-Eicosatetraenoic Acid Induces Important Eosinophil Transmigration through Basement Membrane Components: Comparison of Normal and Asthmatic Eosinophils. Am. J. Resp. Cell Mol. 1999;21:97–104. doi: 10.1165/ajrcmb.21.1.3517. [DOI] [PubMed] [Google Scholar]
- 17.Powell WS, Gravel S, Halwani F. 5-Oxo-6,8,11,14-Eicosatetraenoic Acid Is a Potent Stimulator of L-Selectin Shedding, Surface Expression of Cd11b, Actin Polymerization, and Calcium Mobilization in Human Eosinophils. Am. J. Resp. Cell Mol. 1999;20:163–170. doi: 10.1165/ajrcmb.20.1.3141. [DOI] [PubMed] [Google Scholar]
- 18.Schwenk U, Schroder JM. 5-Oxo-Eicosanoids Are Potent Eosinophil Chemotactic Factors - Functional Characterization and Structural Requirements. J. Biol. Chem. 1995;270:15029–15036. doi: 10.1074/jbc.270.25.15029. [DOI] [PubMed] [Google Scholar]
- 19.O'Flaherty JT, Kuroki M, Nixon AB, Wijkander J, Yee E, Lee SL, Smitherman PK, Wykle RL, Daniel LW. 5-Oxo-Eicosatetraenoate Is a Broadly Active, Eosinophil-Selective Stimulus for Human Granulocytes. J. Immunol. 1996;157:336–342. [PubMed] [Google Scholar]
- 20.Muro S, Hamid Q, Olivenstein R, Taha R, Rokach J, Powell WS. 5-Oxo-6,811,14-Eicosatetraenoic Acid Induces the Infiltration of Granulocytes into Human Skin. J. Allergy Clin. Immun. 2003;112:768–774. doi: 10.1016/s0091-6749(03)01888-8. [DOI] [PubMed] [Google Scholar]
- 21.Patel P, Cossette C, Anumolu JR, Gravel S, Lesimple A, Mamer OA, Rokach J, Powell WS. Structural Requirements for Activation of the 5-Oxo-6e,8z,11z,14z-Eicosatetraenoic Acid (5-Oxo-ETE) Receptor: Identification of a Mead Acid Metabolite with Potent Agonist Activity. J. Pharmacol. Exp. Ther. 2008;325:698–707. doi: 10.1124/jpet.107.134908. [DOI] [PubMed] [Google Scholar]
- 22.Powell WS, Gravel S, Khanapure SP, Rokach J. Biological Inactivation of 5-Oxo-6,8,11,14-Eicosatetraenoic Acid by Human Platelets. Blood. 1999;93:1086–1096. [PubMed] [Google Scholar]
- 23.Khanapure SP, Powell WS, Rokach J. The Total Synthesis of 5-Oxo-12(S)-Hydroxy-6(E),8(Z),10(E),14(Z)-Eicosatetraenoic Acid and Its 8,9-Trans-Isomer and Their Identification of Human Platelets. J. Org. Chem. 1998;63:8976–8982. [Google Scholar]
- 24.Sugiura M, Beierbeck H, Belanger PC, Kotovych G. Conformational Analysis of Leukotriene B4 in Solution Based on High-Field Nuclear Magnetic Resonance Measurements. J. Am. Chem. Soc. 1984;106:4021–4025. [Google Scholar]
- 25.Loftus P, Bernstein PR. A Study of Some Leukotriene A4 and D4 Analogues by Proton Nuclear Magnetic Resonance Spectroscopy. J. Org. Chem. 1983;48:40–44. [Google Scholar]
- 26.Lee K, Foley MA, Chen L, Behnke ML, Lovering FE, Kirincich SJ, Wang W, Shim J, Tam S, Shen MWH, Khor SP, Xu X, Goodwin DG, Ramarao MK, Nickerson-Nutter C, Donahue F, Ku MS, Clark JD, McKew JC. Discovery of Ecopladib, an Indole Inhibitor of Cytosolic Phospholipase A2α. J. Med. Chem. 2007;50:1380–1400. doi: 10.1021/jm061131z. [DOI] [PubMed] [Google Scholar]