

Abstract
This Letter describes the synthesis and structure–activity relationships (SAR) of isoform-selective PLD inhibitors. By virtue of the installation of a 1,3,8-triazaspiro[4,5]decan-4-one privileged structure, PLD inhibitors with nanomolar potency and an unprecedented 40-fold selectivity for PLD2 over PLD1 were developed. Interestingly, SAR for this diverged from our earlier efforts, and dual PLD1/2 inhibitors were also discovered within this series.
Keywords: PLD, Phospholipase, Cancer, Privileged structure
There are two mammalian isoforms of Phospholipase D (PLD), referred to as PLD1 and PLD2. Despite conserved regulatory and catalytic domains between PLD1 and PLD2, studies indicate distinct modes of activation and distinct functional roles for PLD1 and PLD2.1 From a therapeutic perspective, PLD signaling has been implicated in a variety of human cancers (breast, renal, gastric and colorectal), and PLD signaling is thought to regulate actin cytoskeleton reorganization and cell motility.2–8 Thus, small molecules that selectively inhibit PLD1 or PLD2 are potentially a novel approach for the treatment of cancer. However, the PLD field has been hindered for decades by a lack of isoform selective direct-acting small molecule inhibitors.
In recent reports,9,10 we have disclosed the discovery and development of dual PLD1/2 inhibitors, such as 1, and a PLD1 inhibitor 2 with unprecedented selectivity (1700-fold) over PLD2 in a cell based assay (Fig. 1). Moreover, these compounds represent a new class of antimetastatic agents, and data suggest they may be inhibiting PLD by an allosteric mechanism.9–13 After synthesizing ~500 compounds within the (1-(piperidin-4-yl)-1H-benzo[d]imidazol-2(3H)-one privileged structure scaffold represented by 1 and 2, PLD2 selective inhibitors remained elusive until a limited diversity-oriented synthesis campaign identified the 1,3,8-triazaspiro[ 4,5]decan-4-one scaffold as a PLD2-preferring moiety.9 Indeed, compound 3 represents the only known PLD2 preferring (~ninefold versus PLD1) inhibitor. Monovich and co-workers reported on a series of PLD2 inhibitors in 2007, but the compounds proved to be dual PLD1/2 or modestly PLD1 preferring inhibitors.9,10,14 In this Letter, we describe an iterative analog library synthesis approach based on 3,15 coupled with biochemical assays and mass spectrometric lipid profiling of cellular responses,9,10 for the discovery of PLD2 inhibitors with improved potency and PLD2 isoform selectivity.
Figure 1.
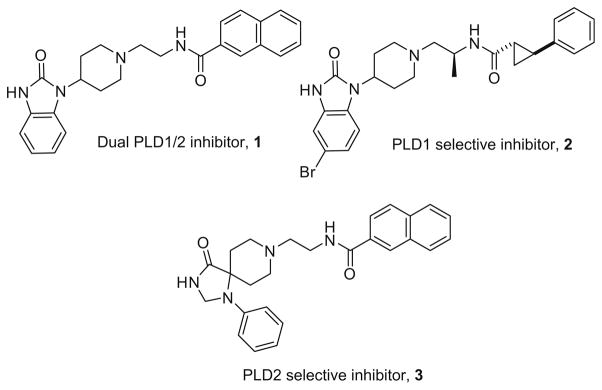
Our recently reported isoform-selective PLD inhibitors: dual PLD1/2 inhibitor 1, PLD1-selective (>1700-fold) inhibitor 2, and PLD2 selective (~ninefold) inhibitor 3.
In order to optimize 3 and evaluate if the 1,3,8-triazaspiro[ 4,5]decan-4-one scaffold would maintain PLD2 selectivity, we employed our iterative parallel synthesis approach,15 and synthesized libraries to address the potential SAR depicted in Figure 2.
Figure 2.
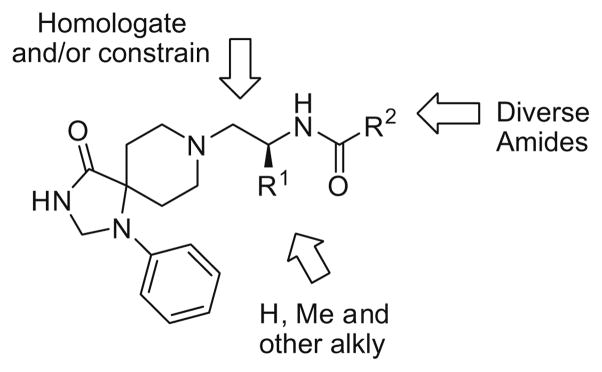
Library strategy to refine PLD inhibitors to improve potency and PLD2 isoform selectivity.
The requisite building blocks were all commercially available and the libraries were prepared according to the general route depicted in Scheme 1. Synthesis begins with a reductive amination with either N-Boc glycinal, a functionalized alinal 5 or a homologated/ cyclic constrained N-Boc amino aldehyde 6 and 1,3,8-triazaspiro[ 4,5]decan-4-one 4 to provide 7 in yields ranging from 72% to 95%. Subsequent removal of the Boc group with 4 N HCl and standard acylation chemistry provides analogs 8. All compounds were then purified to >98% purity by mass-directed preparative HPLC.16
Scheme 1.
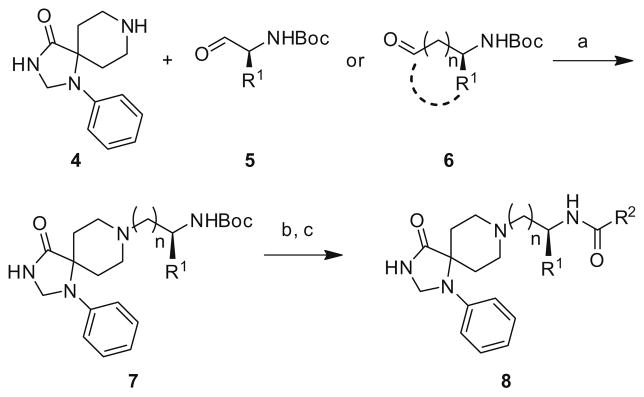
Reagents and conditions: (a) MP-B(OAc)3, DCE, rt, 16 h (72–95%); (b) 4 N HCl/dioxane, MeOH (98%); (c) R2COCl, DCM, DIEA, rt (60–94%) or (i) R2COH, PS-DCC, HOBt, DCM, DIEA; (ii) MP-CO32−(55–90%).
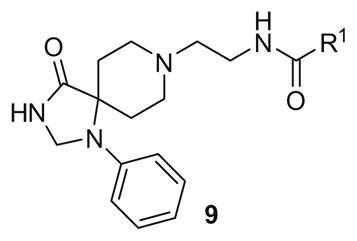
All library members were evaluated for their ability to inhibit PLD1 and PLD2 in a cellular assay (Calu-1 and HEK293-gfpPLD2, respectively) as well as a biochemical assay with purified PLD1 and PLD2 enzymes.9 The cellular assays were the ‘workhorse’ assays which drove the SAR, with routine confirmation in the in vitro biochemical assay. As observed in the previous series of PLD1 selective inhibitors, represented by 2, the ethyl diamino linker was essential—homologation to the corresponding 3- and 4-carbon tethers were inactive, as were cyclic constraints. Table 1 highlights unsubstituted ethyl diamine linker congeners 9 without the (S)-methyl group and examines only alternative amides. Compound 3 was the lead PLD2-preferring compound previously identified in a diversity-oriented synthesis campaign, with a PLD2 IC50 of 110 nM and ~ninefold selectivity versus PLD1 (IC50 = 1000 nM). In general, analogs 9 with PLD inhibitory activity were PLD2-preferring (1.3- to 21.1-fold). Incorporation of the PLD1 preferring trans-phenyl cyclopropane amide moiety of 2, provides 9a, and a complete loss of PLD inhibition. A 4-amino, 5-methoxybenzamide analog 9f (PLD2 IC50 = 550 nM) displayed over 10-fold selectivity for PLD2 over PLD1. The most potent analog in the series, 9d (PLD2 IC50 = 30 nM), incorporated a 2-benzothiophene amide, but displayed only fivefold PLD2 selectivity. A 2-quinoline amide congener, 9b, displayed comparable PLD2 potency (PLD2 IC50 = 90 nM) to the lead 3, but selectivity versus PLD1 (PLD1 IC50 = 1900 nM) was improved (>20-fold). Incorporation of a second nitrogen atom to provide the corresponding quinoxaline derivative, 9g, results in a complete loss of PLD inhibitory activity.
Table 1.
Structures and activities of analogs 9
| ||||
|---|---|---|---|---|
| Compd | R1 | PLD1 IC50a (nM) | PLD2 IC50b (nM) | Fold PLD2-selective |
| 3 |
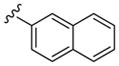
|
1000 | 110 | 9.1 |
| 9a |
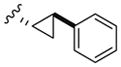
|
>20,000 | >20,000 | – |
| 9b |
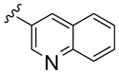
|
1900 | 90 | 21.1 |
| 9c |
|
2100 | 1600 | 1.3 |
| 9d |
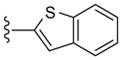
|
150 | 30 | 5.0 |
| 9e |
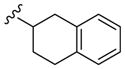
|
4250 | 990 | 7.8 |
| 9f |
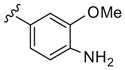
|
5900 | 550 | 10.7 |
| 9g |
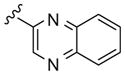
|
>20,000 | >20,000 | – |
Cellular PLD1 assay with Calcu-1 cells.
Cellular assay with HEK293-gfp PLD2 cells. Each IC50 was determined in triplicate. For details see Ref. 9.
The concentration–response-curves (CRCs) for inhibition in PLD1 and PLD2 cellular assay (Calu-1 and 293-PLD2, respectively) for 9f (10.7-fold PLD2 selective) and 9b (21.1-fold PLD2 selective) are shown in Figure 3. In an in vitro biochemical assay using purified PLD1 and PLD2 enzymes, 9b possessed a PLD1 IC50 of >20 μM and a PLD2 IC50 of 500 nM, or >40-fold PLD2 selective. While not at the level of isoform selectivity as the PLD1 inhibitor 2 (1700-fold selective),10 compound 9b represents the most potent and selective PLD2 inhibitor ever described.17
Figure 3.

CRCs for cellular PLD1 ■(Calu-1) assay and PLD2
 (HEK293-gfpPLD2) assay, highlighting the unprecedented PLD2 versus PLD1 selectivity for: (A) 9f (10.7-fold) and (B) 9b (21.1-fold).
(HEK293-gfpPLD2) assay, highlighting the unprecedented PLD2 versus PLD1 selectivity for: (A) 9f (10.7-fold) and (B) 9b (21.1-fold).
While this data was very exciting, the results from a directed library exploring the impact of incorporation of the (S)-methyl group (PLD1-preferring)9,10 on the diamino ethyl linker with the 1,3,8-triazaspiro[4,5]decan-4-one scaffold (PLD2-preferring) were completely unexpected. Incorporation of the (S)-methyl group into analogs 10 had significant impact. While affording a series with shallow SAR and few PLD inhibitors, PLD1 inhibitory activity was dramatically increased with the previously PLD2-preferring 1,3,8-triazaspiro[4,5]decan-4-one privileged structure (Table 2). For example, compound 10a, the (S)-methyl analog of the ~ninefold PLD2 selective 3, displayed comparable PLD2 inhibitory activity (PLD2 IC50 = 140 nM), but PLD1 inhibitory activity increased 40-fold (PLD1 IC50 = 25 nM), relative to 3. Thus, 10a is considered a PLD1/2 dual inhibitor, and the in vitro biochemical assay verified this result (PLD1 IC50 = 299 nM, PLD2 IC50 = 235 nM). Another classical PLD1-preferring moiety, the trans-phenyl cyclopropane moiety of 2, was inactive on both PLD isoforms in analogs such as 9a, but in combination with the (S)-methyl group, congener 10b now possessed measureable PLD1 inhibitory activity (PLD1 IC50 = 2.6 μM) with no effect, relative to 9a, on PLD2 inhibition. A 3,4-difluorobenzamide analog, 10c, also displayed dual PLD1/2 inhibition (PLD1 IC50 = 150 nM, PLD2 IC50 = 200 nM).
Table 2.
Structures and activities of analogs 10

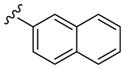
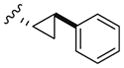
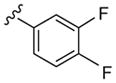
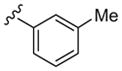
Cellular PLD1 assay with Calu-1 cells.
Cellular assay with HEK293-gfpPLD2 cells. Each IC50 is avg of three determinations. For details see Ref. 9.
The ability of different GPCR privileged structures18 to engender either PLD1- or PLD2-preferring inhibition, and the ability of a single chiral (S)-methyl group to override PLD2-preferring scaffolds to serve as a ‘molecular switch’ to dial-in PLD1 inhibition, led us to evaluate additional GPCR privileged structures (Fig. 4). This effort was disappointing, as 10 new scaffolds were surveyed, but with little success. With the exception of two heteroaryl piperazines 15 and 16 (PLD1 IC50s in the 3.6- to 18 μM range), none of these analogs displayed PLD inhibition.
Figure 4.

Alternative privileged structures evaluated 11–16.
In summary, the discovery of these potent and selective PLD2 inhibitors highlights the power of an iterative analog library synthesis approach for lead optimization, coupled with biochemical assays and mass spectrometric lipid profiling of cellular responses. PLD inhibitor 9b (cellular assay: PLD IC50 = 1900 nM, PLD2 IC50 = 90 nM, 21-fold selective; in vitro biochemical assay: PLD IC50 = >20,000 nM, PLD2 IC50 = 500 nM, >40-fold selective) represents the most potent and selective PLD2 small molecule inhibitor described to date. PLD2 selective inhibitor 9b and the PLD1 selective inhibitor 2 will serve as invaluable tools to study and dissect the role of PLD1 and PLD2 in blocking invasiveness in metastatic beast cancer models and other signaling pathways in which PLD is thought to play key regulatory roles. Work in this field, and additional experiments to confirm an allosteric mode of inhibition are in progress and will be reported in due course.
Acknowledgments
The authors thank the Vanderbilt Department of Pharmacology, the Vanderbilt Institute of Chemical Biology and the A.B. Hancock Family Foundation for Cancer Research and the Sartain-Lanier Family Foundation (C.W.L.) for support of this research. J.A.L. and R.L. are supported by an NIH ITTD training Grant (T90DA022873). P.E.S. is supported by a Pharmacology Training Grant (NIH 5T326M007628-30).
References and notes
- 1.Ponting CP, Kerr ID. Protein Sci. 1996;5:914. doi: 10.1002/pro.5560050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noh DY, Ahn SJ, Lee RA, Park IA, Kim JH, Suh PG, Lee KH, Han JS. Cancer Lett. 2000;161:207. doi: 10.1016/s0304-3835(00)00612-1. [DOI] [PubMed] [Google Scholar]
- 3.Uchida N, Okamura S, Nagamachi Y, Yamshita S. J Cancer Res Clin Oncol. 1997;123:280. doi: 10.1007/BF01208639. [DOI] [PubMed] [Google Scholar]
- 4.Zhao Y, Ehara H, Akao Y, Shamoto M, Nakagawa Y, Banno Y, Deguchi T, Ohishi N, Yagi K, Nozawa Y. Biochem Biophys Res Commun. 2000;278:140. doi: 10.1006/bbrc.2000.3719. [DOI] [PubMed] [Google Scholar]
- 5.Uchida N, Okamura S, Kuwano H. Anticancer Res. 1999;19:671. [PubMed] [Google Scholar]
- 6.Yamada Y, Hamajima N, Kato T, Iwata H, Yamamura Y, Shinoda M, Suyama M, Mitsudomi T, Tajima K, Kusakabe S, Yoshida H, Banno Y, Akao Y, Tanaka M, Nozawa Y. J Mol Med. 2003;81:126. doi: 10.1007/s00109-002-0411-x. [DOI] [PubMed] [Google Scholar]
- 7.Min DS, Kwon TK, Park WS, Chang JS, Park SK, Ahn BH, Ryoo ZY, Lee YH, Lee YS, Rhie DJ, Yoon SH, Hahn SJ, Kim MS, Jo YH. Carcinogenesis. 2001;22:1641. doi: 10.1093/carcin/22.10.1641. [DOI] [PubMed] [Google Scholar]
- 8.Buchanan FG, McReynolds M, Couvillon A, Kam Y, Holla VR, DuBois RN, Exton JA. Proc Natl Acad Sci USA. 2005;102:1638. doi: 10.1073/pnas.0406698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scott SA, Selvy PE, Buck JR, Cho HP, Criswell TL, Thomas AL, Armstrong MD, Arteaga CL, Lindsley CW, Brown HA. Nat Chem Biol. 2009;5:108. doi: 10.1038/nchembio.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lewis JA, Scott SA, Lavieri R, Buck JR, Selvy PE, Stoops SL, Armstrong MD, Brown HA, Lindsley CW. Bioorg Med Chem Lett. 2009;19:1916. doi: 10.1016/j.bmcl.2009.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lewis JA, Lebois EP, Lindsley CW. Curr Opin Chem Biol. 2008;12:269. doi: 10.1016/j.cbpa.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 12.Lindsley CW, Zhao Z, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, Huff JR, Huber HE, Duggan ME. Bioorg Med Chem Lett. 2005;15:761. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 13.Zhao Z, Duggan ME, Barnett SF, Defeo-Jones D, Huber HE, Huff JR, Hartman GD, Leister WH, Kral A, Fu S, Hancock PJ, Jones RE, Robinson R, Lindsley CW. Bioorg Med Chem Lett. 2005;15:761. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 14.Monovich L, Mugrage B, Quadros E, Toscano K, Tommasi R, LaVoie S, Liu E, Du Z, LaSala D, Boyar W, Steed P. Bioorg Med Chem Lett. 2007;17:2310. doi: 10.1016/j.bmcl.2007.01.059. [DOI] [PubMed] [Google Scholar]
- 15.Kennedy JP, Williams L, Bridges TM, Daniels RN, Weaver D, Lindsley CW. J Comb Chem. 2008;10:345. doi: 10.1021/cc700187t. [DOI] [PubMed] [Google Scholar]
- 16.Leister WH, Strauss KA, Wisnoski DD, Zhao Z, Lindsley CW. J Comb Chem. 2003;5:322. doi: 10.1021/cc0201041. [DOI] [PubMed] [Google Scholar]
- 17.Experimental for PLD2 selective inhibitor 9b: N-(2-(4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]decan-8-yl)ethyl)quinoline-3-carboxamide: A flask was charged with 8-(2-aminoethyl)-1-phenyl-1,3,8-triazaspiro[4.5]decan-4-one (7, 695 mg, 2.00 mmol), quinoline-3-carboxylic acid (381 mg, 2.20 mmol), and O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU, 1.14 g, 3.00 mmol) before adding DIPEA (775 mg, 1.045 mL, 6.00 mmol) and diluting with a 2:1 mixture of anhydrous CH2Cl2 (14.0 mL) and anhydrous DMF (7.0 mL) to an amine concentration of 95 mM. The mixture was stirred at room temperature for at least 4 h. The desired product precipitated out of solution as an off-white solid and was isolated by vacuum filtration. Yield: 28% (242mg). 1H NMR (DMSO-d6, 400 MHz, 25 °C): δ 9.26 (d, J = 2.0 Hz, 1H), 8.96 (t, J = 5.2 Hz, 1H), 8.77 (br s, 2H), 8.52 (dd, J = 4.1, 1.0 Hz, 1H), 8.31 (dd, J = 8.2, 1.0Hz, 1H), 8.06 (t, J = 8.4 Hz, 2H), 7.86 (td, J = 7.7, 1.2 Hz, 1H), 7.68 (t, J = 7.6 Hz, 1H), 7.32 (dd, J = 8.4, 4.4 Hz, 1H), 7.09 (t, J = 7.7 Hz, 2H), 6.86 (d, J = 8.2 Hz, 2H), 6.67 (t, J = 7.3 Hz, 1H), 4.58 (s, 2H), 3.61 (q, J = 6.2 Hz, 2H), 3.14 (br s, 1H), 2.93 (br t, J = 6.2 Hz, 2H), 2.70 (m, 2H), 1.70 (d, J = 13.9 Hz, 2H). 13C NMR DMSO-d6, 100 MHz, 25 °C): δ 176.1, 165.4, 149.5, 149.2, 148.8, 143.5, 139.8, 135.7, 134.9, 131.5, 129.5, 129.4, 128.4, 127.3, 120.1, 118.0, 114.5, 59.1, 57.9, 56.6, 49.6, 36.6, 27.8. LCMS, single peak, 2.135min, ES-API: m/z 430.3 [M+H]. ES-HRMS calcd for C25H27N5O2Na (M+Na): 452.2062. Found, 452.2055.
- 18.Patchett AA, Nargund RP. Annu Rep Med Chem. 2000;35:289. and references cited therein. [Google Scholar]