

Abstract
The National Cancer Institute (NCI) Clinical Proteomic Tumor Analysis Consortium (CPTAC) is applying latest generation of proteomic technologies to genomically annotated tumors from The Cancer Genome Atlas (TCGA) program, a joint initiative of the NCI and National Human Genome Research Institute. By providing a fully integrated accounting of DNA, RNA and protein abnormalities in individual tumors, these datasets will illuminate the complex relationship between genomic abnormalities and cancer phenotypes, thus producing biological insights as well as a wave of novel candidate biomarkers and therapeutic targets amenable to verification using targeted mass spectrometry methods.
Keywords: Gene Expression, Cancer Proteomics, Protein Phosphorylation, Mass Spectrometry, Cancer Genome Atlas
Like any good experiment, cancer genomic analysis generates more questions than answers
The generation of comprehensive somatic mutation profiles of human tumors through massively parallel sequencing programs, such as The Cancer Genome Atlas (TCGA), has created an unprecedented opportunity for new biological insights and an expectation that some of these discoveries will be translated into improved clinical outcomes. This promise has already been realized in many excellent examples of precise therapies available for gain-of-function events such as amplifications (HER2 positive breast cancer), point mutations (C-RAF V600E positive melanoma), or translocations (BCR/ABL in chronic myeloid leukemia).
However the deluge of new data generated by DNA and RNA sequencing has often been more confusing than enlightening. The list of “significantly mutated genes” (SMG) that are recurrent at the level of point mutations or small insertions/deletions is startlingly short in many common epithelial cancers, with the genomic landscape dominated by loss-of-function mutations in tumor suppressor genes rather than the gain-of-function mutations in oncogenic protein kinases that have been so central to recent therapeutic successes. High grade serous ovarian cancer is an excellent example of this frustrating pattern, as besides TP53 mutation, which was present in almost all cases, the ovarian SMG list was limited to only nine other genes, none of which was an oncogenic kinase and all of which had low recurrence frequencies.
Basal-like breast cancer is very similar to serous ovarian cancer in this regard, but luminal-type breast cancer has a longer list of higher recurrence rate SMG including an oncogenic lipid kinase (PIK3CA), motivating active exploration of a number of new therapeutic directions (1). More commonly, however, the functions of somatic mutations identified through genomic analysis of common solid malignancies are very poorly understood, particularly in a disease and tissue-specific context and therefore do not drive therapeutic initiatives. Similarly, though chromosomal amplification and deletion events are thought to be critical driver events, the contributions of each of the often multiple genes within amplicons or deletions are often unclear. Complicating matters further is the recognition that in many instances several genes in one region may function in concert.
The vast majority of mutations are actually not in protein coding space but in regulatory sequences or other non-coding regions that could, nonetheless, be functionally relevant. Furthermore, recurrent clustered mutations in long non-coding RNAs such as MALAT1 are also emerging, but again the associated biochemistry is mysterious. One must therefore inescapably conclude that we still do not understand the biochemical and cell biological effects of most genomic alterations in cancer revealed by sequencing studies. Nor do we understand the combinatorial effects of many mutations that are present in any single cancer, as these are very hard to model. Our limited understanding of this complex molecular interplay renders even our therapeutic successes incomplete, since even when proteins harboring significant driver mutations can be successfully targeted, therapeutic resistance often develops because of the development of alternative pathways for cell survival and growth. Unbiased discovery of these resistance mechanisms is a clearly a priority if we are to translate pathway targeted approaches into curative regimens.
Informatics and pathway analyses based on DNA and RNA sequencing data generate hypotheses, not conclusions
Currently the cancer informatics field makes conclusions regarding cancer biology and signaling events by developing databases from a priori understandings of signal transduction or other biochemical studies. The genomic data typically are parsed into networks or “interactomes” and lists of biochemical activities that connect or map into pathways containing over- or under-expressed genes, amplified or deleted genes or mutant genes. These relationships are then interpreted as the identification of specific biological processes that drive clinical phenotypes. While clearly useful, the functional data used to annotate cancer informatics programs nevertheless may not be correct in the biological context of tumors. Since the actual biochemistry is inferred, but not directly observed, the outputs of these programs must be considered hypotheses, not conclusions (2). Thus, biochemical analyses that link genotype to phenotype are critical to the translational success of cancer genomics.
Proteomic technologies can bridge the gap between genotype and phenotype
The central dogma of biology places proteins and their functions as the direct mediators of phenotypic characteristics. Proteomic analyses therefore offer the means to measure the biochemical impact of cancer-related genomic abnormalities, including expression of variant proteins encoded by mutations, protein changes driven by altered DNA copy number, chromosomal amplification and deletion events, epigenetic silencing, and changes in microRNA expression. Analysis of protein post-translational modifications, particularly phosphorylation enables the detection of signaling network adaptations driven by genomic changes.
Proteomic technologies, which are based primarily on mass spectrometry (MS) have fundamentally advanced our knowledge of biochemistry and cell biology, including protein dynamics, multi-protein complexes and signaling networks (3) and have enabled systematic development of targeted assays for precise quantification of proteins in tissues and biofluids (4). MS analytical platforms are capable of providing both global profiles of protein expression and post-translational modification, as well as precise, targeted quantification of proteins and their modified and variant forms.
A complement to MS-based proteomics is the reverse phase protein array (RPPA) platform, which uses antibodies to probe printed arrays of tissue lysates and has been used recently in TCGA tumor analyses (5). The key advantage of RPPA is a small sample requirement and an ability to efficiently probe cancer-relevant signaling pathways. However, RPPA requires highly specific antibodies to reliably detect target analytes in unfractionated lysates. The number of phosphosite-specific antibodies remains very limited and few antibodies can distinguish between members of closely-related protein families that may, nonetheless have very different biological effects, such as AKT1, 2 and 3. Despite these constraints RPPA analyses have demonstrated utility and can be used to inform more in-depth pathway characterization by targeted MS approaches.
CPTAC and clinical proteomic technologies
The NCI launched the initial CPTAC program in 2006 as the Clinical Proteomic Technology Assessment for Cancer, designed to evaluate the performance of proteomic technology platforms for both global profiling and targeted quantitative analysis in tissues and biofluids. Major contributions of the first CPTAC program were:
Demonstration of the intra-laboratory reproducibility of unbiased, data dependent proteomic platforms for biological discovery, together with generation of reference materials and performance metrics for system assessment (6).
Confirmation of the ability of targeted protein quantification by multiple reaction monitoring (MRM) to achieve reproducible, precise quantification of protein levels in tissues and biofluids (7) across multiple laboratories throughout the CPTAC network. This group also developed Skyline, a widely used and community-supported open-source software platform for MRM assay design and data analysis on all commercial instrument platforms (8).
The renewal of the CPTAC program as the Clinical Proteomic Tumor Analysis Consortium shifted the focus from technology assessment to integrated cancer genomics and proteomics. The CPTAC centers are applying standardized proteome analysis platforms to analyze tumor tissues from TCGA, as well as unique cell and xenograft models and other tissue collections, all of which are accompanied by rich genomic datasets. The current CPTAC program is thus a broad, proof of concept initiative to bring high throughput proteomic technologies into the cancer genomics enterprise.
Five multi-institution Proteome Characterization Centers (PCCs) comprise CPTAC: 1) Broad Institute of MIT and Harvard/Fred Hutchinson Cancer Research Center/Massachusetts General Hospital; 2) Johns Hopkins University/Memorial-Sloan Kettering/Stanford University/University of Chicago; 3) Pacific Northwest National Laboratory/Massachusetts Institute of Technology/Oregon Health and Science University/University of Texas MD Anderson Cancer Center; 4) Vanderbilt University/Massachusetts Institute of Technology; 5) Washington University/University of North Carolina. Each team combines required capabilities in MS and related proteomic technologies, bioinformatics and biostatistics, cancer biology, and clinical/translational cancer research.
A CPTAC-Data Coordinating Center (CPTAC-DCC) provides a central repository for mass spectrometry raw data and metadata from biospecimens analyzed by the CPTAC PCCs. These data are available to the research community through the CPTAC Data Portal, which is accessed from the NCI Office of Cancer Clinical Proteomics Research web site (http://proteomics.cancer.gov/). In addition to the original (“raw”) MS datafiles, users can access peptide and protein assemblies, posttranslational modification maps and quantitative assay results processed through a standardized data analysis pipeline. The CPTAC-DCC incorporates standards (metadata, protein names, file formats) established by the Human Proteome Organization (HUPO), the Human Genome Organization (HUGO) and genomics resources to facilitate vertical integration of multiple data types to better define the molecular features of cancer.
Proteomic technology platforms in CPTAC
The CPTAC PCCs employ untargeted proteomics (also referred to as “discovery proteomics” or “shotgun proteomics”) for unbiased, global profiling of both protein expression and posttranslational modifications. Quantitative comparisons in data-dependent MS analyses are made by tagging peptides with isotope-encoded mass tags (e.g., iTRAQ reagents (9) or by a label-free, spectral counting approach (10). The iTRAQ-based approach is also applied for global phosphoproteomic profiling (9) and glycoprotein profiling, and can be extended to N-acetlated and ubiquitylated protein inventories. Shotgun proteomics analyses use a new generation of tandem hybrid Orbitrap and time-of-flight mass analyzers, which provide high resolution MS and tandem MS (MS/MS) analysis of peptides at high scan rates (10–100 Hz), yielding >10 MS/MS spectra per second (11). These instruments, when combined with intelligent sample processing and fractionation, enable both deep inventory and quantitative comparisons of complex peptide mixtures from cell, tissue and biofluid proteomes. For example, recent studies in the Broad and PNNL laboratories have yielded over 11,000 distinct proteins and more than 25,000 phosphosites from individual iTRAQ-labeled tumor samples. The number of distinct proteins identified and quantified in these experiments now rivals the number of transcripts observed in microarray experiments, while delivering valuable additional information on PTS hidden to genomic analyses.
The complementary technology to global profiling is targeted analysis by multiple reaction monitoring (MRM) (also termed selected reaction monitoring or “SRM”) (3, 4), which enables systematic development of quantitative protein assays through measurements of specific peptides in proteolytic digests. MRM technology overcomes a fundamental limitation of immunochemical methods—the availability of specific antibodies. Moreover, MRM enables selective quantification of variant/mutant or post-translationally modified sequences, both or which are difficult to achieve with antibodies.
MRM assays typically employ triple quadrupole instruments, but similar assays are beginning to be deployed on new, hybrid quadrupole-time-of-flight and quadrupole-orbitrap instruments that have higher resolution and mass precision than triple quadrupole instruments (12). A key strength of MRM assays, and one that further distinguishes it from antibody-based approaches, is the ability to monitor multiple peptides in a single analysis, thus providing for multiplexed quantification of as many as a hundred proteins in a single assay. For example, phosphotyrosine capture combined with MRM enabled quantitative and dynamics assessment of growth factor signaling networks (13) and effects of oncogenic mutations on tyrosine kinase networks (14, 15). MRM assays can be applied to simultaneously quantify wild type and variant protein forms (16) and the multiplexing capability supports simultaneous monitoring of multiple components in signaling pathways (17). Combining immunoaffinity enrichment of specific proteins and peptides with MRM enables systematic deployment of highly sensitive, specific assays (18). Similar sensitivities can be attained without immunoaffinity enrichment by combining another stage of HPLC separation with depletion of major serum proteins (e.g., LOD of 50 pg/mL for PSA (19).
Integrating genomic and proteomic data to understand breast, colon and ovarian cancer
Proteomic technologies are analogous to genomic technologies in providing broad inventories of diverse sequences and modified or variant forms, together with quantitative data that indicate expression and dynamics. Proteomics thus can be placed immediately downstream of genomics in a unified analysis scheme that describes the translation of genomic characteristics to functions and phenotypes (Figure 1). This creates important synergies in proteogenomic analysis. RNA sequencing data from individual tissue specimens can be used to generate customized databases for the identification of sequence variants at the protein level. Comparison of the identified sequence variants with dbSNP and cancer mutation databases distinguishes polymorphisms from somatic mutations and identifies peptides that arise from the unique genetic background of each patient. With appropriate alignment tools, variant peptides can indicate expression of novel gene structures associated with cancer-specific events. Determination of which DNA or RNA sequence level variants are expressed as proteins provides a basis for prioritizing mutations for further study of their contributions to cancer phenotypes.
Figure 1.
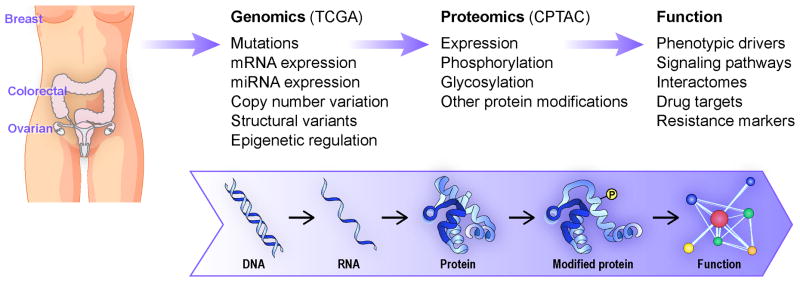
Proteogenomic integrated work flow for the Clinical Proteomic Tumor Analysis Consortium.
Integrated proteogenomics enables key insights into other genomic abnormalities. For example, quantitative comparisons at the copy number, RNAseq and protein expression levels can identify the gene drivers in focally amplified chromosomal segments. An obvious benefit of proteomic analysis—particularly in profiling phosphoproteins—is the direct measurement of the status of cancer signaling pathways (13). However, global protein expression and posttranslational modification profiling can also reveal unanticipated network and pathway adaptations to mutations, amplification and deletion events. Combining global proteome expression profiles with miRNA and mRNA data indicates mechanisms by which tumor-specific alterations in miRNA expression control gene expression (20).
The CPTAC PCCs initially conducted a series of due diligence studies characterizing analysis platform performance, optimizing sample preparation methods, establishing suitable controls, and scrutinizing the effects of ischemia on the stability of protein expression and phosphorylation. Having demonstrated the suitability of platforms, methodologies and samples, the next major objective of the CPTAC PCCs is the analysis of approximately one hundred genome-annotated samples each from breast, ovarian and colon/rectal cancer by global shotgun proteomic, phosphoproteomic and glycoproteomic profiling and targeted MRM analyses. The goals of these analyses are:
To generate rich proteomic datasets for these genome-annotated tumors with state of the art analysis platforms, together with quality control datasets from analyses of a common performance standard.
To identify differential protein expression characteristics that distinguish tumors of known biological subtypes (e.g., microsatellite instability in colon tumors or basal and luminal breast tumors) and potentially provide a new molecular taxonomy that can be compared with DNA and RNA based classification approaches.
To identify variant protein sequences corresponding to somatic mutations and to evaluate the relationship between mutation frequency and variant protein expression.
To determine how copy number variation translates into protein expression differences.
To evaluate the impact of genomic features on the status of signaling networks through direct analysis of phosphoprotein intermediates
To derive preliminary associations with clinical characteristics, such as platinum-resistance in ovarian cancer.
These analyses will comprise a strong proof of concept for the implementation of proteomic technology in tumor tissue analysis, but clearly are only a first step, since discoveries based on proteomics will require validation in independent cohorts. For this purpose, prospective tissue collections are underway for breast, ovarian and colon cancer. A key feature of these verification studies will be thorough documentation and, to the extent possible, control of relevant preanalytical variables, such as ischemia time. Cancer xenografts are available to the consortium and experimental perturbation studies with anticancer drugs are ongoing.
Finally, additional studies in more precise clinical contexts will begin. For example, in collaboration with the NSABP, jointly funded by the Breast Cancer Research foundation and CPTAC, we will accrue rapidly frozen samples from patients with HER2 positive breast cancer, both before and within 48 to 72 hours of receiving a dose of neoadjuvant treatment with paclitaxel and trastuzumab. The objective is to determine if the acute proteomic response to chemotherapeutic stress is predictive of treatment outcome. Similar studies are under consideration for colon and ovarian cancers. Other studies in CPTAC will focus on cancers sampled longitudinally during therapy, using the unique lens of proteomics to detect molecular adaptations that may be silent at the genome level. A baseline sample anchors quantitative comparisons of acute drug responses against the longer-term effects of the treatment. These studies will leverage high precision MRM assays, which may enable detection of important indicator proteins in the limiting protein amounts available from core needle biopsies.
Conclusion
The CPTAC program is deploying rapidly maturing proteomic technologies for analysis of fully genomically characterized tumors, to perform large scale comprehensive gene-to-protein integration in cancer for the first time. The combined use of genomic and proteomic data produces analytical strong synergy; more importantly, proteomic analyses can enable direct detection of the impact of genomic alterations. The emphasis on analysis of human tumors, rather than experimental model systems, is deliberate, minimizing the distance from data to clinical impact. An important lesson from the TCGA program is the need to plan in advance for validation studies by carefully accruing samples through precisely designed protocols suitable to address central unresolved questions in clinical oncology. Effective planning must engage the talents and resources of the wider clinical and scientific community. Thus, the fundamental purpose of this article is a call for collaboration between clinical investigators, basic scientists and computational biologists. All of the data from CPTAC studies will be made available to the research community, so that the value of this investment made by the National Cancer Institute in cancer proteomics will continue to grow.
Acknowledgments
Funding: U24CA160034 (Broad Institute; Fred Hutchinson Cancer Research Center, M.G., S.A.C., A.G.P), U24CA160036 (Johns Hopkins University), U24CA160019 (Pacific Northwest National Laboratory, R.D.S., K.K.R.), U24CA159988 (Vanderbilt University D.C.L.), U24CA160035 (Washington University, St. Louis; University of North Carolina, Chapel Hill, M.J.E., R.R.T).
Footnotes
The authors report no relevant conflicts of interest.
References
- 1.Ellis MJ, Perou CM. The genomic landscape of breast cancer as a therapeutic roadmap. Cancer Discov. 2013;3:27–34. doi: 10.1158/2159-8290.CD-12-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldstein TC, Paull EO, Ellis MJ, Stuart JM. Molecular pathways: extracting medical knowledge from high-throughput genomic data. Clinical cancer: an official journal of the American Association for Cancer Research. 2013;19:3114–20. doi: 10.1158/1078-0432.CCR-12-2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Picotti P, Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nature methods. 2012;9:555–66. doi: 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- 4.Gillette MA, Carr SA. Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nature methods. 2013;10:28–34. doi: 10.1038/nmeth.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Breast Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rudnick PA, Clauser KR, Kilpatrick LE, Tchekhovskoi DV, Neta P, Blonder N, et al. Performance metrics for liquid chromatography-tandem mass spectrometry systems in proteomics analyses. Mol Cell Proteomics. 2010;9:225–41. doi: 10.1074/mcp.M900223-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Addona TA, Abbatiello SE, Schilling B, Skates SJ, Mani DR, Bunk DM, et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat Biotechnol. 2009;27:633–41. doi: 10.1038/nbt.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26:966–8. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mertins P, Udeshi ND, Clauser KR, Mani DR, Patel J, Ong SE, et al. iTRAQ labeling is superior to mTRAQ for quantitative global proteomics and phosphoproteomics. Mol Cell Proteomics. 2012;11:M111 014423. doi: 10.1074/mcp.M111.014423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76:4193–201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 11.Michalski A, Damoc E, Lange O, Denisov E, Nolting D, Muller M, et al. Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol Cell Proteomics. 2012;11:O111 013698. doi: 10.1074/mcp.O111.013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gallien S, Duriez E, Crone C, Kellmann M, Moehring T, Domon B. Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol Cell Proteomics. 2012;11:1709–23. doi: 10.1074/mcp.O112.019802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolf-Yadlin A, Hautaniemi S, Lauffenburger DA, White FM. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc Natl Acad Sci U S A. 2007;104:5860–5. doi: 10.1073/pnas.0608638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang G, Fang B, Liu RZ, Lin H, Kinose F, Bai Y, et al. Mass spectrometry mapping of epidermal growth factor receptor phosphorylation related to oncogenic mutations and tyrosine kinase inhibitor sensitivity. J Proteome Res. 2011;10:305–19. doi: 10.1021/pr1006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai Y, Li J, Fang B, Edwards A, Zhang G, Bui M, et al. Phosphoproteomics identifies driver tyrosine kinases in sarcoma cell lines and tumors. Cancer research. 2012;72:2501–11. doi: 10.1158/0008-5472.CAN-11-3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halvey PJ, Ferrone CR, Liebler DC. GeLC-MRM quantitation of mutant KRAS oncoprotein in complex biological samples. J Proteome Res. 2012;11:3908–13. doi: 10.1021/pr300161j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Gruidl M, Remily-Wood E, Liu RZ, Eschrich S, Lloyd M, et al. Quantification of beta-catenin signaling components in colon cancer cell lines, tissue sections, and microdissected tumor cells using reaction monitoring mass spectrometry. J Proteome Res. 2010;9:4215–27. doi: 10.1021/pr1005197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Razavi M, Frick LE, LaMarr WA, Pope ME, Miller CA, Anderson NL, et al. High-throughput SISCAPA quantitation of peptides from human plasma digests by ultrafast, liquid chromatography-free mass spectrometry. J Proteome Res. 2012;11:5642–9. doi: 10.1021/pr300652v. [DOI] [PubMed] [Google Scholar]
- 19.Shi T, Fillmore TL, Sun X, Zhao R, Schepmoes AA, Hossain M, et al. Antibody-free, targeted mass-spectrometric approach for quantification of proteins at low picogram per milliliter levels in human plasma/serum. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:15395–400. doi: 10.1073/pnas.1204366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Q, Halvey PJ, Shyr Y, Slebos RJ, Liebler DC, Zhang B. Integrative omics analysis reveals the importance and scope of translational repression in microRNA-mediated regulation. Mol Cell Proteomics. 2013 doi: 10.1074/mcp.M112.025783. [DOI] [PMC free article] [PubMed] [Google Scholar]