

CXCL13 in Sjögren's syndrome increases with disease progression in mice and is elevated in human sera and saliva; blockade of CXCL13 ameliorates disease in an SS murine model.
Keywords: B-cell, CXCR5, saliva, sialadenitis
Abstract
SS is an autoimmune disease. pSS affects exocrine glands predominantly, whereas sSS occurs with other autoimmune connective tissue disorders. Currently, care for patients with SS is palliative, as no established therapeutics target the disease directly, and its pathogenetic mechanisms remain uncertain. B-cell abnormalities have been identified in SS. CXCL13 directs B-cell chemotaxis and is elevated in several autoimmune diseases. In this study, we tested the hypothesis that CXCL13 is elevated in SS in mice and humans and that neutralization of the chemokine ameliorates disease in a murine model. We assayed CXCL13 in mouse models and human subjects with SS to determine whether CXCL13 is elevated both locally and systemically during SS progression and whether CXCL13 may play a role in and be a biomarker for the disease. Cxcl13 expression in salivary tissue increases with disease progression, and its blockade resulted in a modest reduction in glandular inflammation in an SS model. We demonstrate that in humans CXCL13 is elevated in serum and saliva, and an elevated salivary CXCL13 level distinguishes patients with xerostomia. These data suggest a role for CXCL13 as a valuable biomarker in SS, as 74% of patients with SS displayed elevated CXCL13 in sera, saliva, or both. Thus, CXCL13 may be pathogenically involved in SS and may serve as a new marker and a potential therapeutic target.
Introduction
SS is an autoimmune disease that affects approximately 0.6% of the general population. pSS affects salivary and lacrimal glands predominantly, whereas sSS is seen with other autoimmune connective tissue diseases. SS-induced xerostomia and xerophthalmia exert a significant negative impact on the patient's quality of life, and serious systemic disease manifestations may also occur, including lymphoma [1–4]. Thus, both forms of SS entail significant morbidity for affected individuals.
SS has several well-characterized clinical manifestations. However, the disease is difficult to diagnose. Like other rheumatologic conditions, diagnosis is based on a compilation of numerous signs and symptoms [5, 6]. These include subjective and objective assessment of salivary and lacrimal gland function. In addition, anti-SSA (Ro) and anti-SSB (La) autoantibody titers are measured, and minor salivary glands are examined for the presence of periductal FLS. Notably, suggested new criteria for SS diagnosis include assessment of antinuclear antibody levels in addition to RF autoantibodies [6]. Despite these detailed criteria, the average patient experiences SS symptoms for 7.1 years before diagnosis [7]. Therefore, the diagnosis of SS is not straightforward, and improved diagnostic criteria for this immune dyscrasia would have significant clinical utility.
Given the debilitating nature of SS, there has been much effort to identify seminal events in disease development and factors that contribute to progression. B cells are implicated in the pathogenesis of SS because they stimulate T cells, secrete inflammatory cytokines, and produce autoantibodies that focus inflammation. Significantly, B cells also influence differentiation of Th cells [8]. In SS, B cells comprise 20% of the lymphocytes present in salivary tissue, and their number increases with disease progression [9]. Patients with SS have aberrant B-cell profiles in peripheral blood [10–14]. Thus, these studies suggest a key role for B cells in SS.
CXCL13 is a homeostatic chemokine that regulates B-cell movement. Aberrant lymphocyte recruitment and subsequent tissue damage have been noted in several autoimmune diseases, including SS [15, 16]. CXCL13 is secreted by follicular stromal cells, antigen-experienced T cells, and Th follicular cells [17–19]. Cxcl13 directs B cells to lymphoid follicles, as mice deficient in Cxcl13 or its receptor, Cxcr5, fail to form these structures [20, 21]. Moreover, overexpression of Cxcl13 causes formation of ectopic lymphoid tissue composed primarily of B cells [22], suggesting the possibility that CXCL13 may recruit B cells to diseased salivary glands.
Thus, CXCL13 may be pathogenically involved in SS progression, and levels of CXCL13 may reflect disease severity. Accordingly, CXCL13 may serve as a biomarker for SS, as suggested for other autoimmune diseases [23–25]. We found that serum and salivary gland Cxcl13 increases with disease progression in several SS murine models. Furthermore, Cxcl13 is elevated before disease becomes evident in one SS model, and administration of neutralizing anti-Cxcl13 antibody diminishes the disease. We found that elevated CXCL13 levels are expressed by 74% of patients with SS in either serum or saliva. Thus, CXCL13 may be valuable as a biomarker for initial diagnosis, in the assessment of disease progression and severity, and as a therapeutic target.
MATERIALS AND METHODS
Mice
BALB/c, MRL/MpJ, NOD/ShiLtJ (NOD), B6.129S-Id3tm1Zhu/J (Id3−/−), and C57BL/6 mice were used (Jackson Laboratory, Bar Harbor ME, USA). The mice were cared for and handled in accordance with institutional animal care and use committee (IACUC) and U.S. NIH guidelines.
Serum collection
Sera from SS mice were harvested by cardiac puncture immediately after euthanasia, before disease developed, and at early- and late-stage time points. Blood was allowed to clot for 2 h and centrifuged at 2000 rpm for 20 min. Serum was removed and stored at −20°C until use. Healthy age and sex-matched BALB/c and C57BL/6 mice were used as controls.
Isolation of salivary tissue
Salivary tissue was removed after euthanasia. Tissue was fixed in 10% buffered formalin or frozen for IHC or mRNA analysis, respectively.
Cell isolation, flow cytometry, and sorting
B cells were isolated from spleens and cervical lymph nodes of age-matched female BALB/c and NOD mice. SMG tissue from NOD animals was isolated as previously described [26]. Splenocytes, cervical lymph node cells, and SMGs were stained by immunofluorescence for B220, CD4, (BD Biosciences, Franklin Lakes, NJ, USA), and F4/80 (Invitrogen, Carlsbad, CA, USA). Enriched B cells (B220+, CD4−, and F4/80−) were sort purified with an Influx instrument (BD Biosciences). T-cells (CD4+, B220−, and F4/80−) and macrophages (F4/80+, B220−) were sort purified similarly. Macrophages in SMG tissue were fluorescently stained with F4/80 (Invitrogen), and flow cytometry was performed on a Gallios (Beckman Coulter, Brea CA, USA). Samples were analyzed using FlowJo software (Tree Star).
RNA isolation and quantitative PCR
RNeasy kits (Qiagen, Valencia, CA, USA) were used to isolate mRNA from sorted cells and salivary tissue. cDNA synthesis was performed with an iScript kit (Bio-Rad, Hercules, CA, USA). Quantitative (q)PCR was performed with SYBR Green (Bio-Rad), as previously described (Applied Biosystems, Foster City, CA, USA) [27]. Primers were as follows: Cxcl13 and Cxcr5 have been published elsewhere [28, 29]. Cd19: forward: 5′-ACCAGTACGGGAATGTGCTC-3′, reverse: 5′-GACTTGAATGCGTGGATTT-3′; and Cd4: forward: 5′-TCGCCTTCGCAGTTTGATC-3′, reverse: 5′-CACCCACAACTCCACCTCCT-3′. PCR settings were as follows: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Relative expression was calculated with the ΔCt method, where R = 2[Ct sample − Ct control]. All samples were analyzed in duplicate, and expression was normalized to actin.
IHC and histopathology
Formalin-fixed, paraffin-embedded tissue was H&E stained on a Symphony automated platform (Ventana, Tuscon, AZ, USA). IHC was performed with an IntelliPATH autostainer (BioCare Medical, Concord, CA, USA), with a Cxcl13 (5 μg/mL) antibody (R&D Systems, Minneapolis, MN, USA).
ELISAs
CXCL13 was quantified by ELISA (R&D Systems). Sera and saliva were harvested as described earlier and diluted according to the manufacturer's instructions. All samples and standards were analyzed in duplicate.
CXCL13 Neutralization
Antibody generation.
Swiss Webster mice were immunized with KLH-conjugated CXCL13 and the spleens harvested. Hybridomas were generated by fusion of splenocytes with SP2/0 myeloma cells according to standard procedures, resulting in production of a monoclonal anti-CXCL13-neutralizing antibody (mAb 5378) that also bound mouse Cxcl13 (anti-CXCL13/Cxcl13). The IgG2a control antibody was generated from hybridoma 2B8 (American Type Culture Collection [ATTC], Manassas, VA, USA).
Antibody specificity.
Plates were precoated with recombinant chemokine (85–125 nM) (PeproTech, Rocky Hill, NJ, USA). Primary antibodies were used as follows: mouse IgG2a isotype control and anti-CXCL13/Cxcl13 (mAb 5378); anti-CXCL13 (mAb801); anti-Cxcl13 (mAb470) (R&D Systems); anti-CXCL8/IL-8; anti-CXCL10/IP-10; anti-CXCL12/SDF-1α; and anti-CXCL9/MIG (PeproTech). HRP-conjugated secondary antibodies (goat anti-mouse or rabbit anti-mouse IgG [Jackson ImmunoResearch, West Grove, PA, USA]rsqb] or goat anti-rat IgG [Bethyl Laboratories, Montgomery, TX, USA]) were added, and samples were developed with TMB (tetramethylbenzidine) substrate (BD Biosciences). Absorbance was read at 450/570 nm.
CXCL13 migration assay.
C57BL/6 splenocytes (1×107 cells/mL) were washed and resuspended with chemotaxis buffer (RPMI 1640 with l-glutamine, 10 mM HEPES, 1% penicillin/streptomycin, and 0.5% BSA). Cxcl13 (5 μg/mL), with anti-CXCL13/Cxcl13 (mAb 5378) or IgG2a isotype control antibody (50 μg/mL), was added to Transwell tissue culture plates (5 μm pore size, 6.5 mm diameter) (Corning Costar, Corning, NY, USA). Splenocytes (1×106) were also added, and the plates were incubated for 2 h at 37° C. The cells were stained with alamar blue (BioSource, Camarillo, CA, USA), and fluorescence was measured at 530/590 nm. Cxcl12 (0.1 μg/mL) was used as a negative control for anti-CXCL13/Cxcl13 binding. The migration index was calculated as follows: [specific migration (chemokine+antibody)]/spontaneous migration.
Antibody administration.
NOD females were given 100 μg anti-CXCL13/Cxcl13 (mAb 5378) or control IgG2a antibody by intraperitoneal injection 3 times weekly for 12 weeks, beginning at 4 weeks of age. This dosage schedule was based on a previously published study in which Cxcl13 neutralization was performed in an arthritis model [30]. Animals were euthanized at 16 weeks of age. Sera and saliva were collected as described earlier. Spleens, cervical lymph nodes, and salivary tissue were harvested.
Assessment of disease development and quantification of FLS.
Disease severity was assessed by lymphocytic infiltration of H&E-stained tissue. The presence of periductal FLS within SMG tissue was considered indicative of SS and was quantified as described elsewhere [5].
Human subject recruitment and specimen collection
Recruitment.
Patients with a confirmed diagnosis of SS according to the European American Consensus Criteria were recruited [5]. Thirty-five (n=27 pSS, n=8 sSS) fulfilled the criteria. Serum and saliva samples were also collected from 21 healthy control subjects. The healthy control group consisted of individuals similar in age and sex to the patients with SS. Control subjects with autoimmune disease and those taking immunosuppressive medications were excluded. All clinical investigation was conducted according to the principles of the Declaration of Helsinki. All human studies were approved by the North Shore-LIJ Health System and SUNY at Buffalo institutional review boards. Written, informed consent was obtained from participants before study inclusion.
Patients' characteristics.
We recruited 27 patients with pSS (age range, 31–84, median, 59, F=25, M=2) and 21 healthy control subjects (age range, 36–70, median, 54; F=20, M=1). While most patients with pSS were not taking immunosuppressants, 7 were taking hydroxychloroquine, and CXCL13 levels in saliva and sera in those patients were similar to levels in other patients with SS included in the study (data not shown). We recruited 8 individuals with sSS whom we included in the salivary CXCL13 studies (age range, 24–70, median, 58, F=8). Four patients had RA, and 2 had SLE. One patient had autoimmune hepatitis, and 1 had ulcerative colitis. Six of 8 individuals with sSS were receiving immunosuppressants. In most of the patients recruited, SS was diagnosed by serologic studies. Only 5 individuals had a salivary gland biopsy performed (n=3 pSS, n=2 sSS), allowing for diagnosis in each case. Five patients with SS produced no unstimulated saliva, and thus salivary CXCL13 levels could not be assessed. The patients' demographics, characteristics, and treatment regimens are shown in Supplemental Table S1.
Serum and saliva collection.
Blood was collected by venipuncture, and the sera were frozen at −80°C until use. Unstimulated whole saliva was collected for 5 min. The patients were instructed to refrain from eating, drinking, or smoking for 1 hour before saliva collection and to discontinue sialogogue use on the day of the appointment. Saliva was placed on ice and frozen at −80°C until use.
Statistical analyses.
The Mann-Whitney U test and Student's t test were used to compare groups. The Spearman correlation coefficient showed correlations between variables. All graphing was performed with GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA).
Methods for online supplemental material
Flow cytometry.
Lymphocytes were isolated from spleens and cervical lymph nodes of age-matched female BALB/c and NOD mice by mechanical disruption. SMG tissue from NOD animals was isolated, as previously described [26]. Splenocytes, cervical lymph node cells, and SMGs were stained by immunofluorescence for B220 and CXCR5 (BD Biosciences). Flow cytometry was performed on a FACSCalibur (BD Biosciences). Samples were analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
Chemotaxis assays.
For chemotaxis assays, splenocytes were isolated from 16-week-old NOD animals and age and sex-matched BALB/c controls. Recombinant murine Cxcl13 (500 nM) (PeproTech) was added to the lower chamber of a Transwell tissue culture plate in the presence or absence of antibody (mAb 5378 or IgG2a isotype control). Splenocytes (1×106) were added to the top chamber, and assays were carried out as described above.
RESULTS
Cxcl13 and its receptor, Cxcr5, are expressed in murine salivary tissue in SS
Initially, we sought to determine whether Cxcl13 is elevated within salivary tissue of diseased mice. Three models of SS were used: one primary (Id3−/−) and two secondary (MRL/MpJ and NOD). SS models display characteristic FLS in advanced disease by histological analysis of SMG tissue, and this is similar to findings in minor salivary glands from patients with SS. Mice and humans have 3 major salivary glands: parotid, sublingual, and SMG. We focused our analyses on the SMG, since this is the largest salivary gland in mice and it shows FLS characteristic of SS [31]. We found that Cxcl13 was expressed in diseased SMG tissue of all SS models examined (Fig. 1). Cxcl13 transcripts were slightly elevated in the SMG, even prior to lymphocytic infiltration, and were significantly elevated at early and late stage disease in NOD animals (Fig. 1C). Finally, we assayed both protein and gene expression of Cxcr5. We found that expression of Cxcr5 also increased with disease progression (Fig. 1D).
Figure 1. Expression of Cxcl13 and its receptor, Cxcr5, increased with disease progression in salivary tissue of SS mice.

(A) Id3−/− mice (n=5, 41–54 wk). (B) MRL/MpJ mice (n=5, 50 wk) with late-stage disease were euthanized and the salivary glands harvested. (C, D) Cxcl13 and Cxcr5 in SMG tissue was quantified from NOD animals before disease onset (pre-disease, n=5, 4 wk) and in early-stage (n=4, 12 wk) and late-stage (n=5, 16 wk) disease. SMGs from age and sex-matched BALB/c and C57BL/6 mice were used as controls. The tissue was homogenized, and relative Cxcl13 and Cxcr5 transcript levels were quantified by qPCR. Each sample was analyzed in duplicate, and expression was normalized to actin. Significance was determined by Student's t test; horizontal line: sample mean. *P<0.05; **P<0.01, ***P<0.001.
To determine whether Cxcr5 protein is also elevated in advanced-stage disease, we examined tissue from NOD spleen, cervical lymph nodes, and SMG. Cxcr5 expression in NOD splenocytes was elevated, when compared with that in BALB/c controls. Moreover, we detected Cxcr5 protein expression in SMG tissue, although we were unable to compare it to levels in the BALB/c animals, as Cxcr5 expression is limited to lymphocytes, and such cells are absent in healthy salivary tissue [32] (Supplemental Fig. S1). These findings suggest that Cxcl13 contributes to the recruitment of lymphocytes to salivary tissue through interactions with Cxcr5-expressing cells in secondary lymphoid organs and salivary tissue.
To determine the cellular source of Cxcl13, we examined salivary tissue from NOD mice by IHC. We found that Cxcl13 was associated with seromucous acini in SMG tissue in early- and late-stage disease. In contrast, much lower levels of Cxcl13 were detectable within the lymphoid infiltrates. BALB/c SMG tissue showed no Cxcl13 staining (Fig. 2A and data not shown). To confirm and extend these findings, B and T cells, macrophages, and stromal/parenchymal cells were isolated from diseased NOD SMG tissue and tested for Cxcl13 gene expression. Cxcl13 transcripts in macrophages and parenchymal/stromal cells were greater than in lymphocytes (Fig. 2B). Moreover, the percentage of macrophages was increased approximately 3-fold in NOD salivary tissue in advanced disease (8.47%), when compared with that in the BALB/c control (2.76%) (Fig. 2C). Taken together, data from Figs. 1 and 2 show that Cxcl13 is elevated within salivary tissue in early-stage disease and increases with disease progression, suggesting that Cxcl13 expression by macrophages and parenchymal tissue may play a crucial role in the initiation and development of SS.
Figure 2. Macrophages and parenchymal cells in SMG salivary tissue expressed Cxcl13 in late-stage disease.
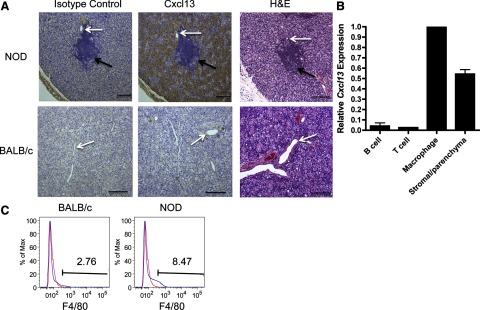
(A) Immunohistochemical staining for salivary gland Cxcl13 was performed on SMG tissue from animals with late-stage disease (16 wk). Brown stain: Cxcl13 expression. White arrows: salivary ducts; black arrows: periductal lymphoid aggregates. Isotype control staining from 3 NOD animals with late-stage disease is shown. All samples were counterstained with hematoxylin (blue); (×100); scale bar = 200 μm. (B) Relative Cxcl13 transcript levels were quantified by qPCR of SMG tissue from female NOD mice with late-stage disease (n=5). Each sample was analyzed in duplicate, and expression was normalized to actin. Cxcl13 levels for each of the populations is shown relative to macrophage expression. Results were compiled from 2 separate experiments. Error bars = sem. (C) Pooled submandibular salivary tissue from BALB/c (n=5, 16 wk) and NOD (n=10, 16 wk) mice was fluorescently stained with isotype control (red trace) or the macrophage marker F4/80 (blue trace).
SS models show elevated CXCL13 in sera
To determine whether circulating Cxcl13 is detectable in SS models and whether it relates to disease progression, we examined serum samples by ELISA at various time points in the disease course. We found notably elevated serum Cxcl13 in early-(MRL/MpJ) or late-stage (NOD) disease in sSS mice (Fig. 3B, C). Moreover, Cxcl13 was elevated in a subset of animals with pSS (Id3−/−), although the significance of this finding is unclear (Fig. 3A). Healthy strain- and age-matched control mice showed slight increases in Cxcl13 levels as they aged, but this increase was significantly less than that in SS models (Fig. 3), suggesting that serum Cxcl13 is a specific marker of SS.
Figure 3. Mouse models of SS expressed Cxcl13 in sera.

Sera were collected as follows: (A) Healthy C57BL/6 controls (8–10 wk, n=5, 48–62 wk, n=5) and Id3−/− mice before disease onset (pre-disease; 12–14 wk n=13) and in early-stage (20–28 wk, n=6) and late-stage (35–46 wk, n=14) disease. (B) Healthy C57BL/6 controls (8–10 wk, n=5, 48–62 wk, n=5) and MRL/MpJ (pre-disease [12 wk, n=4] and early-stage [20 wk, n=5] and late-stage [50 wk, n=5] disease). Scale bar = 200 μm. (C) Healthy BALB/c controls (4 wk, n=5, 16 wk, n=5) and NOD mice (pre-disease [4 wk, n=10] and early-stage [14 wk, n=6] and late-stage [20 wk, n=6] disease). Cxcl13 levels were quantified by ELISA, and each sample was analyzed in duplicate. Note the expanded scales in (A) and (B). Significance was determined with Student's t test; horizontal line: sample mean; *P < 0.05; **P < 0.01; ***P < 0.005; N.S., not significant.
CXCL13 neutralization reduces salivary gland inflammation
To determine whether blockade of Cxcl13 mitigates disease severity, we developed a monoclonal antibody raised against human CXCL13 that recognizes mouse Cxcl13 with similar avidity (Fig. 4A). Dose-dependent neutralization of Cxcl13 by this antibody was confirmed by migration assay (Fig. 4B, C). We also confirmed inhibition of splenocyte migration with this antibody with cells from NOD animals with advanced-stage disease. Of note, the kinetics of inhibition were similar in these animals, as compared with splenocytes from age and sex-matched control mice (Supplemental Fig. S2).
Figure 4. Anti-CXCL13 antibody bound Cxcl13 specifically and inhibited migration in response to Cxcl13.

Binding of the anti-CXCL13/Cxcl13 monoclonal antibody (mAb 5378) to the following ligands was assessed with microtiter ELISA assays: (A) CXCL13, Cxcl13, CXCL8, CXCL10, CXCL12, or CXCL9. Cognate ligands are shown in the top left panel of each graph, and receptors and controls are shown on the x-axes. Each sample was analyzed in duplicate; representative data from 3 comparable experiments are shown. (B) Splenocyte migration in response to mAb 5378 (50 μg/mL) was tested in the presence of Cxcl13 (5 μg/mL) or Cxcl12 (0.1 μg/mL) in Transwell culture plates. Each sample was analyzed in triplicate; representative data from 1 of 3 comparable experiments are shown. Error bars = sem. (C) Splenocytes were incubated with Cxcl13 (5 μg/mL) in the presence of increasing concentrations of mAb 5378 or control IgG2a. The titration curve was fitted by using a 4-parameter sigmoidal curve (R2=0.99).
We then administered this neutralizing antibody 3 times a week starting at 4 weeks of age and assessed SS development. We found that the animals receiving anti-CXCL13/Cxcl13 antibody exhibited significantly fewer lymphocytic foci within SMG tissue than in control-treated animals (Fig. 5A, B). To confirm and extend this finding, we quantified B- and T-cell markers in SMG tissue using Cd19 and Cd4, respectively. Cd19 expression decreased significantly in the animals that received neutralizing antibody, as compared to the control-treated animals (Fig. 5C), presumably reflecting recruitment of fewer salivary B cells. In contrast, Cd4 was not decreased significantly in the animals treated with anti-CXCL13/Cxcl13, as compared with those given control antibody (Fig. 5D). IHC studies of SMG tissue using the B-cell marker B220 revealed equivalent numbers of B cells within the lymphocytic foci of both treatment groups, although the total number of focus scores was reduced in animals receiving Cxcl13 blockade (Fig. 5A, B, and data not shown). Neutralization of Cxcl13 did not affect the total number of B cells or the percentage of B-cell subsets in spleen and cervical lymph nodes, as determined by histological analysis and flow cytometry (data not shown). Thus, Cxcl13 blockade reduced salivary gland inflammation and did not appear to disrupt lymphocytic populations in secondary lymphoid organs.
Figure 5. Cxcl13 blockade reduced salivary gland inflammation.

Female NOD mice were treated with either a control (n=8) or Cxcl13-neutralizing (mAb 5378) (n=9) antibody for 12 wk beginning at 4 wk of age. Animals received 100 μg of antibody 3 times weekly. (A, B) Periductal focal lymphocytic sialadenitis (focus scores) were quantified in control and anti-CXCL13/Cxcl13 (mAb 5378)-treated animals by analysis of H&E-stained SMG tissue. Focus scores were normalized by dividing the number of scores by the average area examined in each treatment group. Representative SMG tissue from a control and an anti-CXCL13/Cxcl13-treated animal is shown (×100). Scale bar = 200 μm. (C, D) Cd19 and Cd4 expression was assessed in SMG tissue. Tissue was homogenized and relative expression quantified by qPCR. Each sample was analyzed in duplicate; normalized expression relative to actin is shown. Significance was determined with the Mann-Whitney U test; horizontal line: sample mean. *P < 0.05; N.S., not significant.
Patients with pSS have elevated serum CXCL13
To determine whether patients with spontaneously appearing pSS, such as genetically determined murine disease, express elevated levels of circulating CXCL13, we analyzed serum with ELISA. Strikingly, we found that these patients registered significantly more serum CXCL13 than the healthy controls (Fig. 6A). In the patients with pSS, the mean CXCL13 level was 162 ± 184 pg/mL (mean ± sd) (range, 12.5–672 pg/mL). In the normal, healthy controls, the mean serum CXCL13 level was 26.8 ± 22.8 pg/mL (range, 0–78.4 pg/mL). The CXCL13 levels did not correlate with years since diagnosis or patients' ages (data not shown).
Figure 6. Serum and salivary CXCL13 levels were elevated in patients with SS, and salivary CXCL13 levels distinguished patients with xerostomia.
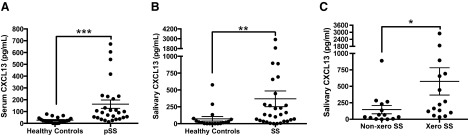
(A) Sera were collected from patients with pSS (n=27) and healthy controls (n=21). Significance was determined with the Mann-Whitney U test. (B) Saliva was collected from patients with SS (pSS, n=23; sSS, n=6) and healthy controls (n=20). Significance was determined with the Mann-Whitney U test. (C) Salivary CXCL13 levels from SS patients with xerostomia (Xero) (n=15, 52%) were compared to SS patients with normal salivary flow (Non-xero) (n=14, 48%). Significance was determined with the Mann-Whitney U test. CXCL13 levels were quantified by ELISA, and each sample was analyzed in duplicate. Horizontal line: sample mean; error bars = sem. *P = 0.0174; **P = 0.0016; ***P < 0.0001.
Serum CXCL13 in healthy controls was consistently low, suggesting that CXCL13 is a specific marker of inflammation and a hallmark of the aberrant immune response in SS, although studies with larger patient cohorts are needed to determine the absolute serum CXCL13 level that is indicative of disease. In the current study, we defined elevated CXCL13 as being at least 2 sd above the mean healthy control level. Thus, levels greater than 72.4 pg/mL were considered elevated. Among the patients with pSS, 56% demonstrated elevated levels of serum CXCL13 (Fig. 6A), although considerable variability was evident at the upper end.
Patients with SS have elevated salivary CXCL13
To determine whether patients with SS express elevated salivary CXCL13, we analyzed unstimulated saliva from diseased and healthy control subjects by ELISA (Fig. 6B). We found that the patients with SS expressed more salivary CXCL13 (mean, 368±631 pg/mL; range, 0–2902) as compared with healthy controls (mean, 26.7±36.8 pg/mL; range, 0–132.8). Two of our healthy control subjects had well-controlled type 2 diabetes. Both individuals had elevated salivary CXCL13 (287.7 and 578.0 pg/mL), although this association is not reported in the literature. Since these values were clearly aberrant, we excluded those individuals from our analyses. Elevated CXCL13 was defined as described for serum. We regarded salivary CXCL13 levels above 100.3 pg/mL as elevated following exclusion of the aforementioned outliers. Of the measured salivary CXCL13 levels, 57% were elevated, by the above criteria. CXCL13 levels in saliva did not correlate with years since diagnosis or with serum CXCL13 levels (data not shown). Of note, the patients with elevated salivary CXCL13 encompass some but not all of the SS patients with elevated serum CXCL13. When both values are considered together, 74% of the patients express elevated CXCL13.
Salivary CXCL13 levels are higher in xerostomic patients with SS
We sought to determine whether elevated levels of either serum or salivary CXCL13 correlate with xerostomia in patients with SS. Patients with unstimulated salivary flow ≤0.15 mL/min were classified as xerostomic in accordance with the literature [33]. Of note, the diagnostic criteria for SS do not require xerostomia [5]. We divided the patients with SS into 2 groups, those with frank xerostomia and those with no xerostomia. Only sera from patients with pSS were analyzed, in order to attribute serum CXCL13 to SS specifically. Xerostomic patients with pSS (P=13, 48%) had similar serum CXCL13 levels compared to patients with pSS who were non-xerostomic (n=14, 52%). However, xerostomic patients with SS (n=15, 52%) demonstrated higher salivary CXCL13 levels than those that had normal salivary production (n=14, 48%) (Figure 6C and data not shown). These data suggest that CXCL13 levels in saliva are reflective of or related to marked salivary gland dysfunction, and may be indicative of a subset of patients with more severe oral manifestations of SS.
DISCUSSION
In this study, CXCL13 increased with disease progression in salivary tissue and serum from SS models, and neutralization of Cxcl13 reduced glandular inflammation in the NOD model. Moreover, work in patients with pSS demonstrates that the disease is reflected in elevated serum and salivary CXCL13 in most patients. Together, these results suggest that CXCL13 may be a novel marker of disease in addition to a potential therapeutic target in the mitigation of glandular inflammation characteristic of SS.
In the current study, we showed significant Cxcl13 expression by macrophages in submandibular salivary glands of mice with advanced disease. Consistent with this, in a recent report, significant macrophage infiltration was recorded in the lacrimal glands of the Aire-deficient SS murine model. Depletion of macrophages improved the ocular phenotype, suggesting an important and previously unappreciated role for macrophages in the pathogenesis of SS [34]. Although the precise role of these cells in SS is poorly understood, macrophages may contribute to disease in several ways. They may become activated through removal of apoptotic debris and may initiate or exacerbate chronic inflammation [35, 36]. Accordingly, it is well established that proapoptotic markers are upregulated in salivary tissue in SS [37]. Thus, therapeutic targeting of monocyte chemotaxis and migration may ameliorate disease through alteration of tissue-localized CXCL13 [29].
In keeping with this, we explored the therapeutic potential of CXCL13 blockade by developing and then administering a Cxcl13-neutralizing antibody to NOD animals. Cxcl13 blockade reduced FLS in SMG tissue, suggesting that Cxcl13 plays an important role in directing B cells to salivary tissue in disease. These findings are further supported by data showing amelioration of SS after neutralization of Ltrβ signaling, which is upstream of Cxcl13 [38–40]. In this study, Cxcl13 levels, along with several other chemokines were reduced after treatment with anti-Ltrβ antibody [39, 40], suggesting that the amelioration of SS by Ltrβ blockade may be due, at least in part, to diminished Cxcl13 expression. These findings are in contrast to those in a similar study in which a Cxcl13-neutralizing antibody was administered to NOD animals, and the effect on development of diabetes was assessed. Blockade of Cxcl13 did not prevent diabetes or diminish the formation of tertiary lymphoid tissue in the pancreas, although B-cell organization in these structures was disrupted [41]. It is important to note that in our study, the animals were injected before disease developed; we were therefore unable to evaluate whether Cxcl13 blockade is an effective therapeutic for the clinical treatment of SS, although the results suggest that Cxcl13 is an integral player in the development of salivary gland inflammation and of B-cell infiltration in particular.
Since Cxcl13 increased with disease progression in salivary tissue and serum in SS and its blockade ameliorated SMG inflammation, we conducted human studies to determine whether CXCL13 may serve as a biomarker in SS. Of note, CXCL13 was elevated in the serum and saliva of most of the patients with SS. CXCL13 is elevated in numerous autoimmune diseases, including MS, RA, and SLE [30, 42–46]. In keeping with these results, previous reports indicate that CXCL13 has clinical utility as a biomarker for assessment of disease activity [23–25]. Thus, while detection of this chemokine in serum is not specific for SS, CXCL13 is clearly emerging as a potential biomarker of immune dysfunction.
Salivary diagnostics is an emerging field, and a point-of-care test to detect proteins indicative of SS would enhance the diagnostic algorithm significantly. Our results indicate that quantification of saliva in conjunction with salivary CXCL13 levels may identify SS in patients, and distinguish between patients with SS-induced xerostomia and those who have medication-induced or perceived xerostomia in the absence of autoimmunity. Moreover, CXCL13 measurements may have predictive value and may mark patients who will go on to develop more serious oral disease manifestations, although further longitudinal studies are necessary to confirm these findings.
Recent studies have detailed significant differences in salivary proteins between healthy individuals and those with SS [47, 48], in addition to detection of anti-Ro and La autoantibodies in saliva [49]. While such results are encouraging, it is still unknown whether patients with other types of autoimmune conditions express elevated inflammatory mediators in saliva and whether salivary proteins can be used to distinguish SS from other rheumatologic diseases. Thus, further studies are needed in a larger and more diverse cohort of patients to determine whether salivary diagnostics provide the specificity and sensitivity necessary to detect SS disease and to distinguish among different immune system dyscrasias.
The discovery of novel markers and diagnostic methods is crucial for the diagnosis and management of SS, as disease-staging criteria are lacking, and therefore there is no standardized way to monitor SS progression. The significant individual variability that is seen in patients with SS at both the clinical and molecular levels further complicates monitoring of the disease. Results from our murine studies suggest that Cxcl13 is expressed relatively early in disease, even prior to lymphocytic infiltration of glandular tissue. Therefore, CXCL13 may be a useful marker to diagnose and stage the disease or to gauge its severity. Such early markers may be particularly beneficial, in that they may allow for early targeting of disease mechanisms before irreversible glandular destruction.
To identify therapeutic targets, it is crucial to elucidate disease mechanisms linked to SS development. Many patients with SS are characterized by an IFN type I gene signature [50–52]. The triggers for this IFNα expression remain poorly understood, but this cytokine upregulates several genes, including CXCL13 and B-cell activating factor of the tumor necrosis factor family (BAFF) [53, 54]. Studies in mice and humans alike point to an important role for BAFF in the pathogenesis of SS [55–57], and BAFF is thought to contribute to the expansion of pathogenic B cells in disease by provision of aberrant survival signals. BAFF also enhances CXCL13-mediated chemotaxis in normal human B cells, and this effect is particularly strong in the memory compartment [58]. Studies of human pSS salivary gland tissue reveal elevated BAFF and CXCL13 [10, 59, 60], and B cells expressing CXCR5 and the BAFF receptor (BAFFR) are also detected in salivary tissue [16, 59]. While CXCL13 directs B-cell recruitment, it is not known whether signals from BAFF and CXCL13 interact to drive pathology in SS. However, it is plausible that BAFF prolongs survival of pathogenic B cells and that CXCL13 serves to recruit these cells to glandular tissue in an aberrant manner, resulting in disease exacerbation. Neutralization of BAFF is currently in clinical trials for treatment of SS [61], and therapeutics that target both mediators concomitantly may be more effective in reducing B-cell-driven pathology than those that target BAFF alone.
In conclusion, we suggest a relationship exists between CXCL13, salivary gland inflammation and loss of salivary function. While the relationship between xerostomia and glandular inflammation is well studied [62], less is known regarding the link between xerostomia and the salivary indicators of disease. In the current study, xerostomic patients with SS had elevated levels of salivary CXCL13, as compared to non-xerostomic patients. Thus, therapeutics directed against CXCL13 and CXCR5 may help to restore or reduce the loss of salivary function in such patients, as suggested by our work in murine models.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the nurses at The Feinstein Institute for Medical Research General Clinic Research Center and Ms. Karen Falkner (Department of Oral Biology, SUNY at Buffalo School of Dental Medicine) for facilitating the collection of blood from research subjects; Dr. Cristina Sison of the Feinstein Institute for Medical Research for help with statistical analyses; Ms. Anne Loosen and Dr. Richard Furie, North Shore-LIJ Health System, for assistance in patient recruitment; Dr. John E. Fantasia for clinical guidance; Dr. Xuemei Zhong (Hematology and Oncology Section, Department of Medicine Core Facilities, Boston University Medical Center, MA) and Yana Moses, Department of Pathology, Long Island Jewish Medical Center, for assistance with IHC. We are indebted to the patients with SS and healthy control subjcts who provided samples that were integral to the study.
This work was supported by a Mentored Clinical Scientist Research Career Development Award (K08 DE020882) from the National Institute of Dental and Craniofacial Research (NIDCR) to J.M.K., grant R01AI29690 from the U.S. National Institutes of Health (NIH), National institute of Allergy and Infectious Diseases (NIAID) awarded to T.L.R., The Feinstein Institute for Medical Research, North Shore-LIJ Health System General Clinical Research Center, and grant M01 RR018535 from the U.S. NIH, National Center for Research Resources (NCRR). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIDCR, NIAID, NCRR, or NIH.
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- BAFF
- B-cell activating factor of the tumor necrosis factor family
- BAFFR
- B-cell activating factor of the tumor necrosis factor family receptor
- FLS
- focal lymphocytic sialadenitis
- IHC
- immunohistochemistry
- KLH
- keyhole limpet hemocyanin
- Ltrβ
- lymphotoxin receptor β
- MS
- multiple Sclerosis
- pSS
- primary Sjögren's syndrome
- qPCR
- quantitative PCR
- RA
- rheumatoid arthritis
- RF
- rheumatoid factor
- SLE
- systemic lupus erythematosus
- SMG
- submandibular gland
- SS
- Sjögren's syndrome
- sSS
- secondary Sjögren's syndrome
AUTHORSHIP
J.M.K., E.K., and T.L.R. designed and executed experiments and contributed to the writing and critical reading of the manuscript.
DISCLOSURES
J.M.K. and T.L.R. have no financial conflicts of interest. E.K. is an employee of Vaccinex, Incorporated, and declares no other financial conflicts of interest.
REFERENCES
- 1. Seror R., Ravaud P., Bowman S. J., Baron G., Tzioufas A., Theander E., Gottenberg J. E., Bootsma H., Mariette X., Vitali C., Force, Eular Sjögren's Task Force. (2010) EULAR Sjögren's syndrome disease activity index: development of a consensus systemic disease activity index for primary Sjögren's syndrome. Ann. Rheum. Dis. 69, 1103–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. García-Carrasco M., Sisó A., Ramos-Casals M., Rosas J., de la Red G., Gil V., Lasterra S., Cervera R., Font J., Ingelmo M. (2002) Raynaud's phenomenon in primary Sjögren's syndrom: prevalence and clinical characteristics in a series of 320 patients. J. Rheumatol. 29, 726–730 [PubMed] [Google Scholar]
- 3. Ekström Smedby K., Vajdic C. M., Falster M., Engels E. A., Martínez-Maza O., Turner J., Hjalgrim H., Vineis P., Seniori Costantini A., Bracci P. M., Holly E. A., Willett E., Spinelli J. J., La Vecchia C., Zheng T., Becker N., De Sanjosé S., Chiu B. C., Dal Maso L., Cocco P., Maynadié M., Foretova L., Staines A., Brennan P., Davis S., Severson R., Cerhan J. R., Breen E. C., Birmann B., Grulich A. E., Cozen W. (2008) Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: a pooled analysis within the InterLymph Consortium. Blood 111, 4029–4038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fox R. I. (2005) Sjögren's syndrome. Lancet 366, 321–331 [DOI] [PubMed] [Google Scholar]
- 5. Vitali C., Bombardieri S., Jonsson R., Moutsopoulos H. M., Alexander E. L., Carsons S. E., Daniels T. E., Fox P. C., Fox R. I., Kassan S. S., Pillemer S. R., Talal N., Weisman M. H. (2002) Classification criteria for Sjögren's syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann. Rheum. Dis. 61, 554–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shiboski S. C., Shiboski C. H., Criswell L., Baer A., Challacombe S., Lanfranchi H., Schiødt M., Umehara H., Vivino F., Zhao Y., Dong Y., Greenspan D., Heidenreich A. M., Helin P., Kirkham B., Kitagawa K., Larkin G., Li M., Lietman T., Lindegaard J., McNamara N., Sack K., Shirlaw P., Sugai S., Vollenweider C., Whitcher J., Wu A., Zhang S., Zhang W., Greenspan J., Daniels T., Groups, Sjögren's Syndrome International Collaborate Clinical Alliance Research Groups. (2012) American College of Rheumatology classification criteria for Sjögren's syndrome: a data-driven, expert consensus approach in the Sjögren's International Collaborative Clinical Alliance cohort. Arthritis Care Res. 64, 475–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Segal B., Bowman S. J., Fox P. C., Vivino F. B., Murukutla N., Brodscholl J., Ogale S., McLean L. (2009) Primary Sjögren's syndrome: health experiences and predictors of health quality among patients in the United States. Health Qual. Life. Outcomes 7, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhong X., Gao W., Degauque N., Bai C., Lu Y., Kenny J., Oukka M., Strom T. B., Rothstein T. L. (2007) Reciprocal generation of Th1/Th17 and T(reg) cells by B1 and B2 B cells. Eur. J. Immunol. 37, 2400–2404 [DOI] [PubMed] [Google Scholar]
- 9. Youinou P. (2008) Sjögren's syndrome: a quintessential B cell-induced autoimmune disease. Joint Bone Spine 75, 1–2 [DOI] [PubMed] [Google Scholar]
- 10. Hansen A., Gosemann M., Pruss A., Reiter K., Ruzickova S., Lipsky P. E., Dorner T. (2004) Abnormalities in peripheral B cell memory of patients with primary Sjögren's syndrome. Arthritis Rheum. 50, 1897–1908 [DOI] [PubMed] [Google Scholar]
- 11. Bohnhorst J. O., Bjorgan M. B., Thoen J. E., Natvig J. B., Thompson K. M. (2001) Bm1-Bm5 classification of peripheral blood B cells reveals circulating germinal center founder cells in healthy individuals and disturbance in the B cell subpopulations in patients with primary Sjögren's syndrome. J. Immunol. 167, 3610–3618 [DOI] [PubMed] [Google Scholar]
- 12. Binard A., Le Pottier L., Devauchelle-Pensec V., Saraux A., Youinou P., Pers J. O. (2008) Is the blood B-cell subset profile diagnostic for Sjögren's syndrome? Ann. Rheum. Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Youinou P., Mackenzie L., le Masson G., Papadopoulos N. M., Jouquan J., Pennec Y. L., Angelidis P., Katsikis P., Moutsopoulos H. M., Lydyard P. M. (1988) CD5-expressing B lymphocytes in the blood and salivary glands of patients with primary Sjögren's syndrome. J. Autoimmun. 1, 185–194 [DOI] [PubMed] [Google Scholar]
- 14. Zeher M., Surányi P., Nagy G., Szegedi G. (1990) B cells expressing CD5 in minor labial salivary glands of patients with primary Sjögren's syndrome. Arthritis Rheum. 33, 453. [DOI] [PubMed] [Google Scholar]
- 15. Rotondi M., Chiovato L., Romagnani S., Serio M., Romagnani P. (2007) Role of chemokines in endocrine autoimmune diseases. Endocr. Rev. 28, 492–520 [DOI] [PubMed] [Google Scholar]
- 16. Hansen A., Reiter K., Ziprian T., Jacobi A., Hoffmann A., Gosemann M., Scholze J., Lipsky P. E., Dorner T. (2005) Dysregulation of chemokine receptor expression and function by B cells of patients with primary Sjögren's syndrome. Arthritis Rheum. 52, 2109–2119 [DOI] [PubMed] [Google Scholar]
- 17. Manzo A., Vitolo B., Humby F., Caporali R., Jarrossay D., Dell'accio F., Ciardelli L., Uguccioni M., Montecucco C., Pitzalis C. (2008) Mature antigen-experienced T helper cells synthesize and secrete the B cell chemoattractant CXCL13 in the inflammatory environment of the rheumatoid joint. Arthritis Rheum. 58, 3377–3387 [DOI] [PubMed] [Google Scholar]
- 18. Gunn M. D., Ngo V. N., Ansel K. M., Ekland E. H., Cyster J. G., Williams L. T. (1998) A B-cell-homing chemokine made in lymphoid follicles activates Burkitt's lymphoma receptor-1. Nature 391, 799–803 [DOI] [PubMed] [Google Scholar]
- 19. Cyster J. G., Ansel K. M., Reif K., Ekland E. H., Hyman P. L., Tang H. L., Luther S. A., Ngo V. N. (2000) Follicular stromal cells and lymphocyte homing to follicles. Immunol. Rev. 176, 181–193 [DOI] [PubMed] [Google Scholar]
- 20. Forster R., Mattis A. E., Kremmer E., Wolf E., Brem G., Lipp M. (1996) A putative chemokine receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell 87, 1037–1047 [DOI] [PubMed] [Google Scholar]
- 21. Ansel K. M., Harris R. B., Cyster J. G. (2002) CXCL13 is required for B1 cell homing, natural antibody production, and body cavity immunity. Immunity 16, 67–76 [DOI] [PubMed] [Google Scholar]
- 22. Luther S. A., Lopez T., Bai W., Hanahan D., Cyster J. G. (2000) BLC expression in pancreatic islets causes B cell recruitment and lymphotoxin-dependent lymphoid neogenesis. Immunity 12, 471–481 [DOI] [PubMed] [Google Scholar]
- 23. Sellebjerg F., Bornsen L., Khademi M., Krakauer M., Olsson T., Frederiksen J. L., Sorensen P. S. (2009) Increased cerebrospinal fluid concentrations of the chemokine CXCL13 in active MS. Neurology 73, 2003–2010 [DOI] [PubMed] [Google Scholar]
- 24. Rioja I., Hughes F. J., Sharp C. H., Warnock L. C., Montgomery D. S., Akil M., Wilson A. G., Binks M. H., Dickson M. C. (2008) Potential novel biomarkers of disease activity in rheumatoid arthritis patients: CXCL13, CCL23, transforming growth factor alpha, tumor necrosis factor receptor superfamily member 9, and macrophage colony-stimulating factor Arthritis Rheum. 58, 2257–2267 [DOI] [PubMed] [Google Scholar]
- 25. Lee H. T., Shiao Y. M., Wu T. H., Chen W. S., Hsu Y. H., Tsai S. F., Tsai C. Y. Serum BLC/CXCL13 concentrations and renal expression of CXCL13/CXCR5 in patients with systemic lupus erythematosus and lupus nephritis. J. Rheumatol. 37, 45–52 [DOI] [PubMed] [Google Scholar]
- 26. Nauntofte B., Dissing S. (1987) Stimulation-induced changes in cytosolic calcium in rat parotid acini. Am. J. Physiol. 253, G290–G297 [DOI] [PubMed] [Google Scholar]
- 27. Zhong X., Tumang J. R., Gao W., Bai C., Rothstein T. L. (2007) PD-L2 expression extends beyond dendritic cells/macrophages to B1 cells enriched for V(H)11/V(H)12 and phosphatidylcholine binding. Eur. J. Immunol. 37, 2405–2410 [DOI] [PubMed] [Google Scholar]
- 28. Lee B. P., Chen W., Shi H., Der S. D., Förster R., Zhang L. (2006) CXCR5/CXCL13 interaction is important for double-negative regulatory T cell homing to cardiac allografts. J. Immunol. 176, 5276–5283 [DOI] [PubMed] [Google Scholar]
- 29. Schiffer L., Bethunaickan R., Ramanujam M., Huang W., Schiffer M., Tao H., Madaio M. P., Madaio M. M., Bottinger E. P., Davidson A. (2008) Activated renal macrophages are markers of disease onset and disease remission in lupus nephritis. J. Immunol. 180, 1938–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zheng B., Ozen Z., Zhang X., De Silva S., Marinova E., Guo L., Wansley D., Huston D. P., West M. R., Han S. (2005) CXCL13 neutralization reduces the severity of collagen-induced arthritis. Arthritis Rheum. 52, 620–626 [DOI] [PubMed] [Google Scholar]
- 31. Delaleu N., Nguyen C. Q., Peck A. B., Jonsson R. (2011) Sjögren's syndrome: studying the disease in mice. Arthritis Res. Ther. 13, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Forster R. (2000) CXCR5. In Cytokine Reference, Vol. 2 (Oppenheim J., Feldmann J., Durum M., Hirano S. K., Vilcek T., Nicos J., eds.), N. A. Academic, New York: 2019–2024 [Google Scholar]
- 33. Sreebny L. M. (2000) Saliva in health and disease: an appraisal and update. Int. Dent. J. 50, 140–161 [DOI] [PubMed] [Google Scholar]
- 34. Zhou D., Chen Y. T., Chen F., Gallup M., Vijmasi T., Bahrami A. F., Noble L. B., van Rooijen N., McNamara N. A. (2012) Critical involvement of macrophage infiltration in the development of Sjögren's Syndrome-associated dry eye. Am. J. Pathol. 181, 753–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moreth K., Brodbeck R., Babelova A., Gretz N., Spieker T., Zeng-Brouwers J., Pfeilschifter J., Young M. F., Schaefer R. M., Schaefer L. (2010) The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. J. Clin. Invest. 120, 4251–4272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sica A., Mantovani A. (2012) Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, 787–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tzioufas A. G., Kapsogeorgou E. K., Moutsopoulos H. M. (2012) Pathogenesis of Sjögren's syndrome: what we know and what we should learn. J. Autoimmun. 39, 4–8 [DOI] [PubMed] [Google Scholar]
- 38. Ngo V. N., Korner H., Gunn M. D., Schmidt K. N., Riminton D. S., Cooper M. D., Browning J. L., Sedgwick J. D., Cyster J. G. (1999) Lymphotoxin alpha/beta and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. J. Exp. Med. 189, 403–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fava R. A., Kennedy S. M., Wood S. G., Bolstad A. I., Bienkowska J., Papandile A., Mavragani C. P., Gatumu M., Skarstein K., Kelly J. A., Browning J. L. (2011) Lymphotoxin-beta Receptor blockade reduces CXCL13 in lacrimal glands and improves corneal integrity in the NOD model of Sjögren's syndrome. Arthritis Res. Ther. 13, R182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gatumu M. K., Skarstein K., Papandile A., Browning J. L., Fava R. A., Bolstad A. I. (2009) Blockade of lymphotoxin-beta receptor signaling reduces aspects of Sjögren syndrome in salivary glands of non-obese diabetic mice. Arthritis Res. Ther. 11, R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Henry R. A., Kendall P. L. (2010) CXCL13 blockade disrupts B lymphocyte organization in tertiary lymphoid structures without altering B cell receptor bias or preventing diabetes in nonobese diabetic mice. J. Immunol. 185, 1460–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aust G., Sittig D., Becherer L., Anderegg U., Schutz A., Lamesch P., Schmucking E. (2004) The role of CXCR5 and its ligand CXCL13 in the compartmentalization of lymphocytes in thyroids affected by autoimmune thyroid diseases. Eur. J. Endocrinol. 150, 225–234 [DOI] [PubMed] [Google Scholar]
- 43. Bagaeva L. V., Rao P., Powers J. M., Segal B. M. (2006) CXC chemokine ligand 13 plays a role in experimental autoimmune encephalomyelitis. J. Immunol. 176, 7676–7685 [DOI] [PubMed] [Google Scholar]
- 44. Meraouna A., Cizeron-Clairac G., Panse R. L., Bismuth J., Truffault F., Tallaksen C., Berrih-Aknin S. (2006) The chemokine CXCL13 is a key molecule in autoimmune myasthenia gravis. Blood 108, 432–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sato T., Ishikawa S., Akadegawa K., Ito T., Yurino H., Kitabatake M., Yoneyama H., Matsushima K. (2004) Aberrant B1 cell migration into the thymus results in activation of CD4 T cells through its potent antigen-presenting activity in the development of murine lupus. Eur. J. Immunol. 34, 3346–3358 [DOI] [PubMed] [Google Scholar]
- 46. Shi K., Hayashida K., Kaneko M., Hashimoto J., Tomita T., Lipsky P. E., Yoshikawa H., Ochi T. (2001) Lymphoid chemokine B cell-attracting chemokine-1 (CXCL13) is expressed in germinal center of ectopic lymphoid follicles within the synovium of chronic arthritis patients. J. Immunol. 166, 650–655 [DOI] [PubMed] [Google Scholar]
- 47. Baldini C., Giusti L., Ciregia F., Da Valle Y., Giacomelli C., Donadio E., Sernissi F., Bazzichi L., Giannaccini G., Bombardieri S., Lucacchini A. (2011) Proteomic analysis of saliva: a unique tool to distinguish primary Sjögren's syndrome from secondary Sjögren's syndrome and other sicca syndromes. Arthritis Res. Ther. 13, R194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ambatipudi K. S., Swatkoski S., Moresco J. J., Tu P. G., Coca A., Anolik J. H., Gucek M., Sanz I., Yates J. R., Melvin J. E. (2012) Quantitative proteomics of parotid saliva in primary Sjögren's syndrome. Proteomics 12, 3113–3120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ching K. H., Burbelo P. D., Gonzalez-Begne M., Roberts M. E., Coca A., Sanz I., Iadarola M. J. (2011) Salivary anti-Ro60 and anti-Ro52 antibody profiles to diagnose Sjögren's Syndrome. J. Dent. Res. 90, 445–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brkic Z., Maria N. I., van Helden-Meeuwsen C. G., van de Merwe J. P., van Daele P. L., Dalm V. A., Wildenberg M. E., Beumer W., Drexhage H. A., Versnel M. A. (2012) Prevalence of interferon type I signature in CD14 monocytes of patients with Sjögren's syndrome and association with disease activity and BAFF gene expression. Ann. Rheum. Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mavragani C. P., Crow M. K. (2010) Activation of the type I interferon pathway in primary Sjögren's syndrome. J. Autoimmun. 35, 225–231 [DOI] [PubMed] [Google Scholar]
- 52. Gottenberg J. E., Cagnard N., Lucchesi C., Letourneur F., Mistou S., Lazure T., Jacques S., Ba N., Ittah M., Lepajolec C., Labetoulle M., Ardizzone M., Sibilia J., Fournier C., Chiocchia G., Mariette X. (2006) Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjögren's syndrome. Proc. Natl. Acad. Sci. U. S. A. 103, 2770–2775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Low H. Z., Witte T. (2011) Aspects of innate immunity in Sjögren's syndrome. Arthritis Res. Ther. 13, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Segal B. M., Nazmul-Hossain A. N., Patel K., Hughes P., Moser K. L., Rhodus N. L. (2011) Genetics and genomics of Sjögren's syndrome: research provides clues to pathogenesis and novel therapies. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 111, 673–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Killedar S. J., Killedar S. Y., Eckenrode S. E., McIndoe R. A., She J. X., Nguyen C. Q., Peck A. B., Cha S. R., Cha S. (2006) Early pathogenic events associated with Sjögren's syndrome (SjS)-like disease of the NOD mouse using microarray analysis. Lab. Invest. 86, 1243–1260 [DOI] [PubMed] [Google Scholar]
- 56. Groom J., Kalled S. L., Cutler A. H., Olson C., Woodcock S. A., Schneider P., Tschopp J., Cachero T. G., Batten M., Wheway J., Mauri D., Cavill D., Gordon T. P., Mackay C. R., Mackay F. (2002) Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjögren's syndrome. J. Clin. Invest. 109, 59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Szodoray P., Jellestad S., Teague M. O., Jonsson R. (2003) Attenuated apoptosis of B cell activating factor-expressing cells in primary Sjögren's syndrome. Lab. Invest. 83, 357–365 [PubMed] [Google Scholar]
- 58. Badr G., Borhis G., Lefevre E. A., Chaoul N., Deshayes F., Dessirier V., Lapree G., Tsapis A., Richard Y. (2008) BAFF enhances chemotaxis of primary human B cells: a particular synergy between BAFF and CXCL13 on memory B cells. Blood 111, 2744–2754 [DOI] [PubMed] [Google Scholar]
- 59. Daridon C., Devauchelle V., Hutin P., Le Berre R., Martins-Carvalho C., Bendaoud B., Dueymes M., Saraux A., Youinou P., Pers J. O. (2007) Aberrant expression of BAFF by B lymphocytes infiltrating the salivary glands of patients with primary Sjögren's syndrome. Arthritis Rheum. 56, 1134–1144 [DOI] [PubMed] [Google Scholar]
- 60. Hansen A., Lipsky P. E., Dörner T. (2007) B cells in Sjögren's syndrome: indications for disturbed selection and differentiation in ectopic lymphoid tissue. Arthritis Res. Ther. 9, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Coca A., Sanz I. (2012) Updates on B-cell immunotherapies for systemic lupus erythematosus and Sjögren's syndrome. Curr. Opin. Rheumatol. 24, 451–456 [DOI] [PubMed] [Google Scholar]
- 62. Daniels T. E., Cox D., Shiboski C. H., Schiødt M., Wu A., Lanfranchi H., Umehara H., Zhao Y., Challacombe S., Lam M. Y., De Souza Y., Schiødt J., Holm H., Bisio P. A., Gandolfo M. S., Sawaki T., Li M., Zhang W., Varghese-Jacob B., Ibsen P., Keszler A., Kurose N., Nojima T., Odell E., Criswell L. A., Jordan R., Greenspan J. S., Groups S., Sjögren's International Collaborative Clinical Alliance Research Groups. (2011) Associations between salivary gland histopathologic diagnoses and phenotypic features of Sjögren's syndrome among 1,726 registry participants. Arthritis Rheum. 63, 2021–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.