

A novel molecular mechanism underlying arrest of monocyte/ macrophage migration important for the physiological maintenance of leukocytes at the site of inflammation.
Keywords: granulocyte, monocyte signaling cascade, chemotaxis, gene expression regulation, cell migration
Abstract
Monocytes and neutrophils infiltrate into tissues during inflammation and stay for extended periods of time until the initial insult is resolved or sometimes remain even longer in the case of chronic inflammation. The mechanism as to why phagocytes become immobilized after the initial cell migration event is not understood completely. Here, we show that overexpression or hyperactivation of Rac2 decreases sustained chemotactic responses of macrophages to MCP-1/CCL2. The resulting leukocyte arrest is not caused by a diminished availability of the cytokine receptor CCR2 that remains intact during MCP-1 stimulation. We show a novel mechanism that links the Rac2-dependent arrest of chemotaxis to decreased expression of PLD2 through the transcription regulator Sp1. Prolonged Rac2 activity leads to nuclear overactivation of Sp1, which acts as a repressor for PLD2. Also, another signaling component plays a regulatory role: β-catenin. Although early times of stimulation (∼20 min) with MCP-1/CCL2 resulted in activation of β-catenin with a positive effect on PLD2, after ∼3 h of stimulation, the levels of β-catenin were reduced and not able to prevent the negative effect of Rac2 on PLD2 activity. This is a novel molecular mechanism underlying immobilization of monocyte/macrophage migration that is important for the physiological maintenance of leukocytes at the site of inflammation. If this immobilization is prolonged enough, it could lead to chronic inflammation.
Introduction
Chemotaxis is the complex physiological process comprising directional movement of cells toward a chemical stimulus present in the immune system to combat against infectious pathogens, tumor cells [1], organogenesis, and homeostasis [2, 3]. Chemokines released by cells in and around damage bind to leukocyte receptors, which activate cell migration toward foreign microorganisms. However, excessive or misdirected cell migration is a major pathological cause for several human diseases and disorders, such as asthma (eosinophils, mast cells), Crohn's disease (TH1 cells), multiple sclerosis (TH1 cells, monocytes), myocardial infarction (neutrophils), cardiac stroke (neutrophils), atherosclerosis (monocytes), rheumatoid arthritis (monocytes, TH1 cells, neutrophils), type I diabetes (TH1 cells, CD8), and hepatitis (CD8) [4].
MCP-1, a member of the small inducible gene family, is also known as CCL2 (C-C motif), which is a small cytokine that mediates monocyte chemotaxis specifically. It plays a key role in the recruitment of monocytes to sites of injury and infectious diseases that have an overabundance of monocyte/macrophage tissue infiltrates, particularly psoriasis, rheumatoid arthritis, and atherosclerosis. MCP-1/CCL2 acts in target cells after binding to its cell membrane receptor, CCR2 [5, 6]. One of the signaling events initiated after receptor activation is the activation of small Rho GTPases, such as Rac [7, 8]. Membrane ruffles and the leading edges of motile cells express a high content of Rac1 and Rac2 and their endogenous GEFs (epidermal growth factor receptor kinase substrate 8, Abelson interactor 1, and son of sevenless homolog 1) [9, 10]. Additionally, Rac2 has been shown to be active in the regulation of gene expression [11].
PLD is a signaling protein involved in leukocyte chemotaxis [12–15]. There are two isoforms of PLD in humans, PLD1 and PLD2. As PLD2 is a membrane-bound protein that has a dual function as a lipase and as a GEF [16, 17], we investigated whether PLD2-derived activation of Rac2 would play a role in long-term chemotaxis and accumulation of leukocytes in chronic inflammation. The function of PLD2 as a lipase is to catalyze the hydrolysis of PC into PA and choline. The PA generated from this reaction is used further as a signaling molecule in cell migration and phagocytosis [16]. Within the PLD2 PH domain, there are two CRIB domains that are responsible for the binding of PLD2 to Rac2 [16, 18]. However, little is known about the regulation of PLD2 expression during extended periods of agonist cell starvation.
Our present study was aimed at understanding the molecular mechanism as to why leukocytes cease to chemotax and are retained in tissues. Although this mechanism is beneficial initially for the leukocytes to localize to tissue and mount an effective inflammatory response, it can also provoke a pathological problem if inflammation is chronically sustained. We chose to study this problem in macrophage cells that were stimulated by MCP-1/CCL2. As MCP-1/CCL2 is implicated in pathogenesis of inflammatory diseases that have an overabundance of monocyte/macrophages infiltrates (such as psoriasis, rheumatoid arthritis, and atherosclerosis), we reasoned that MCP-1/CCL2 could signal to PLD2 through Rac2. Unexpectedly, we found that Rac2 negates gene regulation of PLD2 expression, and this helps to explain why macrophages would not move and probably remain in tissue infiltrates long after the initial chemotactic stimulus has ceased to exist.
MATERIALS AND METHODS
Materials
DMEM was from Mediatech (Manassas, VA, USA). Lipofectamine and Plus reagent were from Invitrogen (Carlsbad, CA, USA). siQUEST transfection reagent was from Mirus (Madison, WI, USA). Mouse anti-PLD2 IgG (clone SKB2) and mouse anti-Rac2 antibodies were from Millipore (Billerica, MA, USA). Rabbit anti-EGFR, anti-JAK3, anti-Fes, anti-β-catenin, and anti-actin antibodies were from Cell Signaling Technology (Danvers, MA, USA). Purified murine rMCP-1 was from PeproTech (Rocky Hill, NJ, USA). [3H]-Butanol was from American Radiolabeled Chemicals (St. Louis, MO, USA). [32P]-γATP was from Perkin-Elmer (Waltham, MA, USA). siNeg and siRac2 were from Applied Biosystems (Foster City, CA, USA).
Cells and cell culture
RAW264.7 macrophages were obtained from American Type Culture Collection (Manassas, VA, USA). The RAW264.7 LR5 macrophage subline was a kind gift from Dianne Cox (Albert Einstein School of Medicine, Bronx, NY, USA). Both cell lines were cultured in DMEM, supplemented with 10% (v/v) FBS at 37°C in a 5% CO2 atmosphere.
Plasmid constructs, DNA transfection, and gene silencing
The plasmids used in these experiments were as follows: pcDNA3.1-mycPLD2-WT and pRK5-mycRac2-WT. Transfections were performed using 2 μg plasmid DNA, 5 μl Lipofectamine (Invitrogen), and 5 μl Plus reagent (Invitrogen) in Opti-MEM medium (Invitrogen), per the manufacturer's protocol. RAW264.7/LR5 macrophages were transfected for 3 h, washed, re-fed with prewarmed complete medium, and maintained for 48 h. Rac2 or Sp1 expression in cells was knocked down using siRNA (200 nM) in combination with the siQUEST transfection reagent (6 μl). For Rac2, we used a pool of three target-specific, 20–25 nt siRNAs designed to knock down Rac2 gene expression. Sequence is not specified for proprietary reasons by the manufacturer (Santa Cruz Biotechnology, Santa Cruz, CA, USA). For Sp1, we used a siRNA that targeted Exons 4–5 (locus 242578; Invitrogen). A Neg for siRNA was also from Invitrogen.
Real-time qRT-PCR
Total RNA was isolated from RAW264.7 macrophages with the RNeasy minikit. RNA concentrations were determined by fluorometric assay with Packard's Fusion and RiboGreen RNA quantitation kit, and samples were normalized to 20 ng/μl RNA. Reverse transcription was performed with 210 ng RNA, 210 ng random hexamers, 500 μM dNTPs, 84 U RNaseOUT, and 210 U MMLV RT and incubated at 42°C for 55 min. qRT-PCR reactions were run with 30 ng total input RNA (5 μl) and 2 μl of the PLD2 gene expression assay (FAM-labeled), multiplexed with the housekeeping gene (β-tubulin, Texas Red-labeled), with the final concentrations at 200 and 400 pmol for the primers and probe, respectively. Primers and fluorescent probes were synthesized by Integrated DNA Technologies (Coralville, IA, USA). qRT-PCR conditions for the Stratagene Cycler were 95°C for 3 min and then 50 cycles of the next three steps: 30 s at 95°C, 1 min at 60°C, and then 1 min at 72°C. The Ct values were chosen from the linear part of the PCR amplification curve, where an increase in fluorescence can be detected at >10 se above the background signal. ΔCt was calculated as: ΔCt = average PLD Ct − average housekeeping Ct, and gene-fold expression was calculated as 2 − (ΔΔCt) = 2 − (experimental condition ΔCt−control ΔCt).
PLD activity assay
RAW264.7/LR5 cell lysates were processed for PLD activity in PC8 liposomes and [3H]-butanol, beginning with the addition of the following reagents (final concentrations): 3.5 mM PC8 phospholipid, 45 mM HEPES (pH 7.8), and 1.0 μCi [3H]-butanol in a liposome form, as indicated in ref. [19], to accomplish the transphosphatidylation reaction of PLD. Samples were incubated for 20 min at 30°C with continuous shaking. Addition of 0.3 ml ice-cold chloroform/methanol (1:2) stopped the reactions. Lipids were then isolated and resolved by thin-layer chromatography. The amount of [3H]-PBut that comigrated with PBut standards (Rf=0.45–0.50) was measured by scintillation spectrometry.
Immunoprecipiation/Western blotting
RAW264.7/LR5 cells were transfected with the expression plasmids pcDNA-HAPLD2 (hemagglutinin-tagged) and/or pRK5-mycRac2 for 2 days. Cells were sonicated in special lysis buffer (5 mM HEPES, 1 mM leupeptin, 768 nM aprotinin, 100 μM sodium orthovanadate, and 0.4% Triton X-100 or 0.5% digitonin). After sonication, aliquots of the supernatant were cleared and immunoprecipitated with anti-actin antibodies. Bead-associated protein complexes were resolved by SDS-PAGE, transferred to a PVDF membrane, followed by immunoblot analysis with anti-HA, anti-myc, or anti-actin antibodies, and used for Western blot via chemiluminescence.
Cell migration assay
RAW264.7/LR5 macrophages were resuspended at a density of 5 × 105 cells/ml in chemotaxis buffer (HBSS, supplemented with 0.5% BSA). A total of 200 μl was placed in the upper chambers (or inserts) of transwell inserts that were separated from the lower wells by a 6.5-mm diameter, 8 μm pore-size polycarbonate membrane. For the study of chemotaxis, MCP-1 was prepared fresh the day of the experiment in 1× PBS-0.5% BSA, pH, 7.2, at a stock concentration of 1 mM. When ready for chemotaxis, MCP-1 was diluted to a 3-nM working concentration in 500 μl chemotaxis buffer and placed into the lower wells of 24-well plates. Cell migration inserts were incubated for 1 h at 37°C under a 5% CO2 atmosphere. The number of cells that migrated to the lower wells was calculated by placing 10 μl aliquots on a hemocytometer and counting four fields in duplicate.
Immunofluorescence of chemotaxing RAW264.7/LR5 macrophages
RAW264.7/LR5 macrophages that were used for chemotaxis, as described above, were fixed onto coverslips using 4% PFA for 10 min at room temperature, permeabilized with 0.5% Triton X-100 in PBS for 10 min at room temperature, and then incubated in 10% FCS-0.1% Triton X-100 in PBS for up to 4 h at room temperature. PLD2 and Rac2 were detected in these cells using a goat anti-PLD2 (N-20 antibody) IgG primary antibody overnight at 4°C, followed by incubation with a donkey anti-goat FITC-conjugated IgG secondary antibody for 1 h at room temperature or a goat anti-Rac2 IgG primary antibody overnight at 4°C, followed by incubation with a donkey anti-goat TRITC-conjugated IgG secondary antibody for 1 h at room temperature. Nuclei were stained using at a 1:2000 dilution of DAPI in PBS for 5 min at room temperature. Coverslips were mounted onto glass microscope slides using VectaShield mounting medium, and cells were visualized using a Nikon 50 Eclipse epifluorescence microscope.
Statistical analysis
Data are presented as mean ± sem. The difference between means was assessed by the single-factor ANOVA test. Probability of P < 0.05 indicated a significant difference.
RESULTS
Rac2 signals a temporal switch that arrests cell chemotaxis
To determine if Rac2 overexpression had a physiological effect on macrophages, cell chemotaxis was measured by transwell assay in cells that had been transfected to overexpress PLD2-WT, Rac2-WT, or PLD2-WT + Rac2-WT (Fig. 1A). Overexpression of PLD2-WT resulted in significantly increased cell migration. However, cells that overexpressed PLD2-WT and Rac2-WT exhibited first, a positive effect on migration at early times (∼20 min), whereas at later times (120 or 150 min), cell migration of these cells was decreased significantly. The positive effect effect of Rac2 on PLD2 observed at 20 min and the negative effect of Rac2 on PLD2 were maintained, for the most part, across a range of MCP-1 concentrations (Fig. 1B and C) with the maximal number of cells migrated for each time-point occurring at ∼5 nM MCP-1, which was used for stimulation of cells in all subsequent experiments. In addition, we observed a slight shift to the right to higher concentrations of MCP-1 to elicit the appropriate effect, in the case of Rac2 + PLD2, at longer times. At any event, the differential effect of Rac2 on PLD2 as a function of time warranted further study, and we set up experiments to investigate the cause of this temporal switch.
Figure 1. Rac2 reduces PLD lipase activity at late times of cell migration.

(A) Migration assay of RAW264.7/LR5 cells as a function of time. Cells were transfected with PLD2-WT, Rac2-WT, or PLD2-WT + Rac2-WT, respectively. The number of cells that migrated was measured in the presence of 3 nM MCP-1. The * symbol is higher; # is lower statistically significant (P < 0.05) differences than controls at zero time by ANOVA. (B and C) Migration assay of RAW264.7/LR5 cells as a function of MCP-1 concentration. Cells were transfected with 2 μg PLD2-WT + 0.5 μg Rac2-WT. The number of cells that migrated was measured at 20 (early time; B) and 150 (later time; C) min toward MCP-1. (D) PLD2 and Rac2 localize in the nucleus, as well as in the cytoplasm, but only at short times after cell stimulation. When ready for immunofluorescence microscopy, cells were treated with 3 nM MCP-1 that was added to a localized region on the coverslip with a micropipette, which forms a temporary chemoattractant gradient.
An inter-regulation between Rac2 and PLD2 was hypothesized, and a visualization of such an interaction was sought out and obtained using immunofluorescence microscopy. myc-tagged Rac2 and HA-tagged PLD2 were found colocalized in the cytoplasm at early times (∼20 min) of cell exposure to MCP1-CCL2 (Fig. 1D), which is in agreement with results presented in Fig. 1A and B. However, as cytokine stimulation time increased, less PLD2 was visible in the cytoplasm, indicating that Rac2 had overcome its initial positive effect on PLD2 at early times of stimulation (Fig. 1C). Figure 1D also documents another important point in that Rac2 and PLD2 are present in the nucleus through all of the times of cytokine stimulation.
We found next that Rac2 and PLD2 are interacting at the level of protein:protein binding. Western blot analyses show that the binding interaction between Rac2 and PLD2 was mediated via actin (Fig. 2A). In cells that were cotransfected with PLD2 and Rac2, immunoprecipitation for either actin and then probing for myc-tagged Rac2 or HA-tagged PLD2 reveal the presence of actin, Rac2, and PLD2 in these samples. All of this indicates a direct protein:protein interaction among actin, Rac2, and PLD2.
Figure 2. A Rac2:PLD2-binding interaction negatively affects PLD activity.
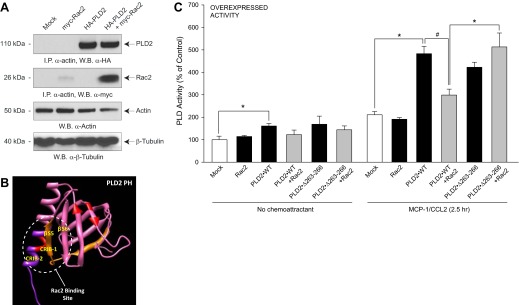
(A) Rac2 and PLD2 form a protein:protein complex with actin. RAW264.7/LR5 cells were transfected with myc-Rac2 or HA-PLD2 alone or in combination, and samples were taken for immunoprecipitation (I.P.) and Western blots (W.B.). (B) Schematic of 3D modeling of the PH domain, a β-sheet composed of four anti-parallel β-strands and one single α-helix, which comprises CRIB domains. This structure was predicted using I-TASSER and viewed using the structure viewer “Chimera” http://www.cgl.ucsf.edu/chimera/download.html. (C) Effect of overexpressed Rac2 on PLD2 lipase activity assay in the presence of MCP-1 and lysates was prepared and then used for the PLD assay. Error bars are sem. Symbols */# denote statistically significant (p < 0.05) differences between means by ANOVA, higher (*) or lower (#) than the indicated controls.
We hypothesized that the CRIB-1 and CRIB-2 domains and β strands 5 and 6, respectively, of the PLD2 PH domain are important for binding of PLD2 to Rac2. Figure 2B shows the scheme of 3D modeling of the PH domain: a β sheet composed of four antiparallel β strands and one single α helix. This PH domain is most likely the site on PLD2 that binds to Rac2 [18, 20, 21].
The effect of this protein:protein association caused a decrease in endogenous PLD enzymatic activity in the presence of MCP-1/CCL2 and Rac2 (Fig. 2C, left group of bars) that was much better observed in cells stimulated with MCP-1 (right group of bars). Overexpression of rPLD2-WT and rRac2 in the presence of MCP-1 for ∼2.5 h resulted in an ∼40% decrease in PLD activity when compared with PLD2-WT alone in the presence of MCP-1. However, this negative effect of Rac2 on PLD activity was reversed when a PLD2 Rac2-binding mutant, PLD2-Δ263–266 that affects the PLD2 CRIB [18], which does not bind to Rac2, was overexpressed simultaneously with Rac2. The results presented in Fig. 2C indicate that Rac2 attenuates the native PLD activity and that the observed changes in PLD activity are mediated by MCP-1 activation of its receptor, CCR2.
The negative effect of Rac2 results in suppression of PLD2 expression
The decrease in PLD activity observed in Fig. 2C could only partially account for the decrease in cell migration (Fig. 1A) at late times. As a negative effect of Rac2 was seen at 3 h or even longer (data not shown), we supposed that Rac2 could have an effect on PLD2 expression. We next determined the levels of endogenous PLD2 protein expression when Rac2 was overexpressed in RAW264.7/LR5 cells (Fig. 3A, left), which shows that macrophage cells that were transfected with Rac2 inhibited endogenous PLD2 protein expression. The negative effect of Rac2 on PLD2 could occur as a change on PLD2 gene or protein expression. As seen in Fig. 3A (right), the negative effect of Rac2 on PLD2 is not a result of protein degradation after translation, as the proteasome inhibitor (MG132) failed to prevent the PLD2 protein decrease by Rac2.
Figure 3. Effect of Rac2 overexpression on PLD2.
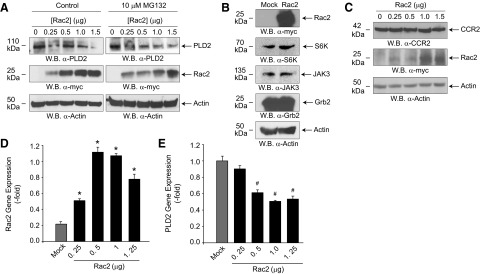
(A) RAW264.7/LR5 cells were transfected with increasing amounts of Rac2 DNA. Twenty-four hours post-transfection, a proteasome inhibitor (MG132) was added to separate sets of cells, and 48 h post-transfection lysates were prepared, and Western blots were performed. (B) Effect of Rac2 overexpression on several signaling proteins. Protein loading control using anti-actin staining (bottom blot) as well as the effective expression of Rac2 (top blot) are included. (C) Effect of Rac2 overexpression on MCP-1/CCL2 receptor, CCR2. Protein loading control using anti-actin staining is included. (D and E) qRT-PCR gene expression of overexpressing Rac2 (D) or endogenous PLD2 (E) in RAW macrophages. Error bars are sem. Symbols */# denote statistically significant (p < 0.05) differences between means by ANOVA, higher (*) or lower (#) than Mock controls.
The effect of Rac2 overexpression seems to be specific for PLD, as it did not have an effect on a variety of signaling proteins, such as S6K, JAK3, and Grb2 (Fig. 3B). Another aspect that we examined was the receptor for MCP-2/CCL2, CCR2. In some cases, chemokines could induce their receptor to be internalized or degraded after cell membrane binding [22–24]. However, the negative effect of Rac2 on PLD2 is not because of a deficit in the expression of the MCP-1R, CCR2, as CCR2 expression remains relatively constant in the presence of increasing Rac2 expression (Fig. 3C).
To further show that the negative effect of Rac2 on PLD2 is a result of a modulation at the level of gene expression, we conducted qRT-PCR. Figure 3C is a positive control, indicting a robust detection of the overexpressing construct Rac2. Figure 3D indicates a decrease in the amount of PLD2 gene expression with concomitant increases in Rac2 protein, which further confirms the negative effect of Rac2 on PLD2.
Silencing Rac2 abrogates the negative effect on PLD2 expression
Another way to understand the negative relationship between Rac2 and PLD2 was done by loss of function experiments. RAW macrophages were silenced with increasing levels of siRNA, specific for Rac2 (Fig. 4A), which resulted in a significant increase in PLD2 protein expression that could be quantified by densitometry (Fig. 4B). Next, we measured the effect of sustained MCP-1 stimulation of RAW24.7/LR5 cells on endogenous PLD2 protein expression levels. As shown in Fig. 4C, sustained MCP-1 stimulation of cells decreased endogenous PLD2 protein expression significantly in the absence of Rac2 silencing (compare yellow ovals). However, if Rac2 is silenced, PLD2 expression is up-regulated (Fig. 4C, blue ovals). qRT-PCR analysis was performed, and these results show that as a result of an acute decrease in Rac2 gene expression (Fig. 4D, open bars) there is a somewhat modest increase in PLD2 gene expression (Fig. 4D, filled bars), which is not seen when Rac2 is overexpressed (Fig. 4D, right-most bars). All of this suggests that Rac2 and PLD2 not only interact at the protein level but also that the Rac2 expression modulates the expression of PLD2.
Figure 4. Effect of silencing Rac2 on PLD2 protein and gene expression.
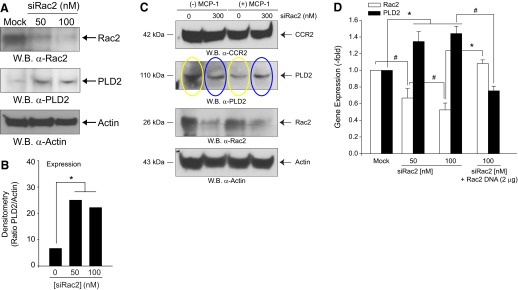
(A) RAW264.7/LR5 cells were silenced with increasing Rac2 siRNA for 3 days, and lysates containing proteins were obtained. Western blots were probed with anti-Rac2 or anti-PLD2 antibodies. (B) Quantification of Western blot results of the ratio of endogenous PLD2 relative to silenced Rac2 expression. Results in this figure are the means ± sem from at least three independent experiments conducted in duplicate. (C) Sustained MCP-1 stimulation decreases endogenous PLD2 protein levels via a mechanism that is rescued by Rac2 siRNA. RAW264.7/LR5 cells were mock-silenced or silenced with increasing Rac2 siRNA for 3 days and simultaneously mock-stimulated or stimulated with 5 nM MCP-1. (D) qRT-PCR RNA expression of endogenous PLD2 and Rac2 in the presence of increasing siRNA, specific for Rac2 and probed with FAM-tagged PLD2 or Rac2 primers. Error bars are sem. Symbols */# denote statistically significant (p < 0.05) differences between means by ANOVA, higher (*) or lower (#) than the indicated controls.
The mechanism of Rac2-induced PLD2 down-regulation is mediated by the Sp1 transcription factor
We investigated next the possible mechanism that could mediate the enhancing effect of Rac2 silencing on PLD2 gene expression (Fig. 4A–C) and concentrated on a specific transcription factor, Sp1. The Sp1 gene on human chromosome arm 12q encodes for the Sp1 transcription factor, which is involved in gene expression during early development. The Sp family members recognize GC/GT boxes in promoter regions of mammalian and viral genes, and the consensus Sp1-binding site (GC-rich) is 5′-(G/T)GGGCGG(G/A) (G/A) (C/T)-3′. Sp1 regulates transcription in a large number of genes involved in differentiation, proliferation, survival, and apoptosis [25–27]. According to refs. [28–30], Sp1 could serve, in general, as a gene enhancer and a gene repressor.
Another new discovery in the present study was that Sp1 is downstream of Rac2 (Fig. 5A), as shown by the increase in Sp1 phosphorylation at threonine453, which is a marker of Sp1 activation. As Fig. 5A indicates that Sp1 is likely a transcription repressor for the PLD2 gene, we performed an in silico search to determine the presence of Sp1-binding sites in the promoter region of PLD2 and found four sequences that fit this criteria (Fig. 5B). In the current scenario, Sp1 activation acts as a repressor for PLD2, as evidenced by the significant decrease in PLD2 protein expression shown in Fig. 5A (middle Western blot).
Figure 5. Effect of Rac2 on Sp1 phosphorylation.

(A) Effect of Rac2 overexpression on Sp1 activation/phosphorylation. RAW264.7/LR5 cells were transfected with increasing Rac2 DNA, and resulting Western blots were performed that were stained with the indicated antibodies. (B) Schematic for potential Sp1 binding to PLD2 promoter. Shaded in yellow are putative Sp1-binding sites in the PLD2 promoter. (C) Effect of silencing Sp1 on PLD2 expression. (D) Effect of prolonged MCP-1 stimulation on Sp1-mediated rescue of PLD2 protein expression. (E) Overexpression of β-catenin reverses the negative effect of Rac2 on PLD2 expression. (F) β-Catenin is activated at early times of incubation with MCP-1/CCL2 but not at later (>1 h) times. RAW264.7/LR5 cells were stimulated with 5 nM MCP-1.
We also document that silencing Sp1 reversed the earlier reduction in PLD2 expression (Fig. 5A, second from bottom Western blot), and it was brought up to level, similar to controls, even at 150 nM siSp1 (Fig. 5C, second Western blot from the top). Figure 5C also includes a Western blot, indicating the robust levels of Rac2 overexpression (second from bottom blot) and a similar protein loading (bottom Western blot). We concluded that knockdown of Sp1 attenuates the Rac2 overexpression-induced decreases in native PLD2 protein levels. An additional experiment shown in Fig. 5D, to address the role of MCP-1 in this new mechanism, indicates: (1) Sp1 silencing was accomplished; (2) silencing Sp1 is partially rescued by MCP-1 cell stimulation (compare yellow ovals); (3) PLD2 is up-regulated with Sp1 silencing (compare blue ovals); and (4) as MCP-1 cell stimulation rescues Sp1 silencing, PLD2 expression is consequently down-regulated (red oval).
The key regulatory role of β-catenin on PLD activity
β-Catenin mediates leukocyte adhesion and is downstream of the Wnt signaling pathway involving GSK-3β [31]. Transcriptional activation of β-catenin, which is defined by nonphosphorylation at S33/37 and T41, occurs when β-catenin translocates to the nucleus and becomes a transcription factor for many different genes, including PLD [32–34]. As a positive control, we present that β-catenin, which is known to be an activator of PLD expression [32, 33], also increases PLD2 expression. However, in the presence of overexpressed β-catenin, Rac2 still exerted its negative effect on PLD2 (Fig. 5E).
To further elucidate the mechanism of an initial positive effect of β-catenin on PLD2, we sought to determine β-catenin activation upon cell stimulation with MCP-1/CCL2. β-Catenin was activated only at early times (<60 min) in nontransfected macrophages stimulated by MCP-1/CCL2 (Fig. 5F). At later times (>60 min), β-catenin was not activated, possibly as a result of degradation. A lack of β-catenin activity could explain its inability to prevent Rac2-initiated down-regulation. Therefore, the negative effect of Rac2 on chemotaxis that we have observed prevails among other positive effects, and macrophages remain effectively arrested in cell migration at late stages of chemokine stimulation.
DISCUSSION
We have shown for the first time a molecular mechanism that generates a “stop” signal that arrests cell migration of monocytes/macrophages. This has an immediate consequence on the physiology, as these cells are required to remain locked in tissue infiltrates at the site of inflammation until the insult (e.g., pathogen) is cleared. It could also have an implication on the pathological stage, as prolonged permanence of leukocytes occurs as a result of chronic inflammation, which further stresses the importance of this molecular mechanism.
This study presents the existence of an interaction between PLD2 and Rac2 that also includes the regulation among Rac2, PLD2, and Sp1. Data presented herein indicate that Rac2 has a negative effect on PLD2 gene expression, which is novel. Sp1 is an essential transcription factor that is involved in cell-cycle progression and enhances transcription in a large number of genes involved in differentiation, proliferation, and apoptosis and links PLD to matrix metalloproteinase-2 up-regulation, which indicates a role for PLD in glioma progression and tumor cell invasion via PKC/PKA/NF-κB/Sp1-mediated signaling pathways [35]. There are several functional domains in Sp1, such as the Sp1 inhibitory domain and the Sp1 zinc-finger DNA-binding domain, which is the most highly conserved domain in the Sp family that can interact with corepressor proteins to repress gene transcription [30, 36]. Although Sp1 has been described as a transcription activator, Sp1 has also been classified as a repressor of gene transcription [29]. Additionally, Sp1 further interacts with growth-suppressor protein p21 through PLD2 to prevent cancer growth [37].
We also report the new finding that MCP-1/CCL2 transiently activates β-catenin, which is elevated after cell adhesion to plastic in human monocytes [38]. β-Catenin was shown to be a transcriptional activator of PLD1 [33]. PA produced by PLD is a factor in the activation of β-catenin [33], which has been shown to be regulated by certain Sp proteins [39]. Interestingly, Rac1 has been found to activate Jnk, Akt, and PKA, which in turn, phosphorylate β-catenin and cause it to translocate to the nucleus, where it acts as a transcription factor [40–43]. Cells transfected with siRNA specific for Rac1 were found to have lower levels of β-catenin [44]. GSK-3β causes degradation of β-catenin, which is decreased significantly in cytosol [45].
We developed a model, as shown in Fig. 6, which explains the negative effect of Rac2 on PLD2. In this model presented herein, we hypothesize that the inter-relation among PLD2, Rac2, and Sp1 is a multistep process. First, the leukocyte is stimulated by MCP-1/CCL2, which is implicated in the pathogenesis of inflammatory diseases that have an overabundance of monocyte/macrophage infiltrates (such as psoriasis, rheumatoid arthritis, and atherosclerosis), and binds to its receptor (CCR2) at the cell membrane. This signaling event allows for GTP-bound Rac2 to bind and interact with PLD2 in the cytoplasm, which allows for the cell to chemotax at early times (∼20 min). We speculate that when Rac2 reaches the nucleus, Sp1 is activated by Rac2-GTP. Subsequent phosphorylation of Sp1 negatively impacts the expression of PLD2 at the protein and mRNA level. This reduces the activity of PLD2, which subsequently reduces the ability of the cells to chemotax.
Figure 6. Model of proposed mechanism for monocyte arresting.

MCP-1/CC12 binds to the CCR2 receptor and stimulates the cell (1). Rac2 alone and Rac2 complexed with PLD2 activate chemotaxis at early times of inflammation (2). At longer times after stimulation, the following mechanism takes place: Rac2 influences Sp1 (3) in a yet-unknown way that makes it translocate from the cytoplasm to the nucleus (4), where it becomes phosphorylated (P in yellow circle) and down-regulates PLD2 expression (5), thus mediating the negative effect of Rac2 on PLD expression. This down-regulation leads to suppressed chemotaxis at later times of cell migration (6). β-Catenin is a regulator of this mechanism, in that initially, and as activated after MCP-1 stimulation, it is a positive effector of early chemotaxis (1b) and can also be a positive PLD2 gene enhancer. However, at later times, as Rac2 and Sp1 accumulate, they overcome the positive action of β-catenin, and the balance is tipped toward leukocyte immobilization (6).
We believe that this is important in the process of slowing down chemotaxis and immobilizing leukocytes during inflammation. The new signaling pathway reported here implies that β-catenin positively enhances PLD2 expression at earlier times of stimulation (∼30 min). However, later on, Rac2 dominates in exerting a negative effect on PLD2 expression. Should this negative effect be sustained, it could help explained prolonged immobilization of leukocytes from normal functionality to pathological stages of chronic inflammation, which could have important clinical consequences.
ACKNOWLEDGMENTS
The following grants to J.G-C. have supported this work: HL056653-14 from the U.S. National Institutes of Health, 229102 from the Boonshoft School of Medicine (BSOM), and 668372 from the State of Ohio Research Incentive.
The authors thank Karen M. Henkels for excellent editorial assistance.
Footnotes
- 3D
- three-dimensional
- CRIB
- Cdc42 and Rac interactive-binding domain
- Ct
- cycle threshold
- GEF
- guanine nucleotide exchange factor
- Grb2
- growth receptor-bound protein 2
- Neg
- negative control
- PA
- phosphatidic acid
- PBut
- phosphatidylbutanol
- PC
- phosphatidylcholine
- PH
- pleckstrin homology
- PLD
- phospholipase D
- qRT-PCR
- quantitative RT-PCR
- Rac2
- Ras-related C3 botulinum toxin substrate 2
- si
- small interfering
- Sp1
- specificity protein 1
AUTHORSHIP
J.G-C. designed research and wrote the paper. M.M. and F.J.S. performed research. M.M., F.J.S., and J.G-C. analyzed data.
DISCLOSURES
There is no known conflict of interest.
REFERENCES
- 1. Koshland D. E. (1982) Special topic: chemotaxis and motility. Annu. Rev. Physiol. 44, 499–500 [Google Scholar]
- 2. Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons J. T., Horwitz A. R. (2003) Cell migration: integrating signals from front to back. Science 302, 1704–1709 [DOI] [PubMed] [Google Scholar]
- 3. Vicente-Manzanares M., Webb D. J., Horwitz A. R. (2005) Cell migration at a glance. J. Cell Sci. 118, 4917–4919 [DOI] [PubMed] [Google Scholar]
- 4. Luster A. D., Alon R., von Andrian U. H. (2005) Immune cell migration in inflammation: present and future therapeutic targets. Nat. Immunol. 6, 1182–1190 [DOI] [PubMed] [Google Scholar]
- 5. Capoccia B. J., Gregory A. D., Link D. C. (2008) Recruitment of the inflammatory subset of monocytes to sites of ischemia induces angiogenesis in a monocyte chemoattractant protein-1-dependent fashion. J. Leukoc. Biol. 84, 760–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Piccinini A.M., Knebl K., Rek A., Wildner G., Diedrichs-Mohring M., Kungl A. J. (2010) Rationally evolving MCP-1/CCL2 into a decoy protein with potent anti-inflammatory activity in vivo. J. Biol. Chem. 285, 8782–8792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Terashima Y., Onai N., Murai M., Enomoto M., Poonpiriya V., Hamada T., Motomura K., Suwa M., Ezaki T., Haga T., Kanegasaki S., Matsushima K. (2005) Pivotal function for cytoplasmic protein FROUNT in CCR2-mediated monocyte chemotaxis. Nat. Immunol. 6, 827–835 [DOI] [PubMed] [Google Scholar]
- 8. Schreiber M. T., Schuler B., Li L., Hall D. J. (2013) Activation of the small G-protein Rac by human rhinovirus attenuates the TLR3/IFN-α axis while promoting CCL2 release in human monocyte-lineage cells. Innate Immun. 19, 278–289 [DOI] [PubMed] [Google Scholar]
- 9. Innocenti M., Frittoli E., Ponzanelli I., Falck J. R., Brachmann S. M., Di Fiore P. P., Scita G. (2003) Phosphoinositide 3-kinase activates Rac by entering in a complex with Eps8, Abi1, and Sos-1. J. Cell Biol. 160, 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krugmann S., Jordens I., Gevaert K., Driessens M., Vandekerckhove J., Hall A. (2001) Cdc42 induces filopodia by promoting the formation of an IRSp53: Mena complex. Curr. Biol. 11, 1645–1655 [DOI] [PubMed] [Google Scholar]
- 11. Gu Y., Byrne M. C., Paranavitana N. C., Aronow B., Siefring J. E., D'Souza M., Horton H. F., Quilliam L. A., Williams D. A. (2002) Rac2, a hematopoiesis-specific Rho GTPase, specifically regulates mast cell protease gene expression in bone marrow-derived mast cells. Mol. Cell. Biol. 22, 7645–7657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou D., Luini W., Bernasconi S., Diomede L., Salmona M., Mantovani A., Sozzani S. (1995) Phosphatidic acid and lysophosphatidic acid induce haptotactic migration of human monocytes. J. Biol. Chem. 270, 25549–25556 [DOI] [PubMed] [Google Scholar]
- 13. Powner D. J., Pettitt T. R., Anderson R., Nash G. B., Wakelam M. J. (2007) Stable adhesion and migration of human neutrophils requires phospholipase D-mediated activation of the integrin CD11b/CD18. Mol. Immunol. 44, 3211–3221 [DOI] [PubMed] [Google Scholar]
- 14. Nishikimi A., Fukuhara H., Su W., Hongu T., Takasuga S., Mihara H., Cao Q., Sanematsu F., Kanai M., Hasegawa H., Tanaka Y., Shibasaki M., Kanaho Y., Sasaki T., Frohman M. A., Fukui Y. (2009) Sequential regulation of DOCK2 dynamics by two phospholipids during neutrophil chemotaxis. Science 324, 384–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lehman N., Di Fulvio M., McCray N., Campos I., Tabatabaian F., Gomez-Cambronero J. (2006) Phagocyte cell migration is mediated by phospholipases PLD1 and PLD2. Blood 108, 3564–3572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gomez-Cambronero J. (2012) Biochemical and cellular implications of a dual lipase-GEF function of phospholipase D2 (PLD2). J. Leukoc. Biol. 92, 461–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jeon H., Kwak D., Noh J., Lee M. N., Lee C. S., Suh P. G., Ryu S. H. (2011) Phospholipase D2 induces stress fiber formation through mediating nucleotide exchange for RhoA. Cell. Signal. 23, 1320–1326 [DOI] [PubMed] [Google Scholar]
- 18. Peng H. J., Henkels K. M., Mahankali M., Dinauer M. C., Gomez-Cambronero J. (2011) Evidence for two CRIB domains in phospholipase D2 (PLD2) that the enzyme uses to specifically bind to the small GTPase Rac2. J. Biol. Chem. 286, 16308–16320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kantonen S., Hatton N., Mahankali M., Henkels K. M., Park H., Cox D., Gomez-Cambronero J. (2011) A novel phospholipase D2-Grb2-WASp heterotrimer regulates leukocyte phagocytosis in a two-step mechanism. Mol. Cell. Biol. 31, 4524–4537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peng H. J., Henkels K. M., Mahankali M., Marchal C., Bubulya P., Dinauer M. C., Gomez-Cambronero J. (2011) The dual effect of Rac2 on phospholipase D2 regulation that explains both the onset and termination of chemotaxis. Mol. Cell. Biol. 31, 2227–2240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mahankali M., Peng H. J., Cox D., Gomez-Cambronero J. (2011) The mechanism of cell membrane ruffling relies on a phospholipase D2 (PLD2), Grb2 and Rac2 association. Cell. Signal. 23, 1291–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gupta S., Rieder S., Richter R., Schulz-Maronde S., Manns J., Escher S. E., Heitland A., Mack M., Forssmann W. G., Elsner J., Forssmann U. (2010) CCR1- and CCR5-mediated inactivation of leukocytes by a nonglycosaminoglycan (non-GAG)-binding variant of n-nonanoyl-CCL14 (NNY-CCL14). J. Leukoc. Biol. 88, 383–392 [DOI] [PubMed] [Google Scholar]
- 23. Hayashi F., Means T. K., Luster A. D. (2003) Toll-like receptors stimulate human neutrophil function. Blood 102, 2660–2669 [DOI] [PubMed] [Google Scholar]
- 24. Tester A. M., Cox J. H., Connor A. R., Starr A. E., Dean R. A., Puente X. S., Lopez-Otin C., Overall C. M. (2007) LPS responsiveness and neutrophil chemotaxis in vivo require PMN MMP-8 activity. PLoS One 2, e312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Szpirer J., Szpirer C., Riviere M., Levan G., Marynen P., Cassiman J. J., Wiese R., DeLuca H. F. (1991) The Sp1 transcription factor gene (SP1) and the 1,25-dihydroxyvitamin D3 receptor gene (VDR) are colocalized on human chromosome arm 12q and rat chromosome 7. Genomics 11, 168–173 [DOI] [PubMed] [Google Scholar]
- 26. Nardelli J., Gibson T. J., Vesque C., Charnay P. (1991) Base sequence discrimination by zinc-finger DNA-binding domains. Nature 349, 175–178 [DOI] [PubMed] [Google Scholar]
- 27. Bouwman P., Philipsen S. (2002) Regulation of the activity of Sp1-related transcription factors. Mol. Cell. Endocrinol. 195, 27–38 [DOI] [PubMed] [Google Scholar]
- 28. Deniaud E., Baguet J., Chalard R., Blanquier B., Brinza L., Meunier J., Michallet M. C., Laugraud A., Ah-Soon C., Wierinckx A., Castellazzi M., Lachuer J., Gautier C., Marvel J., Leverrier Y. (2009) Overexpression of transcription factor Sp1 leads to gene expression perturbations and cell cycle inhibition. PLoS One 4, e7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Law A. Y., Yeung B. H., Ching L. Y., Wong C. K. (2011) Sp1 is a transcription repressor to stanniocalcin-1 expression in TSA-treated human colon cancer cells, HT29. J. Cell. Biochem. 112, 2089–2096 [DOI] [PubMed] [Google Scholar]
- 30. Lee J. A., Suh D. C., Kang J. E., Kim M. H., Park H., Lee M. N., Kim J. M., Jeon B. N., Roh H. E., Yu M. Y., Choi K. Y., Kim K. Y., Hur M. W. (2005) Transcriptional activity of Sp1 is regulated by molecular interactions between the zinc finger DNA binding domain and the inhibitory domain with corepressors, and this interaction is modulated by MEK. J. Biol. Chem. 280, 28061–28071 [DOI] [PubMed] [Google Scholar]
- 31. Lee D. K., Nathan Grantham R., Trachte A. L., Mannion J. D., Wilson C. L. (2006) Activation of the canonical Wnt/β-catenin pathway enhances monocyte adhesion to endothelial cells. Biochem. Biophys. Res. Commun. 347, 109–116 [DOI] [PubMed] [Google Scholar]
- 32. Kang D. W., Min do S. (2010) Positive feedback regulation between phospholipase D and Wnt signaling promotes Wnt-driven anchorage-independent growth of colorectal cancer cells. PLoS One 5, e12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kang D. W., Lee S. H., Yoon J. W., Park W. S., Choi K. Y., Min do S. (2010) Phospholipase D1 drives a positive feedback loop to reinforce the Wnt/β-catenin/TCF signaling axis. Cancer Res. 70, 4233–4242 [DOI] [PubMed] [Google Scholar]
- 34. Du C., Zhang C., Li Z., Biswas M. H., Balaji K. C. (2012) β-Catenin phosphorylated at threonine 120 antagonizes generation of active β-catenin by spatial localization in trans-Golgi network. PLoS One 7, e33830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Park M. H., Ahn B. H., Hong Y. K., Min do S. (2009) Overexpression of phospholipase D enhances matrix metalloproteinase-2 expression and glioma cell invasion via protein kinase C and protein kinase A/NF-κB/Sp1-mediated signaling pathways. Carcinogenesis 30, 356–365 [DOI] [PubMed] [Google Scholar]
- 36. Gao S., Murakami M., Ito H., Furuhata A., Yoshida K., Tagawa Y., Hagiwara K., Takagi A., Kojima T., Suzuki M., Banno Y., Nozawa Y., Murate T. (2009) Mutated ras induced PLD1 gene expression through increased Sp1 transcription factor. Nagoya J. Med. Sci. 71, 127–136 [PMC free article] [PubMed] [Google Scholar]
- 37. Kwun H. J., Lee J. H., Min D. S., Jang K. L. (2003) Transcriptional repression of cyclin-dependent kinase inhibitor p21 gene by phospholipase D1 and D2. FEBS Lett. 544, 38–44 [DOI] [PubMed] [Google Scholar]
- 38. Thiele A., Wasner M., Muller C., Engeland K., Hauschildt S. (2001) Regulation and possible function of β-catenin in human monocytes. J. Immunol. 167, 6786–6793 [DOI] [PubMed] [Google Scholar]
- 39. Pathi S., Jutooru I., Chadalapaka G., Nair V., Lee S. O., Safe S. (2012) Aspirin inhibits colon cancer cell and tumor growth and downregulates specificity protein (Sp) transcription factors. PLoS One 7, e48208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Crampton S. P., Wu B., Park E. J., Kim J. H., Solomon C., Waterman M. L., Hughes C. C. (2009) Integration of the β-catenin-dependent Wnt pathway with integrin signaling through the adaptor molecule Grb2. PLoS One 4, e7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. He X. C., Yin T., Grindley J. C., Tian Q., Sato T., Tao W. A., Dirisina R., Porter-Westpfahl K. S., Hembree M., Johnson T., Wiedemann L. M., Barrett T. A., Hood L., Wu H., Li L. (2007) PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat. Genet. 39, 189–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Taurin S., Sandbo N., Qin Y., Browning D., Dulin N. O. (2006) Phosphorylation of β-catenin by cyclic AMP-dependent protein kinase. J. Biol. Chem. 281, 9971–9976 [DOI] [PubMed] [Google Scholar]
- 43. Fang D., Hawke D., Zheng Y., Xia Y., Meisenhelder J., Nika H., Mills G. B., Kobayashi R., Hunter T., Lu Z. (2007) Phosphorylation of β-catenin by AKT promotes β-catenin transcriptional activity. J. Biol. Chem. 282, 11221–11229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou L., Ercolano E., Ammoun S., Schmid M. C., Barczyk M. A., Hanemann C. O. (2011) Merlin-deficient human tumors show loss of contact inhibition and activation of Wnt/β-catenin signaling linked to the PDGFR/Src and Rac/PAK pathways. Neoplasia 13, 1101–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Van Noort M., Meeldijk J., van der Zee R., Destree O., Clevers H. (2002) Wnt signaling controls the phosphorylation status of β-catenin. J. Biol. Chem. 277, 17901–17905 [DOI] [PubMed] [Google Scholar]