

Abstract
Lipidic mesophases are a class of highly ordered soft materials that form when certain lipids are mixed with water. Understanding the relationship between the composition and the microstructure of mesophases is necessary for fundamental studies of self-assembly in amphiphilic systems and for applications, such as crystallization of membrane proteins. However, the laborious formulation protocol for highly viscous mesophases and the large amounts of material required for sample formulation are significant obstacles in such studies. Here we report a microfluidic platform that facilitates investigations of the phase behavior of mesophases by reducing sample consumption, and automating and parallelizing sample formulation. The mesophases were formulated on-chip using less than 40 nL of material per sample and their microstructure was analyzed in situ using small-angle X-ray scattering (SAXS). The 220 μm-thick X-ray compatible platform was comprised of thin polydimethylsiloxane (PDMS) layers sandwiched between cyclic olefin copolymer (COC) sheets. Uniform mesophases were prepared using an active on-chip mixing strategy coupled with periodic cooling of the sample to reduce the viscosity. We validated the platform by preparing and analyzing mesophases of lipid monoolein (MO) mixed with aqueous solutions of different concentrations of β-octylglucoside (βOG), a detergent frequently used in membrane protein crystallization. Four samples were prepared in parallel on chip, by first metering and automatically diluting βOG to obtain detergent solutions of different concentration, then metering MO, and finally mixing by actuation of pneumatic valves. Integration of detergent dilution and subsequent mixing significantly reduced the number of manual steps needed for sample preparation. Three different types of mesophases typical for monoolein were successfully identified in SAXS data from on-chip samples. Microstructural parameters of identical samples formulated in different chips showed excellent agreement. Phase behavior observed on-chip corresponded well with that of samples prepared via the traditional coupled-syringe method (“off-chip”) using 300-fold larger amount of material, further validating the utility of the microfluidic platform for on-chip characterization of mesophase behavior.
Introduction
Lipidic mesophases are soft materials whose structure consists of highly ordered arrangements of continuous lipid bilayers interpenetrated with aqueous channels.1 Lipidic mesophases form when certain lipids are mixed with water and have been used in a number of applications such as drug delivery2 and biosensing.3 Most importantly, lipidic mesophases serve as matrices for stabilizing and crystallizing integral membrane proteins that are otherwise very difficult to handle.4,5 In the in meso crystallization approach, membrane proteins are reconstituted into the lipid bilayers of the mesophase that create a membrane-like environment and prevent the proteins from unfolding. Crystallization is triggered by adding a multi-component precipitant solution, with protein crystals growing within the lipidic mesophase in a successful crystallization trial. To date this method accounts for ~10% of all structures of membrane proteins available in the public domain5 and counts the structures of the human β2 adrenergic receptor,6 the dopamine D3 receptor,7 and the κ-opioid receptor8 among its recent successes.
The lipid most commonly used for in meso crystallization is monoolein (MO).4 The phase type, i.e., the arrangement of lipid bilayers in MO mesophases is highly sensitive to the composition of the mixture as well as temperature, and can be reliably identified from small-angle X-ray scattering (SAXS) data.4,9 Under crystallization-relevant conditions it usually forms three types of mesophases: lamellar phase Lα and two cubic phases of different symmetries referred to as Pn3m and Ia3d.10–15 The lamellar phase consists of stacks of lipid bilayers with one-dimensional spatial periodicity, whereas Pn3m and Ia3d phases contain curved lipid bilayers in complex geometries, are bicontinuous and periodic in three dimensions.9–11,16,17
The type of mesophase is crucial for the success of crystallization trials: protein crystal growth has only been reported from bicontinuous mesophases.15,18 The components of the crystallization mixture affect phase behavior in a non-trivial way.19 In particular, detergents used to stabilize the protein prior to its incorporation in the mesophase have a profound effect on the phase behavior and tend to promote the formation of lamellar mesophases that are unsuitable for crystallization.13,14,20
Exploration of phase diagrams to unravel trends in phase behavior is an arduous task that requires the preparation and analysis of a large number of samples. For lipidic mesophases the situation is exacerbated by their toothpaste-like consistency, and the state of the art procedure is laborious and time consuming.21 Presently each mesophase sample is prepared by mixing the lipid with water in a coupled-syringe mixer on a milligram (microliter) scale.4 The resulting mixture is then dispensed into a glass capillary for SAXS analysis.4 Reduction of sample consumption coupled with automated and parallelized sample formulation would greatly facilitate the studies of the phase behavior of mesophases, especially for novel, scarcely available lipids.
Several macroscale approaches that address the limitations of the standard formulation approach have been reported. Recently, mixing of lipid and an aqueous component was automated in a 96-well plate format, but still 30 μL of MO and solution had to be metered manually for each sample.22 While only 50 nL of mesophase is required for analysis in another recently published approach for high-throughput SAXS analysis of lipidic mesophases, the sample had to be formulated on a bulk scale in a coupled-syringe mixer and dispensed in small boluses using an expensive robotic system.23 Furthermore, the high-throughput SAXS methods reported to date are limited to the formulation of lipidic mesophases in equilibrium with a large excess of aqueous solutions, excluding a large region of the phase diagram.23,24
Microfluidics offers the potential to automate metering and simultaneous preparation of multiple samples at nanoliter scales, making it very attractive for the studies of the phase behavior. Although microfluidic platforms for phase behavior studies have been demonstrated,25–41 reported systems are only applicable to low-viscosity solutions and typically rely on phase transitions driven by composition changes due to diffusion of solutes,25,26,33–36 evaporation of water,28–32,40–42 and osmotic stress.37,38 Such platforms are not suitable for the formulation of viscous mesophases with strict pre-set ratios of volume or weight of components.
In previous work43 we demonstrated a microfluidic platform (lipid mixer) capable of preparing uniformly mixed mesophases on a ~20 nL scale via an active mixing strategy and validated the chip by the in meso crystallization of membrane protein bacteriorhodopsin. However, the original mixer, as well as the majority of platforms with monolithic pneumatic valves are incompatible with SAXS due to signal attenuation44–47 by thick layers of polydimethylsiloxane (PDMS) and glass used for device fabrication.48 On the other hand, X-ray compatible microfluidic platforms reported for various applications39–42,44–47,49–58 have very limited fluid handling capabilities (with one exception45). Although these microfluidic devices have been successfully used for SAXS analysis of protein folding56,58 and aggregation,57 and for the studies of complex fluids,39–42 they are unsuitable for the formulation of lipidic mesophases.
Here we report an X-ray transparent microfluidic platform with an array of lipid mixers for the analysis of the phase behavior of lipidic mesophases. The platform was specifically designed to assess the effect of additives such as detergents on the phase behavior and is capable of simultaneously preparing four mesophase samples with different compositions by performing dilutions on-chip. The platform uses less than 200 nL of detergent solution and lipid in total, which represents a 300-fold reduction compared to the standard method. To demonstrate the utility of this platform we mapped a section of a phase diagram for MO mixed with solutions of β-octylglucoside (βOG), a detergent commonly used in the isolation and purification of membrane proteins.4,13
Materials and Methods
Device fabrication
Hybrid microfluidic chips consisted of a flat cyclic olefin copolymer (COC) top layer (4 mil, 5013 or 6013, TOPAS Advanced Polymers), a thin PDMS (RTV 650, Momentive Performance Adhesives) control layer, a thin PDMS fluid layer, and a flat COC bottom substrate, Figure 1. The PDMS fluid layer was fabricated using standard soft lithographic procedures.48,59 Photoresist-on-silicon masters were created using SPR-200-7 photoresist (Shipley) for the fluid layer with a feature height of ~14 μm, and SU8-25 photoresist (Microchem) for the control layer with a feature height of ~25 μm. Positive photoresist was reflowed by heating at 120 °C for 2 min to ensure complete closure of channels upon valve actuation.48 PDMS fluid layers were prepared by spin-coating PDMS with a monomer: cross-linker ratio of 5:1 followed by curing at 90 °C for 7–9 min to obtain a 25 μm-thick film. PDMS control layers with a thickness of 45 μm were prepared by spin-coating PDMS with a monomer : crosslinker ratio of 12:1 and curing at 80 °C for 3 min.
Figure 1.

(a) Superimposed control (orange, blue, red) and fluid (black) layers of the microfluidic platform for screening of lipid phase behavior. Circles at ends of lines designate the locations of inlet ports. Control layer: orange, normally open routing valves; red, normally closed routing valves; blue, injection valves. The inset shows the layout of fluid layer with compartment designations for the three components required for filling the chip: lipid (blue outline), detergent solution (red outline), and diluent for detergent solution (purple outline). (b) Schematic of the layers that comprise the thin part of the chip. (c) Photograph of an assembled chip mounted for SAXS data collection. Sample compartments are located in the thin part of the chip, with the fluid and control layer inlets connected via the thick PDMS block. Figure S1 in the ESI shows individual layouts of control and fluid layers.
The surface of COC sheets was smoothed by placing the sheets between glass slides at 177 °C and a load of 150–200 kg in a laminating press (Carver, Model 3851) prior to use. PDMS/PDMS bonding was achieved using standard multilayer soft lithography48 by placing layers of PDMS with different monomer to cross-linker ratios in conformal contact followed by heating at 70 °C for 2 h. Amine/epoxy chemistry45,60 with 3-aminopropyltrimethoxysilane (Sigma-Aldrich, >97%) and 3-glycidyloxypropyltrimethoxysilane (GPTMS, Sigma-Aldrich, 98%) was used for all PDMS/COC bonding.
The chips were assembled as follows: (i) inlet holes were drilled in the 4–7 mm-thick PDMS block and in the top COC sheet using a 750 μm drill bit; (ii) the PDMS block was bonded to the top COC layer to provide support for tubing; (iii) the PDMS-COC assembly was bonded to the PDMS control layer; (iv) the resulting PDMS-COC-PDMS assembly was bonded to the PDMS fluid layer; and (v) the assembled chip was bonded to the bottom COC substrate and kept at 70 °C for 1 h to facilitate bonding. To prevent permanent closure of normally-closed valves (Figure S2) in the final permanent bonding step, the corresponding control lines were actuated using a vacuum pump, prior to bringing the two parts of the device in contact, and remained actuated at all times during curing. After at least 3 h, the fluid layer was filled with a solution of 1M TRIS buffer, pH 8.0, to neutralize any remaining active epoxy groups of the GPTMS layer on the inner surface of fluid channels and chambers. After at least 1 h the fluid layer was rinsed with de-ionized water and dried with nitrogen gas. At this time the valves were allowed to close.
Device operation
During filling and mixing, the microfluidic devices were monitored using an upright microscope (Leica MDG33) equipped with a macro lens and a digital camera (Leica DFC295). Prior to filling and mixing operations, fluid-routing control lines (Figure 1a) were filled with Fluorinert FC-40 (3M) to prevent gas leakage from the control layer through valve membranes to the fluid layer and subsequent bubble formation in the samples.
For pneumatic actuation we used a 32-line solenoid valve manifold (Fluidigm). The pressure in the manifold and the sequence of valve actuations were controlled by Genie V2 software (Fluidigm). The dead-ended fluid chambers were filled with samples by applying a pressure of 38 kPa to the filling fluid to displace air from the chambers. A pressure of 155 kPa was used to actuate fluid-routing valves. A pressure of 90–117 kPa was used to actuate injection valves positioned over the mixer chambers (Figure 1a) during mixing steps. During mixing, an alternating series of forward and reverse sequences of lipid mixer valve actuations60 was used to avoid formation of stagnation zones in the samples.
Chamber volume measurements
We used one of the chips to determine the ratio of volumes for the aqueous and lipidic components of the mesophase. Here we measured the ratio of fluorescence intensities of the two small chambers and the large chamber of the lipid mixer filled with the same solution of a fluorophore. The chambers were filled with a saturated solution of fluorescein (Acros Organics) in deionized water under the pressure of 38 kPa and the isolation valves were closed without releasing the pressure from the fluid lines. Inter-chamber isolation valves were closed during filling to simulate formulation conditions of lipidic mesophases. Fluorescent images were collected with a Leica N PLAN 2.5× (0.07 NA) objective on a Leica DMI 4000B microscope equipped with a xenon lamp. All images were collected with a charge-coupled device camera (Hamamatsu ORCA-ER, Model C4742-80). Images were processed using ImageJ software (v. 1.46i, Wayne Rasband, NIH, USA) by performing a background correction and a flat-field correction. The ratio of volumes of the lipid compartment and the aqueous solution compartment was calculated as the ratio of cumulative fluorescence intensities of the respective chambers after corrections, resulting in the value of 55 (lipid) : 45 (aqueous solution) v/v. Because the illumination intensity in the images was visibly non-uniform, the accuracy of background and flat-field correction was assessed by comparing the ratio of intensities of chambers in a mixer rotated by 180° in the field of view relative to the original position. After background and flat-field correction the ratio of intensities of the chambers in the original mixer and the rotated mixer differed by less than 0.5%, confirming the validity of the analysis protocol. Furthermore, the fluorescence intensity profile in the rounded features of the fluid layer corresponded to that obtained from profilometry.
The volume ratio of 55 (lipid) : 45 (aqueous solution) obtained from fluorescence intensity measurements was identical for all mixers in the chip. Although only a single chip was tested and was not re-used for mesophase formulation, excellent reproducibility of SAXS results between different chips and agreement between samples of identical compositions on-chip and in glass capillaries confirmed the consistency of metering between different chips.
Sample preparation
MO (Sigma Aldrich, 99%) was used as received. A 20% v/w βOG (Anatrace, Anagrade) solution in 25 mM NaH2PO4, pH 5.5 was diluted with a detergent-free solution of 25 mM NaH2PO4, pH 5.5, to obtain solutions with concentrations of 5% v/w and 10% v/w of βOG. NaH2PO4 was obtained from EMD Chemicals. Several drops of red food coloring solution (McCormick) were added to the 25 mM NaH2PO4, pH 5.5 buffer solution without detergent for visualization used for the preparation of on-chip samples. In preliminary off-chip studies we analyzed mesophases with identical lipid:water ratios prepared with and without food coloring in water and established that addition of the food coloring did not affect the phase behavior of MO mesophases.
On-chip samples were prepared 1–4 days prior to the measurements. After preparation, the fluid layer inlets were sealed with Crystal Clear Tape (Hampton Research) and devices were stored at −80 °C to avoid water evaporation. Freezing also eliminated potentially metastable behavior of mesophases.10–15 Prior to measurements, the samples were defrosted and held at data collection temperature (25 ± 0.5 °C) for at least 2 h. Due to chaotic fluid movement during defrosting of the sample, control lines for chamber inlet valves were actuated at a pressure of 208 kPa throughout defrosting and equilibration.
Samples prepared using coupled-syringe mixers were formulated gravimetrically for a total sample amount of 30–40 mg. Although samples on-chip were metered by volume, weight and volume compositions were nearly identical because densities of both MO and βOG solutions are very close to 1 g/mL.61–63 Each sample was dispensed into four thin-walled 0.9 mm glass capillaries (Charles Supper), and sealed with CritoSeal (Leica Microsystems) and QuickSet Epoxy (Henkel). The capillaries were stored at −80 °C and equilibrated at data collection temperature (25 ± 0.5 °C) for at least 2 h prior to data collection.
SAXS data collection
SAXS data collection was performed on a protein crystallography beamline 21-ID-D at the Advanced Photon Source (APS), Argonne National Lab. Beam of X-rays of 1.55 Å wavelength was focused by mirrors and collimated by slits to a size of 20 μm and a flux of 1.3·1012 photons/s on the sample.64 For SAXS measurements, a 850 mm vacuum flight tube and a 5 mm beam stop were installed between the sample and the CCD detector (Rayonix MX300), providing a sample-to-detector distance of 885 mm. The beamline was equipped with a microdiffractometer (MAATEL MD2) consisting of an on-axis video microscope, a goniometer, and an XYZ micropositioner. A detailed description of the setup is available elsewhere.64
Data were collected in a grid of points spaced 200–250 μm apart (Figure S3 in the ESI), preventing any overlapping of exposed sample volumes.65,66 Exposure times from 1 to 5 seconds and beam attenuations of 80–95% were tested to monitor radiation damage of the stationary mesophase samples. We established that phase assignments and lattice parameters of monoolein mesophases were not affected by radiation damage12,65,67 at exposure conditions selected for our measurements (beam attenuation of 80%, exposure time of 2 s). These parameters corresponded to the accumulated radiation dose of 200 Mrad at each measurement point,68 which was within the range reported previously for SAXS analysis of stationary lipidic mesophases performed with a microbeam at APS.12
SAXS data processing
Raw diffractograms were integrated in Fit2D software (v. 12.077, A.P. Hammersley, ESRF). Integrated diffractograms were processed in MATLAB (R2008a, v. 7.6.0.324, The MathWorks Inc., Natick, MA). Publicly available MATLAB code (findpeaks.m, T.C. O’Haver, v2, revised Oct 27, 2006) was used for finding peak positions. A series of MATLAB scripts was developed for baseline correction, and phase assignment for integrated diffractograms. Scattering profiles from empty microfluidic chips were used for background correction. All calculations below were performed using background-corrected diffractograms. Accuracy of all phase assignments was verified manually. For phase assignment diffraction angles were converted into d-spacings using Bragg’s law:69
| (1) |
where λ is the wavelength of the incident X-ray beam, h, k, and l are the Miller indices of a given family of crystal planes, dhkl is the corresponding interplanar spacing, and θhkl is the scattering angle. The scattering vector q is related to the scattering angle
| (2) |
The cubic space groups Pn3m (Q224) and Ia3d (Q230) were identified based on characteristic sequences of allowed reflections. Allowed reflections for the Pn3m space group are hkl = 110, 111, 200, 211, 220, 221/300, 310, etc. The corresponding ratio of inverse d-spacings is , etc. For the Ia3d space group, allowed hkl reflections are 211, 022, 321, 004, 042/332, 422, 431, etc., resulting in the ratio of inverse d-spacings of , etc. The diffraction pattern of lamellar phases consists of a series of peaks at the ratio of inverse d-spacings of 1:2:3, etc.
Peak positions and intensities of highest-intensity reflections (110 for Pn3m, 211 for Ia3d, and 100 for Lα) were used to calculate lattice parameters and the relative amounts of mesophases. The standard crystallographic relationship between d-spacings and the lattice parameter for cubic phases was used to calculate lattice parameters:69
| (3) |
where a is the lattice parameter. Bilayer repeat distance (lattice parameter) for the lamellar phase obeys the same equation. Relative amounts of Ia3d and Lα mesophases in mixed samples were calibrated using the average intensities in data points where only a single mesophase type was observed. For Pn3m phases we used theoretical relationships70,71 to estimate the intensity for calibration because diffractograms with Pn3m-only samples were not found in our measurements. The complete procedure used to calculate relative amounts of mesophases is described in the ESI.
We report average lattice parameters and relative amounts of mesophases for each mixer (sample). In the calculations of average values we systematically excluded certain points based on their location in the mixers (Figure S3). This was done to avoid artifacts related to cross-talk between sample compartments and fluidic lines and systematic error due to slight dehydration of mixer chambers closest to edges of the chip.
Results and Discussion
Design and operation of microfluidic platform
The microfluidic platform reported here is capable of screening the phase behavior of lipidic mesophases by simultaneously preparing samples of different composition from only three stock solutions. The platform formulates mesophases by mixing a lipid with detergent solutions. In all on-chip samples the lipid/solution ratio was kept constant while the detergent concentration in solution was varied for different samples via automated on-chip dilution. The formulation with a constant lipid/solution ratio and different detergent concentrations is typical of membrane protein crystallization trials. These trials usually follow the same mesophase formulation protocol, but the detergent concentration may vary significantly from one batch of protein solution to another.4,15
Chip fabrication
The microfluidic platform fabricated here met the two main requirements for the on-chip preparation and on-chip analysis of lipidic mesophases: (i) active mixing capabilities and (ii) X-ray transparency. The platform was fabricated using soft lithography methods with several modifications (see Materials and Methods). The original lipid mixer43 relied on actuation of elastomeric pneumatic microvalves fabricated in PDMS. To retain mixing capabilities while ensuring X-ray transparency only thin PDMS control and fluid layers were retained in the device. These thin PDMS layers were sandwiched between thin COC layers (Figure 1c) that provided rigidity and served as a water evaporation barrier due to a significantly lower water permeability of COC72 compared to PDMS.73 The overall thickness of the device was ~220 μm, significantly thinner than traditional PDMS-on-glass microfluidic devices. Sample compartments had a nominal height of ~15 μm. The cumulative thicknesses of PDMS and COC layers above and below sample compartments were 30 μm and 150 μm, respectively, with a calculated X-ray transmission factor of 87% at a wavelength of 1.55 Å. All layers were irreversibly bonded to each other to enable active mixing, during which high pressures are applied.
Chip design
The chip was designed to screen the effect of an additive such as a detergent on the phase behavior of lipidic mesophases. The chip was capable of preparing several different samples from just three stock solutions. Due to space constraints during X-ray data collection the size of the sample area was limited to 8 × 4 mm, and the overall size of the chip to 12 × 30 mm. Up to four sample preparation units could be incorporated per chip under those limitations (Figure 1a, b and Figure S1). Identical design principles can be used to scale out the number of samples per chip.
The lipid mixer was described in detail in our previous work.43 Briefly, the mixer consisted of three chambers that were filled with desired components and were connected with each other via short, narrow channels. Each mesophase sample was prepared in its own lipid mixer by combining a lipid with detergent solutions. The central chambers of the mixers were designated for lipid, and the side chambers for detergent solution (Figure 1b). The lipid to solution ratio was thus fixed at 55 : 45 v/v, but the detergent concentration in solutions could be varied using round dilution chambers adjacent to the side chambers of respective mixers (Figures 1b and 2a). The dilution ratio for detergent was controlled by the different sizes of the round chambers at each mixer.
Figure 2.
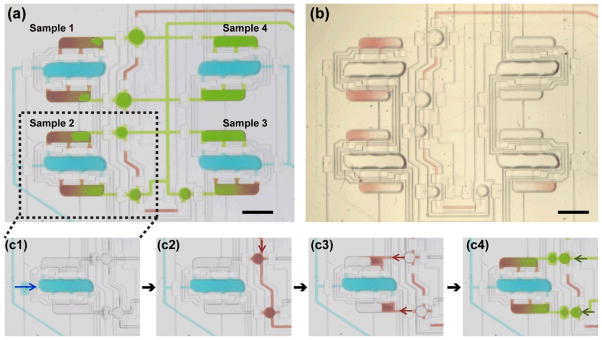
On-chip sample formulation. (a) Optical micrograph of a chip filled with solutions of food coloring to illustrate the composition of samples formulated on-chip. Blue color represents lipid; green, concentrated detergent solution; red, diluent for detergent solution. (b) Optical micrograph of a chip filled with components for mesophase preparation. Lipid: MO (colorless); concentrated detergent solution: 20% βOG in 25 mM NaH2PO4, pH 5.5 (colorless); diluent: 25 mM NaH2PO4, pH 5.5 (red). (c1–c4) Sample formulation protocol. Arrows indicate the direction of fluid movement. (c1) Central chamber of the mixer is filled with lipid (blue). (c2) Auxiliary round dilution chambers are filled with a diluent for detergent solution (red). (c3) Diluent is transferred into side chambers of the mixer by actuating injection valves on top of the round chambers. (c4) Side chambers are filled to volume with concentrated detergent solution (green). Scale bar: 1 mm.
For fluid routing during filling and mixing, the platform relied on normally open valves that were controlled pneumatically.48 To minimize the number of control lines each set of valves performing identical functions were connected using a single control line. Additionally, normally closed valves74 were incorporated at the inlets of all lipid mixer chambers to minimize cross-talk during X-ray data collection when pneumatic actuation was not possible. Schematic operation of normally open and normally closed valves is shown in Figure S2 in the ESI.
Chip operation: filling and metering
A typical filling protocol is illustrated in Figure 2(c1)–(c4). The central chambers of the mixer were filled with lipid, Figure 2(c1). To control the dilution of the detergent solution, the round chambers were filled with a diluent, Figure 2(c2), which was then transferred to adjacent chambers of lipid mixers by actuating valve membranes on top of the round chambers, Figure 2(c3). The partially filled chambers of lipid mixers were then filled to volume with a concentrated detergent solution through a separate fluid line, Figure 2(c4). In this last step the small amount of diluent remaining in the round chambers was also moved to the lipid mixer chambers by the flow of the detergent solution.
All chambers, including round dilution chambers, were dead-ended and filling was carried out by pushing the filling fluid into the chambers and slowly displacing the air75 under a pressure of 38 kPa. The inlet valves for each chamber of the mixer were closed without releasing the pressure from the fluidic lines. The applied pressure caused tenting of the valve membranes on top of each lipid mixer chamber, altering the volume of each chamber from the originally specified design. The ratio of chamber volumes estimated from fluorescence intensity measurements was 55 (lipid) : 45 (solution) v/v, which deviated from the chamber footprint ratio of 1:1 v/v, presumably due to the tenting. However, releasing the pressure before closing the inlet valves would have resulted in less accurate and reproducible metering due to uncontrollable sagging of valve membranes caused by the large aspect ratio of our features. The accuracy of our metering method was further corroborated by the excellent agreement of SAXS data between different chips. We also established that the ratio of volumes of small and round chambers that determined dilution of detergent on-chip were accurately represented by the ratio of footprints of respective chambers based on SAXS data. In this case identical lattice parameters of samples obtained with (Table 1, Chips A–D) and without (Table 1, Chips E–F) dilutions of detergent solution on-chip indicated that footprint-based estimate of dilution was correct and did not require further characterization.
Table 1.
Average lattice parameters and standard deviations (in Å) of mesophases at 25 ± 1 °C in samples prepared and analysed on-chip and in samples prepared using coupled syringes and analysed in glass capillaries. Chips A–D are the same as those in Figure 5.
| Sample composition
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MO/5% βOG 55:45 v/v |
MO/6.7% βOG 55:45 v/v |
MO/8.3% βOG 55:45 v/v |
MO/10% βOG 55:45 v/v |
|||||||||
|
| ||||||||||||
| Detergent solutions and mesophases formulated on-chipa
| ||||||||||||
| Pn3m | Ia3d | Lαb | Pn3m | Ia3d | Lαb | Pn3m | Ia3d | Lαb | Pn3m | Ia3d | Lαb | |
| Chip A | 182 ± 2 | 185 ± 3 | 49.2 | 136 ± 1 | 215 ± 12 | 49.9 | 144 ± 3 | 49.8 | ||||
| Chip B | 187 ± 3 | 49.1 | 183 ± 6 | 48.9 | 136 ± 1 | 190 | 49.7 | 146 ± 2 | 49.7 | |||
| Chip C | 181 ± 2 | 48.9 | 198 ± 1 | 49.5 | 143 ± 3 | 191 | 49.9 | 144 ± 2 | 49.7 | |||
| Chip D | 179 ± 1 | 193 ± 2 | 49.8 | 141 ± 2 | 215 ± 3 | 50.1 | 145 ± 2 | 50.0 | ||||
|
| ||||||||||||
|
Detergent solutions formulated off-chip, mesophases formulated on-chipc
| ||||||||||||
| Chip E | 181 ± 2 | 48.6 | ||||||||||
| Chip F | 182 ± 3 | 48.9 | ||||||||||
| Chip G | 148 ± 3 | 49.8 | ||||||||||
| Chip H | 148 ± 8 | 49.7 | ||||||||||
|
| ||||||||||||
|
Samples prepared in coupled syringes and analyzed in glass capillaries
| ||||||||||||
| CS-1 | 177 ± 2 | 48.4 | ||||||||||
| CS-2 | 173 ± 1 | 49.9 | ||||||||||
Lattice parameters were averaged for each of the four samples on-chip
Standard deviations of lattice parameters for Lα phases were <0.5 Å in all cases.
Four identical samples were prepared on a single chip. The reported value is the average for the four samples.
The chip required a total volume of ~200 nL of each material (lipid, detergent solution, diluent) for filling, a 300-fold reduction compared to preparing four samples in coupled syringes. The nominal chamber volume calculated based on the footprint of the chamber and the height of photoresist was 13.2 nL for lipid chambers and 13.7 nL for detergent solution chambers. However, the actual volume was probably larger due to the membrane tenting described above, but should not exceed 36.8 nL and 38.2 nL for lipid and detergent solution chambers, respectively, as estimated from the footprint of the chambers and the sum of heights of chambers in the fluid and the control layer.
Chip operation: mixing
Mixing was achieved by pneumatic actuation of injection valves located over each chamber. These valves drove the fluid from one chamber to another (Figure 1a). The valve actuation sequence used in this work was identical to that used for the original lipid mixer as we described previously along with the mechanisms governing the mixing process.43 Available data21 indicate that at shear rates relevant for our devices (~0.001 s−1), viscosities of lamellar, cubic Pn3m, and cubic Ia3d phases may be as high as 103, 106, and 107 Pa·s, respectively. In our mixing tests we established that samples designed to produce lamellar phases were easily mixed and routed in the mixer. The more viscous cubic phases, however, could not be manipulated once formed, leading to partially mixed, non-uniform samples (Figure 3a). To circumvent this problem, we exploited the phase behavior of lipidic mesophases, which are known for their propensity to form lamellar structures when cooled. Cooling was achieved by placing a piece of dry ice on the device to let the sample material freeze, inducing a cubic-to-lamellar phase transition. Active mixing was started as soon as the material thawed and was carried out for 1–2 min, after which the freeze-thaw-mix cycle was repeated. Because the chips were thin, freezing and thawing itself took under 1 minute. Uniformly mixed lamellar samples were obtained after 5–7 cycles (Figure 3b). Although only tested for MO mesophases here, this strategy should be applicable to other monoacylglycerols, especially when mixed with detergent solutions, due to their increased propensity for the formation of lamellar phases over a wide range of compositions at low temperatures.13,14,76
Figure 3.

Optical micrographs of mesophase samples during mixing. (a) Non-uniform mixtures of cubic (light) and lamellar (dark) mesophases at ambient temperature. (b) Fully mixed mesophases after several freeze-thaw-mix cycles; the dark color indicates a predominantly lamellar state in all mixers. (c) Samples from (b) after 30 min at 23 °C. Changes in the appearance of the samples indicate a phase transition from a lamellar phase to cubic phases. (d) Same as panel (c), under cross-polarized light. Numbers indicate initial βOG concentrations in aqueous compartments and are the same for all panels. Scale bar: 1 mm.
The uniformly lamellar state did not represent the equilibrium phase behavior of our mesophase samples at ambient temperature. After letting the samples rest at room temperature we observed changes in sample appearance indicative of phase transitions (Figure 3c, d). The mesophase samples with the two lowest detergent concentrations appeared predominantly cubic, and the ones with higher detergent concentrations appeared predominantly lamellar. Regions of equilibrium phase coexistence are found in MO/water systems with13,14,77,78 and without10,11 detergent, depending on the composition. Therefore, interspersed lamellar and cubic regions seen in Figure 3d were not indicative of poor mixing of the samples.
Because SAXS analysis could not be performed immediately after preparation, all chips were frozen using dry ice with all chamber inlet valves in an actuated state to minimize evaporation of water from the samples. Since the control lines were filled with a fluid, the samples remained sealed in their respective compartments during storage. The samples were transferred to a freezer without allowing them to defrost and were stored at −80 °C for 1–3 days prior to the measurements. Prior to SAXS analysis, the samples were equilibrated at the data collection temperature (25 ± 1 °C) for 2 h.
Analysis of the phase behavior of mesophases
A high-intensity X-ray source was required for the analysis of on-chip mesophase samples because of their small path length. SAXS data collection was performed at beamline 21-ID-D, of the Advanced Photon Source at Argonne National Lab. A total of 48 locations were probed within each sample (lipid mixer) using a 20 μm X-ray beam (Figure S3). An XY-positioner was used to move the chip in the X-ray beam, and exact beam position was monitored in an on-axis video microscope (Figure 4a). Although only a small part of the chip was visible in the microscope, the beam position could be easily mapped to the location within the device (Figure 4a).
Figure 4.
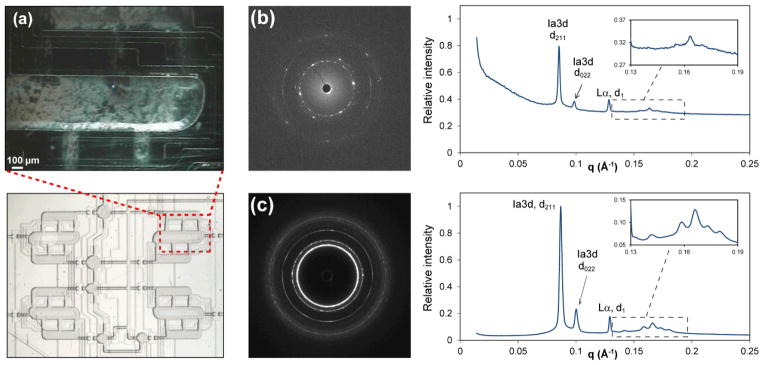
(a) Section of a sample compartment seen in the on-axis video microscope during SAXS data collection (top) and the corresponding sample location (bottom). The bright dot in the center of the top image corresponds to the footprint of the X-ray beam, 20 μm. (b, c) Comparison of diffraction patterns from matching samples prepared on-chip and using a coupled-syringe mixer. The composition of both samples was MO/detergent solution ratio 55:45 v/v; detergent solution: 5% βOG in 25 mM NaH2PO4, pH 5.5. (b) Raw diffractogram of a 15-μm thick on-chip sample and a corresponding integrated diffractogram with peak assignments for prominent reflections. The inset shows a magnified view of higher-order hkl reflections from the Ia3d phase. Lattice parameters: Ia3d, 180 Å, and Lα, 48.9 Å. (c) Raw diffractogram of a ~0.9 mm-thick sample in a glass capillary and a corresponding integrated diffractogram with peak assignments for the most prominent reflections. The inset shows a magnified view of higher-order hkl reflections from the Ia3d phase. Lattice parameters: Ia3d, 177 Å, and Lα, 48.7 Å.
Phase type identification
Comparison of data for samples prepared and analyzed on-chip to those prepared using the standard method (mixing in coupled syringes, analysis in glass capillaries) showed that the microfluidic platform reported here was suitable for analysis of the nanostructure of lipidic mesophases (Figures 4b,c). Integrated diffraction patterns (Figure 4b,c) and, consequently, the structural characteristics, were very similar for the two samples that had near-identical compositions. The main difference between the two types of samples was the signal/noise ratio and the noticeable background scattering at low q values observed only for on-chip samples. This background scattering resulted from PDMS present in the devices.64
The difference in the signal/noise ratio was not unexpected given the ~60-fold smaller sample path length on-chip. On the other hand, X-ray attenuation by device materials or by walls of glass capillaries was unlikely to contribute significantly to the observed differences in the signal/noise ratio. Calculated transmission factors45,47 at an X-ray wavelength of 1.55 Å for materials in the chips (PDMS, 30 μm; COC, 150 μm) and in X-ray glass capillaries (glass, 20 μm) were 87 and 95%, respectively.
The higher signal/noise ratio of samples in capillaries compared to on-chip samples was especially evident in the appearance of higher-hkl reflections of the Ia3d phase (insets in Figures 4b and 4c). However, signal intensity in the on-chip data was sufficient for identifying the types of lipidic mesophases present in these samples. For example, both samples in Figure 4 contained a mixture of the Ia3d cubic phase and a lamellar Lα phase in equilibrium coexistence. Table 1 summarizes phase assignments and lattice parameters of all on-chip samples tested in this work. Examples of diffractograms of samples containing mixtures of Pn3m and Lα phases are shown in Figures S4–S6.
Another noticeable difference was the presence of spotty rings in the raw diffraction pattern of the on-chip samples (Figure 4b) compared to mostly uniform rings of the sample in a capillary (Figure 4c). The spottiness was the result of the path length of the on-chip sample being of comparable size to the monocrystalline domains of the mesophase (up to 200 μm for cubic phases).12 The few monocrystalline domains in the X-ray beam path in the microfluidic device were insufficient to produce uniform powder diffraction rings that require the presence of a large number of randomly oriented domains. The number of domains in the beam path may be expected to vary depending on the location probed in the chip. Similar spotty patterns were also observed for samples in capillaries after maturation for several days.
Trends in phase behavior of lipidic mesophases
Figure 5 shows the relative amounts of various phase types identified for identical samples in four different chips. The excellent reproducibility of the phase behavior data between different devices illustrates the robustness of the microfluidic platform reported here. The phase type, the lattice parameters, and the relative amount of phases agreed well between different chips, indicating the accuracy of on-chip metering to formulate samples with identical compositions.
Figure 5.
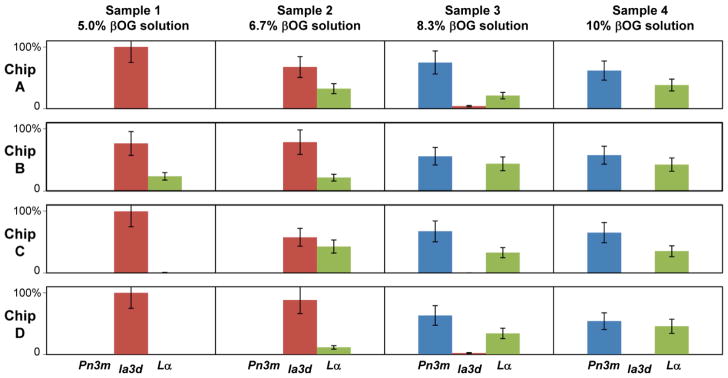
Relative amounts of different phases in samples prepared and analyzed on-chip using four different microfluidic devices at 25 ± 1 °C. The amount of each phase was estimated from intensities of corresponding reflections in SAXS diffractograms. Relative amounts were obtained by normalizing data for each sample to yield the combined amount of all phases of 100%..
At the lowest βOG concentration tested in this work we observed predominantly Ia3d cubic phases. As the amount of detergent progressively increased the following was observed: (i) increasing amounts of the Lα phase; (ii) replacement of the Ia3d cubic phase with the Pn3m cubic phase; and (iii) increasing amount of Lα phase at the expense of the Pn3m cubic phase. The trends are in general agreement with those reported for samples prepared with the standard method and result from the flattening of lipid bilayers upon increasing the MO to detergent ratio due to the differences in molecular shapes of amphiphiles.13,14,77,78
Lattice parameters of mesophases
The lattice parameters of the lipidic mesophases observed on-chip and in capillaries are reported in Table 1. The lattice parameters of cubic phases are highly sensitive to their composition and are important for understanding the properties that govern phase transformations as well as for assessing the suitability of certain mesophases for applications such as membrane protein crystallization or drug delivery. To assess the accuracy of metering on-chip we prepared a set of samples where detergent solution was diluted off-chip on a milliliter scale, and then formulated samples on-chip without dilution, i.e., skipping steps c2 and c3 in Figure 2. The results for samples prepared with 5% βOG solution and 10% βOG solution are also presented in Table 1. Lattice parameters of both Pn3m and Ia3d cubic phases were identical for samples prepared with and without on-chip dilutions, highlighting the metering accuracy of the microfluidic platform and the filling method.
Lattice parameters: comparison of the on-chip and the standard method
To benchmark mesophase microstructural data obtained from on-chip samples we prepared identical mesophase samples of MO mixed with 5% or 10% βOG solution (MO : solution ratio 55:45 w/w) using the standard method. The samples were prepared gravimetrically on a milligram (microliter) scale in a coupled-syringe mixer, followed by dispensing into 0.9 mm glass capillaries and SAXS data collection.
Table 1 shows phase assignments and corresponding lattice parameters obtained from samples in capillaries as well as from on-chip samples. Data of samples prepared with the 5% βOG solution were in very good agreement with respect to phase types and lattice parameters. In the samples prepared with the 10% βOG solution, the phase types for the on-chip and macroscopic samples were the same, Pn3m and Lα, but the lattice parameters for the Pn3m phase were noticeably different, 144–148 Å on-chip vs. 173 Å off-chip. Hence, our results likely illustrate the difficulty of reproducibly preparing and analyzing mesophase samples containing cubic phases with large lattice parameters observed previously. For example, Caffrey et al.14 reported that samples with putatively identical compositions of MO to water ratios of 60:40 w/w may result in either Pn3m or Ia3d phases. Similar phenomena were also observed for detergent-containing samples.14 Batch-to-batch variations in the lattice parameters of MO mesophases have also been reported.13 We established that the discrepancy between on-chip and macroscopic sample behavior observed here did not result from potential inaccuracies in the ratio of volumes of chambers. At the same time, the value of 173 Å is at variance with previously reported lattice parameters of Pn3m cubic phases of MO mixed with detergent solutions,13,14,33 which typically do not exceed 135–145 Å under conditions similar to ours.
To further clarify the origin of mesophase behavior observed here, we performed a new SAXS measurement of samples containing MO and 10% βOG solution in the 55 : 45 w/w ratio after 24 h of storage at 23 °C. We observed changes in the lattice parameters of the Pn3m phase in the four sealed capillaries that initially contained identical samples dispensed from the same coupled-syringe mixer. All samples contained the Lα phase (48.9 – 49.1 Å) coexisting with the Pn3m phase. The lattice parameters of the Pn3m phase varied significantly between capillaries, with one at the original value of 173 Å, one sample at 152 Å, and two samples at 144 Å. The likeliest cause of such changes is minute loss of water from the samples in sealed capillaries, indicating a very strong dependence of the Pn3m lattice parameter on mesophase hydration. Additional tests showed that the loss of 2% of water from the sample was sufficient to drive the Pn3m/Ia3d transition, providing the upper estimate for water loss from devices. The latter may account for previous lack of observations of Pn3m phases with very large lattice parameters of over 170 Å, since consistent observations of such values require very fresh samples. Indeed, the values of lattice parameters in slightly dehydrated samples measured here, 144–152 Å, were much closer to those reported previously and to our on-chip data. Given the aforementioned difficulties, the excellent reproducibility of phase types and lattice parameters for the mesophase samples containing 10% βOG solution between different chips serves as a proof of the robustness of our platform and the validity of our results, even though the lattice parameters are only in qualitative agreement with the initial values for samples in capillaries. The good agreement between on-chip and macroscopic samples containing 5% βOG solution with respect to both phase types and lattice parameters also validates our approach.
Conclusions
The 220-μm thick COC/PDMS platform developed in this work is, to our knowledge, the first example of a microfluidic device that combines metering and active mixing capabilities with X-ray transparency for on-chip SAXS analysis. We demonstrated the capabilities of the platform by mapping a section of the phase diagram of lipid MO mixed with solutions of detergent β-octylglucoside of different detergent concentrations, mimicking conditions of membrane protein crystallization trials. Four samples with different compositions were simultaneously prepared on-chip. Despite the samples being only 15–40 μm thick, the three phase types typical for lipidic mesophases at ambient temperature could be identified in on-chip SAXS data. Analysis of SAXS data revealed excellent reproducibility of the phase types and lattice parameters of lipidic mesophases between different chips, confirming the high accuracy of sample formulation on-chip. Similarly, data from on-chip samples agreed well with those for samples prepared using the standard method by mixing in coupled syringes.
The platform developed here is a viable alternative to the standard method of sample preparation. The platform reduces the amount of material required for sample formulation and analysis 300-fold, which makes it a valuable tool for analyzing the suitability of scarcely available novel lipids for membrane protein crystallization. Design principles used in this work can be used to scale out the number of samples per chip. With small modifications the platform could also be used to screen the phase behavior of lipidic mesophases in a wide range of lipid/solution ratios in order to gain understanding of driving forces behind phase transitions in liquid crystalline systems.
Supplementary Material
Acknowledgments
This work was funded by NIH through its roadmap for membrane protein structure program (R01 GM086727). Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the University of Illinois as well as the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). We thank Jay Von Osinski for his assistance in the development of the SAXS setup, and Sudipto Guha for his help with SAXS data collection.
Footnotes
Electronic Supplementary Information (ESI) available: layouts of the contol and fluid layer (Figure S1), schematic operation of pneumatic microvalves (Figure S2), locations probed in on-chip SAXS data collection (Figure S3), examples of diffractograms of samples with Pn3m and Lα phases (Figures S4–S6), and details of the calculations of relative amounts of the mesophases in Figure 5. See DOI: 10.1039/b000000x/
Contributor Information
Keith Brister, Email: brister@northwestern.edu.
Paul J.A. Kenis, Email: kenis@illinois.edu.
Notes and references
- 1.Kulkarni CV, Wachter W, Iglesias-Salto G, Engelskirchen S, Ahualli S. Phys Chem Chem Phys. 2011;13:3004–3021. doi: 10.1039/c0cp01539c. [DOI] [PubMed] [Google Scholar]
- 2.Guo C, Wang J, Cao F, Lee RJ, Zhai G. Drug Discov Today. 2010;15:1032–1040. doi: 10.1016/j.drudis.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 3.Nazaruk E, Bilewicz R, Lindblom G, Lindholm-Sethson B. Anal Bioanal Chem. 2008;391:1569–1578. doi: 10.1007/s00216-008-2149-y. [DOI] [PubMed] [Google Scholar]
- 4.Caffrey M, Cherezov V. Nat Protocols. 2009;4:706–731. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caffrey M. Biochem Soc Trans. 2011;39:725–732. doi: 10.1042/BST0390725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chien EYT, Liu W, Zhao Q, Katritch V, Won Han G, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larsson K. Nature. 1983;304:664–664. [Google Scholar]
- 10.Briggs J, Chung H, Caffrey M. J Phys II. 1996:723–751. [Google Scholar]
- 11.Qiu H, Caffrey M. Biomaterials. 2000;21:223–234. doi: 10.1016/s0142-9612(99)00126-x. [DOI] [PubMed] [Google Scholar]
- 12.Cherezov V, Caffrey M. Faraday Discuss. 2007;136:195–212. doi: 10.1039/b618173b. [DOI] [PubMed] [Google Scholar]
- 13.Misquitta Y, Caffrey M. Biophys J. 2003;85:3084–3096. doi: 10.1016/S0006-3495(03)74727-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ai X, Caffrey M. Biophys J. 2000;79:394–405. doi: 10.1016/S0006-3495(00)76301-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nollert P, Qiu H, Caffrey M, Rosenbusch JP, Landau EM. FEBS Lett. 2001;504:179–186. doi: 10.1016/s0014-5793(01)02747-8. [DOI] [PubMed] [Google Scholar]
- 16.Caffrey M. Curr Opin Struct Biol. 2000;10:486–497. doi: 10.1016/s0959-440x(00)00119-6. [DOI] [PubMed] [Google Scholar]
- 17.Longley W, McIntosh TJ. Nature. 1983;303:612–614. [Google Scholar]
- 18.Sparr E, Wadsten P, Kocherbitov V, Engstrom S. Biochim Biophys Acta. 2004;1665:156–166. doi: 10.1016/j.bbamem.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 19.Cherezov V, Fersi H, Caffrey M. Biophys J. 2001;81:225–242. doi: 10.1016/S0006-3495(01)75694-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Angelov B, Ollivon M, Angelova A. Langmuir. 1999;15:8225–8234. [Google Scholar]
- 21.Mezzenga R, Meyer C, Servais C, Romoscanu AI, Sagalowicz L, Hayward RC. Langmuir. 2005;21:3322–3333. doi: 10.1021/la046964b. [DOI] [PubMed] [Google Scholar]
- 22.Liang Y, Yuan FZ, Weidong C. Rev Sci Instrum. 2010;81:054301. doi: 10.1063/1.3422244. [DOI] [PubMed] [Google Scholar]
- 23.Joseph JS, Liu W, Kunken J, Weiss TM, Tsuruta H, Cherezov V. Methods. 2011;55:342–349. doi: 10.1016/j.ymeth.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conn CE, Darmanin C, Mulet X, Le Cann S, Kirby N, Drummond CJ. Soft Matter. 2012;8:2310–2321. [Google Scholar]
- 25.Shi F, Han Z, Li J, Zheng B, Wu C. Macromolecules. 2011;44:686–689. [Google Scholar]
- 26.Park S, Wijethunga PAL, Moon H, Han B. Lab Chip. 2011;11:2212–2221. doi: 10.1039/c1lc20111e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laval P, Lisai N, Salmon JB, Joanicot M. Lab Chip. 2007;7:829–834. doi: 10.1039/b700799j. [DOI] [PubMed] [Google Scholar]
- 28.Moreau P, Dehmoune J, Salmon JB, Leng J. Appl Phys Lett. 2009;95:033108. [Google Scholar]
- 29.Salmon JB, Leng J. Compt Rend Chim. 2009;12:258–269. [Google Scholar]
- 30.Leng J, Joanicot M, Ajdari A. Langmuir. 2007;23:2315–2317. doi: 10.1021/la063169k. [DOI] [PubMed] [Google Scholar]
- 31.Leng J, Lonetti B, Tabeling P, Joanicot M, Ajdari A. Phys Rev Lett. 2006;96:084503. doi: 10.1103/PhysRevLett.96.084503. [DOI] [PubMed] [Google Scholar]
- 32.Zhou X, Li J, Wu C, Zheng B. Macromol Rapid Comm. 2008;29:1363–1367. [Google Scholar]
- 33.Sugiyama M, Gasperino D, Derby JJ, Barocas VH. Cryst Growth Design. 2008;8:4208–4214. [Google Scholar]
- 34.Anderson MJ, Hansen CL, Quake SR. Proc Natl Acad Sci USA. 2006;103:16746–16751. doi: 10.1073/pnas.0605293103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sommer MOA, Larsen S. J Synchrotron Rad. 2005;12:779–785. doi: 10.1107/S0909049505002621. [DOI] [PubMed] [Google Scholar]
- 36.Hansen CL, Sommer MOA, Quake SR. Proc Natl Acad Sci USA. 2004;101:14431–14436. doi: 10.1073/pnas.0405847101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shim JU, Cristobal G, Link DR, Thorsen T, Jia YW, Piattelli K, Fraden S. J Am Chem Soc. 2007;129:8825–8835. doi: 10.1021/ja071820f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Selimovic S, Gobeaux F, Fraden S. Lab Chip. 2010;10:1696–1699. doi: 10.1039/b925661j. [DOI] [PubMed] [Google Scholar]
- 39.Barrett R, Faucon M, Lopez J, Cristobal G, Destremaut F, Dodge A, Guillot P, Laval P, Masselon C, Salmon JB. Lab Chip. 2006;6:494–499. doi: 10.1039/b517055a. [DOI] [PubMed] [Google Scholar]
- 40.Merlin A, Angly J, Daubersies L, Madeira C, Schöder S, Leng J, Salmon JB. Eur Phys J E. 2011;34:1–7. doi: 10.1140/epje/i2011-11058-y. [DOI] [PubMed] [Google Scholar]
- 41.Brennich ME, Nolting JF, Dammann C, Noding B, Bauch S, Herrmann H, Pfohl T, Koster S. Lab Chip. 2011;11:708–716. doi: 10.1039/c0lc00319k. [DOI] [PubMed] [Google Scholar]
- 42.Daubersies L, Leng J, Salmon JB. Lab Chip. 2013;13:910–919. doi: 10.1039/c2lc41207a. [DOI] [PubMed] [Google Scholar]
- 43.Perry SL, Roberts GW, Tice JD, Gennis RB, Kenis PJA. Cryst Growth Design. 2009;9:2566–2569. doi: 10.1021/cg900289d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dhouib K, Khan Malek C, Pfleging W, Gauthier-Manuel B, Duffait R, Thuillier G, Ferrigno R, Jacquamet L, Ohana J, Ferrer J-L, Theobald-Dietrich A, Giege R, Lorber B, Sauter C. Lab Chip. 2009;9:1412–1421. doi: 10.1039/b819362b. [DOI] [PubMed] [Google Scholar]
- 45.Guha S, Perry SL, Pawate AS, Kenis PJA. Sensors Actuators B. 2012:1–9. doi: 10.1016/j.snb.2012.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hansen CL, Classen S, Berger JM, Quake SR. J Am Chem Soc. 2006;128:3142–3143. doi: 10.1021/ja0576637. [DOI] [PubMed] [Google Scholar]
- 47.Greaves ED, Manz A. Lab Chip. 2005;5:382–391. doi: 10.1039/b415836a. [DOI] [PubMed] [Google Scholar]
- 48.Unger MA, Chou HP, Thorsen T, Scherer A, Quake SR. Science. 2000;288:113–116. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- 49.Zheng B, Tice JD, Roach LS, Ismagilov RF. Angew Chem Int Ed. 2004;43:2508–2511. doi: 10.1002/anie.200453974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steinert CP, Mueller-Dieckmann J, Weiss M, Roessle M, Zengerle R, Koltay P. 20th IEEE Intl. Conf. on Micro Electro Mechanical Systems (MEMS 2007); Hyogo, Japan. 2007. [Google Scholar]
- 51.Ng JD, Clark PJ, Stevens RC, Kuhn P. Acta Crystallogr D. 2008;64:189–197. doi: 10.1107/S0907444907060064. [DOI] [PubMed] [Google Scholar]
- 52.Sauter C, Dhouib K, Lorber B. Cryst Growth Design. 2007;7:2247–2250. [Google Scholar]
- 53.Gerdts CJ, Elliott M, Lovell S, Mixon MB, Napuli AJ, Staker BL, Nollert P, Stewart L. Acta Crystallogr D. 2008;64:1116–1122. doi: 10.1107/S0907444908028060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weinhausen B, Koster S. Lab Chip. 2013;13:212–215. doi: 10.1039/c2lc41014a. [DOI] [PubMed] [Google Scholar]
- 55.Stehle R, Goerigk G, Wallacher D, Ballauff M, Seiffert S. Lab Chip. 2013 doi: 10.1039/c3lc41291a. [DOI] [PubMed] [Google Scholar]
- 56.Russell R, Millett IS, Tate MW, Kwok LW, Nakatani B, Gruner SM, Mochrie SGJ, Pande V, Doniach S, Herschlag D, Pollack L. Proc Natl Acad Sci USA. 2002;99:4266–4271. doi: 10.1073/pnas.072589599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dootz R, Evans H, Köster S, Pfohl T. Small. 2007;3:96–100. doi: 10.1002/smll.200600288. [DOI] [PubMed] [Google Scholar]
- 58.Wu Y, Kondrashkina E, Kayatekin C, Matthews CR, Bilsel O. Proc Natl Acad Sci USA. 2008;105:13367–13372. doi: 10.1073/pnas.0802788105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xia Y, Whitesides GM. Angew Chem Int Ed. 1998;37:550–575. doi: 10.1002/(SICI)1521-3773(19980316)37:5<550::AID-ANIE550>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 60.Tang L, Lee NY. Lab Chip. 2010;10:1274–1280. doi: 10.1039/b924753j. [DOI] [PubMed] [Google Scholar]
- 61.Antonelli ML, Bonicelli MG, Ceccaroni G, Mesa C, Sesta B. Colloid Polymer Sci. 1994;272:704–711. [Google Scholar]
- 62.Kraineva J, Narayanan RA, Kondrashkina E, Thiyagarajan P, Winter R. Langmuir. 2005;21:3559–3571. doi: 10.1021/la046873e. [DOI] [PubMed] [Google Scholar]
- 63.Vacklin H, Khoo BJ, Madan KH, Seddon JM, Templer RH. Langmuir. 2000;16:4741–4748. [Google Scholar]
- 64.Kondrashkina E, Khvostichenko D, Perry SL, von Osinski J, Kenis PJA, Brister K. J Phys Conf Ser. doi: 10.1088/1742-6596/425/1/012013. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cherezov V, Riedl KM, Caffrey M. J Synchrotron Rad. 2002;9:333–341. doi: 10.1107/s0909049502014528. [DOI] [PubMed] [Google Scholar]
- 66.Sanishvili R, Yoder DW, Pothineni SB, Rosenbaum G, Xu S, Vogt S, Stepanov S, Makarov OA, Corcoran S, Benn R, Nagarajan V, Smith JL, Fischetti RF. Proc Natl Acad Sci USA. 2011;108:6127–6132. doi: 10.1073/pnas.1017701108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cheng A, Caffrey M. Biophys J. 1996;70:2212–2222. doi: 10.1016/S0006-3495(96)79787-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kuwamoto S, Akiyama S, Fujisawa T. J Synchrotron Rad. 2004;11:462–468. doi: 10.1107/S0909049504019272. [DOI] [PubMed] [Google Scholar]
- 69.Sands DE. Introduction to Crystallography. W.A. Benjamin, Inc; New York, NY: 1969. [Google Scholar]
- 70.Garstecki P, Hołyst R. Langmuir. 2002;18:2519–2528. [Google Scholar]
- 71.Garstecki P, Hołyst R. Langmuir. 2002;18:2529–2537. [Google Scholar]
- 72.http://www.topas.com/sites/default/files/files/topas_product-brochure_english.pdf.
- 73.Zhang Y, Ishida M, Kazoe Y, Sato Y, Miki N. IEEJ Trans Electrical Electron Eng. 2009;4:442–449. [Google Scholar]
- 74.Lee CC, Sui G, Elizarov A, Shu CJ, Shin YS, Dooley AN, Huang J, Daridon A, Wyatt P, Stout D, Kolb HC, Witte ON, Satyamurthy N, Heath JR, Phelps ME, Quake SR, Tseng HR. Science. 2005;310:1793–1796. doi: 10.1126/science.1118919. [DOI] [PubMed] [Google Scholar]
- 75.Hansen CL, Skordalakes E, Berger JM, Quake SR. Proc Natl Acad Sci USA. 2002;99:16531–16536. doi: 10.1073/pnas.262485199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Misquitta Y, Cherezov V, Havas F, Patterson S, Mohan JM, Wells AJ, Hart DJ, Caffrey M. J Struct Biol. 2004;148:169–175. doi: 10.1016/j.jsb.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 77.Persson G, Edlund H, Amenitsch H, Laggner P, Lindblom G. Langmuir. 2003;19:5813–5822. [Google Scholar]
- 78.Persson G, Edlund H, Lindblom G. Progr Colloid Polym Sci. 2004;123:36–39. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.