

Abstract
Objective
Peroxisome proliferator-activated receptor α agonists reduce blood pressure in rodents, but clinical trials provide conflicting data regarding their effects in humans. We tested the hypothesis that the effect of fenofibrate on blood pressure depends on salt sensitivity.
Methods
Thirty-one hypertensive volunteers (17 salt-resistant, 14 salt-sensitive) completed a randomized, crossover, double-blind protocol with three dietary phases: low salt diet (10 mmol/day) followed by two consecutive high salt diets (200 mmol/day), each for 6 days. During high salt, volunteers were randomized to fenofibrate 160 mg/day or placebo. Hemodynamic and metabolic parameters were measured on the last morning of each treatment arm.
Results
Fenofibrate reduced triglycerides similarly in salt-sensitive and salt-resistant volunteers. Fenofibrate did not affect blood pressure in salt-resistant volunteers. In salt-sensitive volunteers, fenofibrate significantly decreased diastolic (P =0.02 versus placebo) and mean arterial (P = 0.04 versus placebo) blood pressure during high salt. In all volunteers, the decrease in systolic pressure during fenofibrate correlated inversely with the salt sensitivity of mean arterial pressure as a continuous variable. Fenofibrate significantly decreased heart rate, plasma renin activity, and renal vascular resistance during high salt in salt-sensitive volunteers, but not salt-resistant volunteers. Fenofibrate did not affect sodium excretion or weight gain during high salt. The effect of salt intake and fenofibrate on plasma and urine epoxyeicosatrienoic acid concentrations differed in salt-resistant and salt-sensitive volunteers.
Conclusion
Fenofibrate reduces blood pressure, heart rate and renal vasoconstriction in salt-sensitive volunteers, but not in salt-resistant volunteers. These findings have implications for the treatment of hyperlipidemia in hypertensive individuals.
Keywords: blood pressure, fenofibrate, hypertension, peroxisome proliferator-activated receptor α, salt
INTRODUCTION
Hypertension, obesity and the metabolic syndrome have reached epidemic proportions in developed countries and convey increased risk of cardiovascular mortality [1]. Pharmacological strategies to reduce mortality are targeted at reducing individual risk factors such as hyperlipidemia and blood pressure. Fenofibrate, a peroxisome proliferator-activated receptor (PPAR) α agonist, lowers triglycerides and raises high density lipoprotein (HDL) cholesterol. In the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study, treatment with fenofibrate reduced the incidence of cardiovascular events and reduced the incidence of retinopathy requiring laser therapy in patients with type 2 diabetes mellitus [2,3].
Clinical trials provide conflicting information regarding the effect of fibrates on blood pressure. SBP and DBP were similar in patients treated with fenofibrate and placebo in the FIELD study, but patients randomized to placebo were more likely to be using concurrent angiotensin-converting enzyme (ACE) inhibitors, β-blockers and diuretics at the end of the study [2]. Fenofibrate reduced SBP (−3.1 mmHg, 95% CI −6.3 to 0.1 mmHg) and DBP (−2.9 mmHg, −4.9 to 0.08 mmHg, P <0.05 versus placebo) in a multicenter study in patients with dyslipidemia [4]. Bezafibrate has also been reported to reduce blood pressure in a small study of patients with hyperlipidemia [5]. Other clinical studies have suggested that fenofibrate decreases [6], increases [7], or has no effect on [8] blood pressure.
The effect of PPARα agonists on blood pressure may depend on the mechanism of high blood pressure. In rodent models, PPARα agonists improve endothelial function and increase the renal expression of cytochrome P450 enzymes responsible for the formation of epoxyeico-satrienoic acids (EETs) and 20-hydroxyeicosatetraenoic acid (20-HETE) [9–15]. EETs cause vasodilation and both EETs and 20-HETE promote natriuresis by inhibiting tubular sodium transport [16]. Thus, PPARα agonists induce natriuresis and reduce blood pressure in rodent models of salt-sensitive hypertension [10,12,17,18]. Conversely, PPARα deficient mice develop salt-sensitive hypertension [19].
In this study, we tested the hypothesis that the PPARα agonist fenofibrate would reduce blood pressure in humans with salt-sensitive hypertension.
METHODS
Volunteers with mild-to-moderate essential hypertension were studied. The study protocol was approved by the Vanderbilt Institutional Review Board, registered at ClinTrials.gov (NCT00872599), and conducted in accordance with the Declaration of Helsinki. All volunteers provided written, informed consent and underwent a history and physical examination, screening electrocardiogram and laboratory assessment. Hypertension was defined as an untreated, seated SBP of 140 mmHg or greater on three separate occasions; an untreated, seated DBP of 90 mmHg or greater on three separate occasions; or the use of an antihypertensive agent(s) for a minimum of 6 months. Volunteers with secondary forms of hypertension, significant cardiovascular disease (other than essential hypertension and left ventricular hypertrophy), pulmonary and neurological disorders, diabetes, anemia, overt renal insufficiency and/or any other relevant diagnoses which could interfere with the volunteer’s ability to comply with the study protocol were excluded. All antihypertensive medications were discontinued for three weeks prior to the study. Concomitant medications which could potentially affect the study outcomes (e.g. hormone replacement therapy, oral contraceptives, statins or fibrates, glucocorticoids, nonsteroidal anti-inflammatory drugs and anticoagulants) were also discontinued.
Volunteers participated in a randomized, crossover, double-blind protocol with three dietary phases beginning with a low salt diet (10 mmol/day) followed by two consecutive high salt diets (200 mmol/day) each for 6 days (Fig. 1). Low and high sodium diets contained 100 mmol/day potassium, 1000–1350 mg/day calcium and were caffeine-and alcohol-free. After the first day of high salt diet, participants were randomized to receive fenofibrate 160 mg daily (high salt fenofibrate arm, indicated in figures as ‘Feno’) or matching placebo (high salt placebo arm, indicated as ‘Pla’) for 5 days.
FIGURE 1.
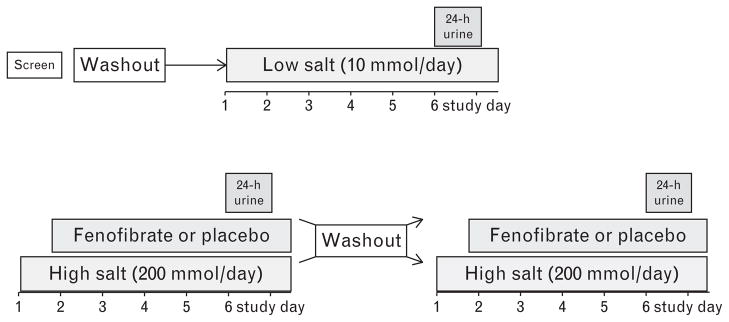
Study protocol. All antihypertensive medications were washed out at least 3 weeks prior to the first study day. Volunteers underwent a washout period of at least 1 week between study arms.
At the end of each dietary phase, volunteers collected their urine for 24 h for measurement of electrolytes, and creatinine. On the last day of each treatment arm, volunteers reported to the Vanderbilt General Clinical Research Center after an overnight fast. That morning, volunteers were weighed and urine samples were collected for measurement of creatinine, catecholamines, 20-HETE, EET and dihydroxyeicosatrienoic acid (DHET) concentrations. Volunteers were then studied in the supine position. An intravenous catheter was placed in each arm, one for medication infusion and the other for blood sampling. Para-aminohippuric acid (PAH) was administered as a continuous, steady-state infusion for 1 h for calculation of renal plasma flow (RPF) as previously described [20]. Blood pressure and heart rate were measured in triplicate at 45 min and 1 h, using an automated device (DINAMAP: Critikon, Carlsbad, California, USA). Blood was then drawn at 45 min and 1 h for measurement of plasma renin activity (PRA), aldosterone, glucose, insulin, plasma EETs, lipid profile (1 h only), catechol-amines (1 h only) and PAH. During high salt study days volunteers also completed an intravenous glucose tolerance test, using a previously described protocol [21].
After completing the first high salt arm, volunteers underwent a washout period of at least 1 week before crossing-over to the second 6-day high salt arm, during which they received the opposite drug (placebo or fenofibrate) for the last 5 days. At the conclusion of the study, volunteers were classified as salt-sensitive if the average study day mean arterial pressure (MAP) was at least 5 mmHg higher during the high salt placebo arm than during low salt intake.
Laboratory analysis
Blood samples were collected on ice and centrifuged for 20 min. All samples were frozen and stored at −80°C until assay. Total cholesterol, HDL-cholesterol and triglycerides were determined using ACE cholesterol, HDL-cholesterol and triglyceride reagent, respectively, and performed on the Alfa Wasserman ACE clinical chemistry system (ALFA Wasserman, West Caldwell, New Jersey, USA). Urine sodium and potassium concentrations were measured by flame photometry and creatinine by the sodium picrate method. PRA was determined by radioimmunoassay (Dia-Sorin, Stillwater, Minnesota, USA). Aldosterone was determined using a radioimmunoassay with 125I-aldosterone (MP Biomedicals, Irvine, California, USA), a primary antibody to aldosterone (National Institute of Diabetes and Digestive and Kidney Diseases National Hormone and Peptide Program, Torrance, California, USA), and a secondary antirabbit γ-globulin antibody (Linco Research, St Charles, Missouri, USA). PAH concentrations were measured by spectrophotometry and PAH clearance was calculated as previously reported [20]. Renal vascular resistance (RVR) was calculated as MAP/RPF. Urine catechol-amines were batch extracted over alumina measured by high pressure liquid chromatography with electrochemical detection using a method modified from Holmes et al. [22]. Urine 20-HETE, EETs and DHETs and plasma EETs were quantified using high pressure liquid chromatography/ tandem mass spectrometry as previously described [23–25].
Statistical analysis
Data are presented as mean ± SD, unless otherwise noted. A paired t-test or signed rank test was used to test within volunteer differences (e.g. fenofibrate versus placebo). A two-sample t-test or Wilcoxon rank sum test was used to test between groups (between salt-sensitive and salt-resistant). Correlations between changes in endpoints were measured using Spearman’s correlation coefficient. A two-sided P value 0.05 or less was considered statistically significant.
Because this is a crossover study, we tested for carry-over or period effects on SBP, DBP, MAP and triglycerides using the t-test approach proposed by Jones and Kenward [26]. There was no evidence for carry-over or period effect on any of these parameters.
To test whether adequate washout of antihypertensive medication was achieved prior to the first (low salt) study day, we assessed whether there was any relationship between prestudy antihypertensive medication [ACE inhibitor or angiotensin receptor blocker (ARB), β-blocker and thiazide diuretic] and volunteer characteristics measured during low salt intake. We found no relationship between prestudy medication and blood pressure, heart rate, RPF, RVR or plasma or urine EETs measured during the first low salt study day. In three volunteers (two salt-resistant, one salt-sensitive) who were treated with a β-blocker prior to washout, urine sodium excretion was significantly higher (37.6 ± 9.9 versus 21.9 ± 19.9 mmol/day) and PRA was significantly lower (0.7 ± 0.4 versus 3.4 ± 2.6 ng Ang I/ml per h) during low salt intake compared to 28 volunteers who had not taken a β-blocker. Volunteers who had been treated with a thiazide diuretic prior to the 3-week washout period had a lower PRA during low salt intake compared with those who had not (2.0 ± 2.0 versus 3.8 ± 2.8 ng Ang I/ml per h).
RESULTS
Thirty-one volunteers completed the study. Seventeen (59%) were salt-resistant and fourteen (41%) were salt-sensitive, as defined as having a 5 mmHg or greater difference between MAP measured during low salt and high salt intake. Volunteer characteristics appear in Table 1. Salt-resistant and salt-sensitive volunteers did not differ with respect to these baseline measurements.
TABLE 1.
Volunteer characteristics in salt-resistant and salt-sensitive subjects at screening
| Salt-resistant (N=17) | Salt-sensitive (N=14) | |
|---|---|---|
| Age (years) | 45.7 ± 11.4 | 42.1 ± 11.8 |
|
| ||
| Gender (male: female) | 6 : 11 | 9 : 5 |
|
| ||
| Race (white: black) | 12 : 5 | 9 : 5 |
|
| ||
| Body mass index (kg/m2) | 29.3 ± 4.8 | 29.9 ± 6.6 |
|
| ||
| Waist to hip ratio | 0.90 ± 0.11 | 0.92 ± 0.15 |
|
| ||
| Fasting glucose (mg/dl) | 90.5 ± 10.6 | 88.2 ± 12.5 |
|
| ||
| Triglycerides (mg/dl) | 102.7 ± 73.4 | 103.3 ± 55.9 |
|
| ||
| Cholesterol-HDL (mg/dl) | 54.1 ± 20.0 | 52.1 ± 15.8 |
|
| ||
| Mean arterial pressure (mmHg) | 96.1 ± 8.6 | 95.9 ± 7.0 |
|
| ||
| Systolic blood pressure (mmHg) | 129.8 ± 14.4 | 129.9 ± 13.5 |
|
| ||
| Diastolic blood pressure (mmHg) | 79.2 ± 7.5 | 78.9 ± 5.5 |
|
| ||
| Antihypertensive medications | ||
| 0 [N (%)] | 3 (18%) | 4 (29%) |
| 1 [N (%)] | 8 (47%) | 8 (57%) |
| 2 [N (%)] | 6 (37%) | 2 (14%) |
|
| ||
| Type of antihypertensive medication | ||
| ACE inhibitor or ARB [N (%)] | 12 (71%) | 6 (43%) |
| β-Blocker [N (%)] | 2 (12%) | 1 (7%) |
| Thiazide diuretic [N (%)] | 6 (35%) | 5 (36%) |
Screening measurements were made prior to the washout of antihypertensive medication. ACE, angiotensin-converting enzyme; HDL, high-density lipoprotein. Data presented as N, % or mean ± SD.
Within salt-sensitive volunteers, DBP (93.8 ± 5.0 versus 81.8 ± 6.8 mmHg, P =0.005) and MAP (109.3 ± 5.5 versus 100.8 ± 6.3 mmHg, P = 0.03) were significantly higher in African–Americans than in Whites during high salt placebo. There were no other effects of race or sex on blood pressure during any treatment arm.
Effect of salt intake and fenofibrate on blood pressure and heart rate
There was no effect of fenofibrate on SBP, DBP, or MAP during high salt intake in salt-resistant volunteers. In contrast, fenofibrate significantly decreased DBP (P = 0.02 versus placebo) and MAP (P = 0.04 versus placebo) during high salt intake in salt-sensitive volunteers after controlling for race (Table 2). The decrease in DBP and MAP during fenofibrate versus placebo was significantly greater in salt-sensitive than in salt-resistant volunteers (Fig. 2a and b). This was true even after adjusting for prestudy antihypertensive medication (P >0.035 for effect of prestudy ACEi or ARB, β-blocker or thiazide diuretic). Although fenofibrate did not significantly reduce SBP in salt-sensitive volunteers compared with salt-resistant volunteers, the decrease in SBP during fenofibrate treatment varied significantly inversely with salt sensitivity of MAP as a continuous variable in all volunteers (Fig. 2d). Heart rate was decreased during high salt placebo compared with low salt intake in all volunteers combined and in salt-resistant volunteers (Table 2). Compared with high salt intake placebo, fenofibrate significantly decreased heart rate further in salt-sensitive volunteers only; moreover, heart rate was significantly lower in salt-sensitive compared with salt-resistant volunteers during fenofibrate (Table 2).
TABLE 2.
Hemodynamic measures by treatment and salt sensitivity
| Low salt | High salt placebo | High salt fenofibrate | |
|---|---|---|---|
| Mean arterial pressure (mmHg) | |||
| Salt-resistant | 98.2 ± 9.8 | 95.5 ± 10.4 | 97.5 ± 10.4 |
| Salt-sensitive | 93.2 ± 5.5 | 103.8 ± 7.2a,e | 100.5 ± 5.8b,c |
| All | 95.9 ± 8.4 | 99.3 ± 9.9b | 98.8 ± 8.7b |
|
| |||
| SBP (mmHg) | |||
| Salt-resistant | 135.6 ± 13.2 | 132.2 ± 13.8 | 134.5 ± 10.2 |
| Salt-sensitive | 124.0 ± 8.8a | 139.4 ± 10.9b | 136.8 ± 12.6b |
| All | 130.4 ± 12.7 | 135.5 ± 12.9b | 135.5 ± 11.2b |
|
| |||
| DBP (mmHg) | |||
| Salt-resistant | 79.5 ± 9.1 | 77.1 ± 9.8 | 78.9 ± 11.1 |
| Salt-sensitive | 77.8 ± 5.5 | 86.1 ± 8.5a,b | 82.4 ± 5.8b,c |
| All | 78.8 ± 7.6 | 81.2 ± 10.1 | 80.5 ± 9.1 |
|
| |||
| Heart rate (bpm) | |||
| Salt-resistant | 69.0 ± 8.0 | 63.6 ± 9.4b | 66.2 ± 8.6b |
| Salt-sensitive | 64.2 ± 9.0 | 62.9 ± 13.8 | 59.2 ± 7.6a,b,c |
| All | 66.9 ± 8.7 | 63.3 ± 11.4b | 63.1 ± 8.8b |
|
| |||
| Renal plasma flow (ml/min per 1.73 m2) | |||
| Salt-resistant | 750.4 ± 171.0 | 794.5 ± 190.6 | 800.2 ± 152.8 |
| Salt-sensitive | 675.7 ± 149.8 | 711.3 ± 171.4 | 716.0 ± 168.7 |
| All | 716.6 ± 163.5 | 756.9 ± 184.1 | 763.7 ± 162.7b |
|
| |||
| Renal vascular resistance (U) | |||
| Salt-resistant | 0.137 ± 0.030 | 0.127 ± 0.032 | 0.126 ± 0.0280 |
| Salt-sensitive | 0.144 ± 0.031 | 0.154 ± 0.038a | 0.147 ± 0.031c |
| All | 0.140 ± 0.030 | 0.139 ± 0.037 | 0.135 ± 0.031 |
Data presented as N or mean ± SD.
P <0.05 versus salt-resistant.
P <0.05 versus low salt intake.
P <0.05 versus high salt intake placebo, controlling for race in the case of blood pressure.
FIGURE 2.

Effect of salt intake and fenofibrate on mean arterial pressure (MAP), SBP and DBP. Changes from low salt to high salt intake and placebo (ΔPla-Low), from low salt to high salt intake and fenofibrate (ΔFeno-Low), and during high salt and fenofibrate compared with high salt intake and placebo (ΔFeno-Pla) are indicated. Volunteers were classified as salt-sensitive if their MAP was at least 5 mmHg higher during high salt and placebo compared with during low salt intake. Panel (d) shows the effect of fenofibrate treatment on SBP during high salt intake (Δ SBPFeno-Pla) as a function of salt sensitivity of blood pressure defined as the difference in MAP between low salt and high salt intake during placebo (Δ MAPPla-Low). Grey filled circles indicate those volunteers defined as salt-sensitive.
Renal plasma flow increased during high salt intake in all volunteers, although this was significant only during fenofibrate. The increment in RPF during high salt intake and fenofibrate treatment compared with low salt intake was similar in salt-resistant and salt-sensitive volunteers. RVR, however, was significantly higher in salt-sensitive compared with salt-resistant volunteers during high salt intake; in addition, fenofibrate significantly reduced RVR in salt-sensitive volunteers (Table 2).
Effect of salt intake and fenofibrate on metabolic, electrolyte, and endocrine parameters
During high salt intake, fenofibrate reduced plasma triglycerides significantly and similarly in salt-resistant and salt-sensitive volunteers (Table 3). Fenofibrate also increased the acute insulin response in both groups, but this reached significance only in the combined groups (from 401.5 to 497.6 mU/l per min, P =0.04).
TABLE 3.
Metabolic and endocrine parameters by treatment and salt sensitivity
| Low salt | High salt placebo | High salt fenofibrate | |
|---|---|---|---|
| Triglycerides (mg/dl) | |||
| Salt-resistant | 74.2 ± 34.1 | 76.6 ± 35.8 | 56.6 ± 28.3a,b |
| Salt-sensitive | 83.9 ± 31.8 | 82.4 ± 25.6 | 59.4 ± 18.1a,b |
| All | 78.6 ± 32.9 | 79.1 ± 31.4 | 57.8 ± 24.1a,b |
|
| |||
| Fasting glucose (mg/dl) | |||
| Salt-resistant | 92.4 ± 10.4 | 94.5 ± 12.6 | 93.3 ± 10.5 |
| Salt-sensitive | 95.6 ± 11.4 | 92.6 ± 10.8 | 88.8 ± 6.9a |
| All | 93.8 ± 10.8 | 93.5 ± 11.6 | 91.3 ± 9.3 |
|
| |||
| Weight (kg) | |||
| Salt-resistant | 82.6 ± 17.3 | 83.0 ± 17.0 | 83.0 ± 17.4 |
| Salt-sensitive | 91.5 ± 27.1 | 92.5 ± 26.3 | 92.1 ± 26.5 |
| All | 86.6 ± 22.3 | 87.3 ± 21.9a | 87.1 ± 22.0a |
|
| |||
| Urinary sodium (mmol/day) | |||
| Salt-resistant | 28.0 ± 24.5 | 187.1 ± 74.5a | 181.8 ± 74.2a |
| Salt-sensitive | 21.4 ± 13.8 | 204.9 ± 73.5a | 216.0 ± 70.5a |
| All | 25.0 ± 20.3 | 195.1 ± 73.4a | 197.2 ± 73.4a |
|
| |||
| Urinary potassium (mmol/day) | |||
| Salt-resistant | 79.6 ± 16.8 | 90.2 ± 30.9 | 101.3 ± 31.5a |
| Salt-sensitive | 67.5 ± 19.9 | 96.2 ± 40.8a | 97.9 ± 40.3a |
| All | 74.1 ± 18.9 | 92.9 ± 35.2a | 99.8 ± 35.1a |
|
| |||
| Urine creatinine (g/day) | |||
| Salt-resistant | 1.41 ± 0.45 | 1.33 ± 0.36 | 1.26 ± 0.44 |
| Salt-sensitive | 1.66 ± 0.57 | 1.50 ± 0.71 | 1.40 ± 0.62 |
| All | 1.53 ± 0.51 | 1.41 ± 0.55 | 1.32 ± 0.52a |
|
| |||
| Aldosterone (pg/ml) | |||
| Salt-resistant | 164.7 ± 75.8 | 76.8 ± 32.7a | 81.2 ± 40.5a |
| Salt-sensitive | 141.6 ± 59.9 | 64.6 ± 20.6a | 63.8 ± 19.3a |
| All | 154.2 ± 68.9 | 71.3 ± 28.1a | 73.6 ± 33.7a |
|
| |||
| PRA (ng Ang l/ml per h) | |||
| Salt-resistant | 3.7 ± 3.1 | 0.78 ± 0.60a | 0.67 ± 0.41a |
| Salt-sensitive | 2.5 ± 1.7 | 0.71 ± 0.65a | 0.51 ± 0.46a,b |
| All | 3.2 ± 2.6 | 0.75 ± 0.62a | 0.60 ± 0.44a |
|
| |||
| Plasma norepinephrine (pg/ml) | |||
| Salt-resistant | 244.1 ± 83.5 | 173.8 ± 79.3a | 192.5 ± 112.2a |
| Salt-sensitive | 260.1 ± 94.5 | 176.6 ± 93.9a | 176.2 ± 80.0a |
| All | 251.3 ± 87.5 | 175.1 ± 84.9a | 185.4 ± 98.3a |
|
| |||
| Urine DHPG (μg/day) | |||
| Salt-resistant | 134.7 ± 118.3 | 40.1 ± 23.9a | 64.5 ± 57.1a,b |
| Salt-sensitive | 85.1 ± 45.8 | 47.9 ± 31.1a | 43.8 ± 32.9a |
| All | 112.3 ± 94.9 | 43.6 ± 27.2a | 55.1 ± 48.1a |
|
| |||
| Urine norepinephrine (μg/day) | |||
| Salt-resistant | 133.1 ± 141.2 | 33.8 ± 34.0a | 58.4 ± 88.8a |
| Salt-sensitive | 93.4 ± 117.9 | 33.2 ± 33.3a | 34.5 ± 46.8a |
| All | 115.2 ± 130.6 | 33.5 ± 33.1a | 47.6 ± 72.8a |
|
| |||
| Urine epinephrine (μg/day) | |||
| Salt-resistant | 17.3 ± 35.4 | 4.1 ± 4.0 | 8.1 ± 8.4b |
| Salt-sensitive | 8.3 ± 7.2 | 6.6 ± 11.3 | 3.8 ± 3.9a |
| All | 13.2 ± 26.7 | 5.2 ± 8.1 | 6.2 ± 7.0 |
|
| |||
| Urine dopamine (μg/day) | |||
| Salt-resistant | 560.0 ± 717.1 | 167.3 ± 113.7a | 234.3 ± 171.9 |
| Salt-sensitive | 346.0 ± 249.5 | 200.9 ± 118.8a | 166.0 ± 129.9a |
| All | 463.3 ± 559.4 | 182.5 ± 115.4a | 203.5 ± 115.8a |
Data presented as N or mean ± SD. DHPG, dihydroxylphenylglycol; PRA, plasma renin activity.
P <0.05 versus low salt.
P <0.05 versus high salt placebo.
Twenty-four hour urine excretion indicated dietary compliance and was similar in salt-resistant and salt-sensitive volunteers (Table 3). Fenofibrate did not affect steady-state 24-h urine sodium excretion. Volunteers gained weight during high salt intake. During high salt intake there was no difference in weight between placebo and fenofibrate treatment. Salt-sensitive volunteers weighed more than salt-resistant volunteers under all conditions. The increment in weight gain during high salt intake was not significantly different in salt-sensitive and salt-resistant volunteers.
PRA and aldosterone concentrations decreased significantly during high salt intake compared with low salt intake, and were comparable in salt-sensitive and salt-resistant volunteers during low salt intake and high salt intake placebo (Table 3). There was no effect of fenofibrate on aldosterone concentrations during high salt intake. Fenofibrate did not affect PRA in salt-resistant volunteers during high salt intake. In contrast, fenofibrate reduced PRA compared with placebo in salt-sensitive volunteers during high salt intake.
There was no influence of prestudy antihypertensive medication on the effect of fenofibrate on heart rate, RVR or PRA (all P >0.30 for ACE inhibitor or ARB, β-blocker or thiazide diuretic).
Plasma norepinephrine and 24-h urine catecholamine excretion (Table 3) were significantly lower during high salt intake compared with low salt intake, and were similar in salt-sensitive and salt-resistant volunteers. During high salt fenofibrate, urine dihydroxylphenylglycol (DHPG) and epinephrine excretion were increased compared with high salt placebo in salt-resistant volunteers, but not in salt-sensitive volunteers. There was a similar nonsignificant trend for plasma norepinephrine and 24-h urine nor-epinephrine excretion. During fenofibrate, heart rate correlated with plasma norepinephrine (r = 0.502, P = 0.005) and with urine norepinephrine excretion (r = 0.368, P = 0.04) and tended to correlate with urine DHPG (P = 0.06) and with total catecholamine (P = 0.08) excretion. During fenofibrate, PRA correlated with the increase in urine norepinephrine (r =0.520, P = 0.003), epinephrine (r = 0.442, P = 0.01), and DHPG (r = 0.379, P = 0.04) from high salt alone.
Effect of salt intake and fenofibrate on plasma and urine epoxyeicosatrienoic acids
The effect of salt intake and fenofibrate on plasma and urine EETs differed in salt-resistant and salt-sensitive volunteers. During low salt intake, plasma total EET, 11,12-EET and 8,9-EET concentrations were significantly higher in salt-resistant compared with salt-sensitive volunteers (Fig. 3). In salt-resistant volunteers, high salt and fenofibrate treatment decreased plasma total EET concentrations and 8,9-EET concentrations compared with low salt intake. The decrease in plasma total EETs and 8,9-EET concentrations during high salt fenofibrate was significantly greater in salt-resistant compared with salt-sensitive volunteers (Fig. 3). During high salt intake, the change in plasma 8,9-EET concentration during fenofibrate compared with placebo correlated inversely with the change in DBP (r = −0.386, P = 0.04).
FIGURE 3.

Effect of salt intake and fenofibrate on plasma epoxyeicosatrienoic acid concentrations. Changes in epoxyeicosatrienoic acid (EET) concentrations from low salt to high salt intake and placebo (Δ Pla-Low), from low salt to high salt intake and fenofibrate (Δ Feno-Low), and during high salt and fenofibrate compared with high salt intake and placebo (Δ Feno-Pla) are also shown. *P <0.05 versus low salt intake.
14,15-EET and 11,12-EET were the prominent isomers excreted in the urine (Fig. 4). During low salt intake, concentrations of total EETs, 14,15-EET and 11,12-EET were significantly higher in the urine of salt-resistant compared with salt-sensitive volunteers. In salt-resistant volunteers, high salt and fenofibrate treatment decreased total EET concentrations compared with low salt intake; this was not the case in salt-sensitive volunteers. The decline in urinary total EET and 11,12-EET concentrations during fenofibrate high salt compared with low salt was significantly greater in salt-resistant compared with salt-sensitive volunteers (Fig. 4).
FIGURE 4.

Effect of salt intake and fenofibrate on urine epoxyeicosatrienoic acid (EET) concentrations and soluble epoxide hydrolase (sEH) activity. Changes in EET concentrations from low salt to high salt intake and placebo (Δ Pla-Low), from low salt to high salt intake and fenofibrate (Δ Feno-Low), and during high salt and fenofibrate compared with high salt intake and placebo (Δ Feno-Pla) are also shown. *P <0.05 versus low salt intake, †P = 0.07 versus low salt intake.
Soluble epoxide hydrolase degrades EETs to corresponding DHETs. Soluble epoxide hydrolase activity, calculated as the ratio of urinary DHETs to DHETs and EETs, trended higher in salt-sensitive compared with salt-resistant volunteers during low salt intake (Fig. 4). In salt-resistant volunteers, sEH activity was decreased during high salt intake placebo treatment compared with during low salt intake but not during high salt intake and fenofibrate treatment.
Urine 20-HETE excretion was not significantly different between salt-resistant and salt-sensitive patient and did not differ significantly among treatment arms.
DISCUSSION
The PPARα agonist fenofibrate has been reported to reduce [4,6], increase [7] or have no effect [8] on blood pressure in clinical trials. These inconsistent findings could result from the study of heterogeneous populations, as well as from differences in concurrent treatments or dietary salt intake. In the present study, we assessed the effect of fenofibrate on blood pressure in individuals who were phenotyped for the sensitivity of their blood pressure to salt intake. We report for the first time that, during high salt intake, fenofibrate reduces blood pressure in individuals with salt-sensitive hypertension, but not in those with salt-resistant hypertension. In salt-sensitive hypertensives, blood pressure reduction by fenofibrate was accompanied by decreases in PRA, heart rate and RVR.
Fenofibrate reduced triglycerides similarly in salt-sensitive and salt-resistant volunteers, suggesting that the antihypertensive effect of fenofibrate in salt-sensitive volunteers resulted from an action downstream from PPARα activation. Fenofibrate could reduce blood pressure in this group by decreasing volume or by reducing vascular resistance. The lack of effect of fenofibrate on weight gain during high salt intake indicates that fenofibrate did not alter volume. Rather, the observation that fenofibrate reduced PRA and heart rate in salt-sensitive volunteers suggests that fenofibrate reduced blood pressure by reducing vasoconstriction mediated by the renin–angiotensin and sympathetic nervous systems. Compatible with this, fenofibrate significantly reduced RVR in salt-sensitive volunteers.
At least two other groups have reported that PPARα agonists reduce heart rate. Chew et al. observed that treatment with fenofibrate decreased 24-h heart rate in patients with type 2 diabetes and left ventricular dysfunction [6]. Jonkers et al. [5] found that bezafibrate reduced heart rate in patients with hypertriglyceridemia, without reducing circulating catecholamines. In addition, fenofibrate has been reported to improve sympathetic tone and increase baroreflex sensitivity in obese volunteers [27]. We did not find that fenofibrate decreased plasma or urine catecholamines in salt-sensitive volunteers, but rather that fenofibrate increased activation of the sympathetic nervous system in salt-resistant volunteers and that this effect was absent in salt-sensitive volunteers. Moreover, we found significant relationships between the plasma and urine markers of sympathetic activity and heart rate or PRA, suggesting that diminished activation of the sympathetic nervous system may have contributed to decreased heart rate and PRA in salt-sensitive volunteers during fenofibrate.
On the basis of rodent models, we predicted that plasma and urine EETs would be higher during high salt intake in salt-resistant compared with salt-sensitive volunteers. Surprisingly, we found that both plasma and urine EETs were significantly higher in salt-resistant volunteers compared to salt-sensitive volunteers during low salt intake. As activation of the renin–angiotensin system induces soluble epoxide hydrolase expression [28], we hypothesize that during low salt intake the degradation of EETs by sEH is enhanced in salt-sensitive volunteers; this is supported by the observation that the ratio of urine DHETs/DHETs+EETs tended to be higher in salt-sensitive volunteers during low salt intake.
In rodent models, PPARα agonists induce expression of CYP2C in the kidney [12,15]. Concomitant with the antihypertensive effect of fenofibrate in salt-sensitive volunteers, we found a significantly greater increase in urine EET excretion during high salt intake fenofibrate versus low salt intake in salt-sensitive compared with salt-resistant volunteers. Fibrates also increase the expression of sEH [29], and it is possible that induction of soluble epoxide hydrolase and EET degradation offset any effect of fenofibrate on renal CYP2C expression in salt-resistant volunteers. Of note, in contrast to the effects of PPARα agonists on CYP2C and CYP4A expression in the kidney [9,17], PPARα agonists reduce vascular CYP2C expression and plasma EETs in rodents [30]. In the present study, we found that plasma EETs were decreased during fenofibrate compared with low salt-intake in salt-resistant volunteers, whereas there was no effect of fenofibrate on plasma EETs in salt-resistant volunteers.
PPARα agonists have also been reported to induce renal ω-hydroxylase activity in rodents in some studies but not in all [10,11]. Fenofibrate did not significantly alter urine 20-HETE excretion in either salt-resistant or salt-sensitive volunteers in this study. As reported previously by Laffer et al. [31], urine 20-HETE excretion did not differ significantly in salt-resistant and salt-sensitive volunteers.
In conclusion, hypertension, obesity and the metabolic syndrome convey increased risk of cardiovascular mortality [1]. Fenofibrate reduces triglycerides and the incidence of cardiovascular events in diabetics [2], but studies provide conflicting information regarding the effect of fibrates on blood pressure. We report for the first time that, during high salt intake, the PPARα agonist fenofibrate reduces blood pressure in salt-sensitive hypertensive individuals but not salt-resistant hypertensives. In addition, fenofibrate reduces heart rate, PRA and renal vasoconstriction in salt-sensitive individuals, whereas fenofibrate increased activity of the sympathetic nervous system in salt-resistant volunteers.
Reviewers’ Summary Evaluations.
Reviewer 1
The major strength of the article is that it provides clinical evidence demonstrating that fenofibrate treatment reduces blood pressure in the theg subgroup of salt-sensitive hypertensive patients but not in the subgroup of salt-resistant hypertensive patients. This may help to clarify the conflicting information published previously on the effects of fibrates on blood pressure in hypertensive patients considered as a group. The major limitation of the article is that it does not provide information robust enough to identify the precise molecular mechanism through which fenofibrate treatment reduces blood pressure in salt-sensitive hypertensive patients.
Reviewer 2
Fibrates have an heterogeneous impact on blood pressure. The authors of the present study have investigated the interaction between salt-sensitivity of blood pressure and the renal, hormonal and hemodynamic response to fenofibrate in subjects with mild to moderate hypertension. The strength of the study is a very careful design of the study using a crossover design in which patients were investigated under various salt regimens and medications. The results show clear differences in the response to fenofibrate according to the salt-sensitivity pattern. The major limit of this study, which provides some interesting insights on the action of fibrates, is of course the ability to identify salt sensitive patients in the real life. The complex protocol applied in this study is unfortunately not applicable in clinical practice.
Acknowledgments
This work was funded by the following grants from the National Institutes of Health: DK038226, DK081662, HL060906, and TR000445. K.G. was also funded by a fellowship from the National Kidney Foundation.
Abbreviations
- 20-HETE
20-hydroxyeicosatetraenoic acid
- DHET
dihydroxyeicosatrienoic acid
- EET
epoxyeicosatrienoic acid
- FIELD
Fenofibrate Intervention and Event Lowering in Diabetes
- HDL
high density lipoprotein
- MAP
mean arterial pressure
- PAH
para-aminohippurate
- PPAR
peroxisome proliferator-activated receptor
- PRA
plasma renin activity
- RPF
renal plasma flow
- RVR
renal vascular resistance
Footnotes
Conflicts of interest
There are no conflicts of interest.
References
- 1.Mozumdar A, Liguori G. Persistent increase of prevalence of metabolic syndrome among U.S. adults: NHANES III to NHANES 1999–2006. Diabetes Care. 2011;34:216–219. doi: 10.2337/dc10-0879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keech A, Simes RJ, Barter P, Best J, Scott R, Taskinen MR, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366:1849–1861. doi: 10.1016/S0140-6736(05)67667-2. [DOI] [PubMed] [Google Scholar]
- 3.Keech AC, Mitchell P, Summanen PA, O’Day J, Davis TM, Moffitt MS, et al. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet. 2007;370:1687–1697. doi: 10.1016/S0140-6736(07)61607-9. [DOI] [PubMed] [Google Scholar]
- 4.Nissen SE, Nicholls SJ, Wolski K, Howey DC, McErlean E, Wang MD, et al. Effects of a potent and selective PPAR-alpha agonist in patients with atherogenic dyslipidemia or hypercholesterolemia: two randomized controlled trials. JAMA. 2007;297:1362–1373. doi: 10.1001/jama.297.12.1362. [DOI] [PubMed] [Google Scholar]
- 5.Jonkers IJ, de Man FH, van der LA, Frolich M, Gevers Leuven JA, Kamper AM, et al. Bezafibrate reduces heart rate and blood pressure in patients with hypertriglyceridemia. J Hypertens. 2001;19:749–755. doi: 10.1097/00004872-200104000-00012. [DOI] [PubMed] [Google Scholar]
- 6.Chew GT, Watts GF, Davis TM, Stuckey BG, Beilin LJ, Thompson PL, et al. Hemodynamic effects of fenofibrate and coenzyme Q10 in type 2 diabetic subjects with left ventricular diastolic dysfunction. Diabetes Care. 2008;31:1502–1509. doi: 10.2337/dc08-0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Subramanian S, DeRosa MA, Bernal-Mizrachi C, Laffely N, Cade WT, Yarasheski KE, et al. PPARalpha activation elevates blood pressure and does not correct glucocorticoid-induced insulin resistance in humans. Am J Physiol Endocrinol Metab. 2006;291:E1365–E1371. doi: 10.1152/ajpendo.00230.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diabetes Atherosclerosis Intervention Study Investigators. Effect of fenofibrate on progression of coronary-artery disease in type 2 diabetes: the Diabetes Atherosclerosis Intervention Study, a randomised study. Lancet. 2001;357:905–910. [PubMed] [Google Scholar]
- 9.Roman RJ, Ma YH, Frohlich B, Markham B. Clofibrate prevents the development of hypertension in Dahl salt-sensitive rats. Hypertension. 1993;21:985–988. doi: 10.1161/01.hyp.21.6.985. [DOI] [PubMed] [Google Scholar]
- 10.Zhou Y, Luo P, Chang HH, Huang H, Yang T, Dong Z, et al. Colfibrate attenuates blood pressure and sodium retention in DOCA-salt hypertension. Kidney Int. 2008;74:1040–1048. doi: 10.1038/ki.2008.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee DL, Wilson JL, Duan R, Hudson T, El-Marakby A. Peroxisome proliferator-activated receptor-alpha activation decreases mean arterial pressure, plasma interleukin-6, and COX-2 While increasing renal CYP4A expression in an acute model of DOCA-salt hypertension. PPAR Res. 2011;2011:502631. doi: 10.1155/2011/502631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakagawa K, Holla VR, Wei Y, Wang WH, Gatica A, Wei S, et al. Salt-sensitive hypertension is associated with dysfunctional Cyp4a10 gene and kidney epithelial sodium channel. J Clin Invest. 2006;116:1696–1702. doi: 10.1172/JCI27546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banks T, Oyekan A. Peroxisome proliferator-activated receptor alpha activation attenuated angiotensin type 1-mediated but enhanced angiotensin type 2-mediated hemodynamic effects to angiotensin II in the rat. J Hypertens. 2008;26:468–477. doi: 10.1097/HJH.0b013e3282f2f0f3. [DOI] [PubMed] [Google Scholar]
- 14.Diep QN, Benkirane K, Amiri F, Cohn JS, Endemann D, Schiffrin EL. PPAR alpha activator fenofibrate inhibits myocardial inflammation and fibrosis in angiotensin II-infused rats. J Mol Cell Cardiol. 2004;36:295–304. doi: 10.1016/j.yjmcc.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 15.Muller DN, Theuer J, Shagdarsuren E, Kaergel E, Honeck H, Park JK, et al. A peroxisome proliferator-activated receptor-alpha activator induces renal CYP2C23 activity and protects from angiotensin II-induced renal injury. Am J Pathol. 2004;164:521–532. doi: 10.1016/s0002-9440(10)63142-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao X, Imig JD. Kidney CYP450 enzymes: biological actions beyond drug metabolism. Curr Drug Metab. 2003;4:73–84. doi: 10.2174/1389200033336892. [DOI] [PubMed] [Google Scholar]
- 17.Sankaralingam S, Desai KM, Glaeser H, Kim RB, Wilson TW. Inability to upregulate cytochrome P450 4A and 2C causes salt sensitivity in young Sprague-Dawley rats. Am J Hypertens. 2006;19:1174–1180. doi: 10.1016/j.amjhyper.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 18.Wilson TW, Alonso-Galicia M, Roman RJ. Effects of lipid-lowering agents in the Dahl salt-sensitive rat. Hypertension. 1998;31:225–231. doi: 10.1161/01.hyp.31.1.225. [DOI] [PubMed] [Google Scholar]
- 19.Obih P, Oyekan AO. Regulation of blood pressure, natriuresis and renal thiazide/amiloride sensitivity in PPARalpha null mice. Blood Press. 2008;17:55–63. doi: 10.1080/08037050701789278. [DOI] [PubMed] [Google Scholar]
- 20.Shoback DM, Williams GH, Moore TJ, Dluhy RG, Podolsky S, Hollenberg NK. Defect in the sodium-modulated tissue responsiveness to angiotensin II in essential hypertension. J Clin Invest. 1983;72:2115–2124. doi: 10.1172/JCI111176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ayers K, Byrne LM, DeMatteo A, Brown NJ. Differential effects of nebivolol and metoprolol on insulin sensitivity and plasminogen activator inhibitor in the metabolic syndrome. Hypertension. 2012;59:893–898. doi: 10.1161/HYPERTENSIONAHA.111.189589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holmes C, Eisenhofer G, Goldstein DS. Improved assay for plasma dihydroxyphenylacetic acid and other catechols using high-performance liquid chromatography with electrochemical detection. J Chromatogr. 1994;653:131–138. doi: 10.1016/0378-4347(93)e0430-x. [DOI] [PubMed] [Google Scholar]
- 23.Karara A, Wei S, Spady D, Swift L, Capdevila JH, Falck JR. Arachidonic acid epoxygenase: structural characterization and quantification of epoxyeicosatrienoates in plasma. Biochem Biophys Res Commun. 1992;182:1320–1325. doi: 10.1016/0006-291x(92)91877-s. [DOI] [PubMed] [Google Scholar]
- 24.Capdevila JH, Wei S, Kumar A, Kobayashi J, Snapper JR, Zeldin DC, et al. Resolution of dihydroxyeicosanoates and of dihydroxyeicosatrienoates by chiral phase chromatography. Anal Biochem. 1992;207:236–240. doi: 10.1016/0003-2697(92)90006-s. [DOI] [PubMed] [Google Scholar]
- 25.Falck JR, Anjaiah S, Tuniki VR, Gopal VR, Capdevila JH. Convenient syntheses of [20,20,20-H(3)]-arachidonic acid and [20,20-H(2)]-20-hydroxyeicosatetraenoic acid. J Labelled Comp Radiopharm. 2006;49:245–252. doi: 10.1002/jlcr.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones B, Kenward MG. Design and analysis of crossover trials. Boca Raton, FL: CRC Press LLC; 2003. [Google Scholar]
- 27.Melenovsky V, Wichterle D, Simek J, Malik J, Haas T, Ceska R, et al. Effect of atorvastatin and fenofibrate on autonomic tone in subjects with combined hyperlipidemia. Am J Cardiol. 2003;92:337–341. doi: 10.1016/s0002-9149(03)00643-x. [DOI] [PubMed] [Google Scholar]
- 28.Ai D, Fu Y, Guo D, Tanaka H, Wang N, Tang C, et al. Angiotensin II up-regulates soluble epoxide hydrolase in vascular endothelium in vitro and in vivo. Proc Natl Acad Sci U S A. 2007;104:9018–9023. doi: 10.1073/pnas.0703229104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pinot F, Grant DF, Spearow JL, Parker AG, Hammock BD. Differential regulation of soluble epoxide hydrolase by clofibrate and sexual hormones in the liver and kidneys of mice. Biochem Pharmacol. 1995;50:501–508. doi: 10.1016/0006-2952(95)00167-x. [DOI] [PubMed] [Google Scholar]
- 30.Pozzi A, Ibanez MR, Gatica AE, Yang S, Wei S, Mei S, et al. Peroxisomal proliferator-activated receptor-alpha-dependent inhibition of endothelial cell proliferation and tumorigenesis. J Biol Chem. 2007;282:17685–17695. doi: 10.1074/jbc.M701429200. [DOI] [PubMed] [Google Scholar]
- 31.Laffer CL, Laniado-Schwartzman M, Wang MH, Nasjletti A, Elijovich F. 20-HETE and furosemide-induced natriuresis in salt-sensitive essential hypertension. Hypertension. 2003;41:703–708. doi: 10.1161/01.HYP.0000051888.91497.47. [DOI] [PubMed] [Google Scholar]