

Abstract
A one-pot hydroamination of alkenes is reported. The synthesis of primary and secondary amines from unactivated olefins was accomplished in the presence of a variety of functional groups. Hydrozirconation, followed by amination with nitrogen electrophiles, provides exclusive anti-Markovnikov selectivity, and most products are isolated in high yields without the use of column chromatography.
The conversion of alkenes to alkylamines is a challenging, but synthetically important transformation.1, 2 This transformation traditionally has been conducted by hydroboration, oxidation, and conversion of the alcohol or aldehyde to the amine. A more efficient method would be the combination of hydrometallation and direct conversion of the alkylmetal intermediate to the amine. The combination of hydroboration and reaction of the alkylboron intermediate with nitrogen electrophiles to form the corresponding amine products was described as early as 19643 (Scheme 1a), but these reactions are limited to the formation of primary amines and occurred in modest yields. A more general method to perform anti-Markovnikov amination of alkenes in high yields with readily available reagents and wide functional group tolerance has not been reported.1, 4
Scheme 1.

Anti-Markovnikov amination of alkenes (yields based on 1 equiv ov alkene).
The combination of hydroboration and amination of the resulting alkylboron compound has been revisited since the initial report.5-8 Recently, amination of alkyl boranes to form tertiary amines has been accomplished with copper catalysts.9-12 In addition, three equivalents of n-butyl lithium and MeONH2 have been shown to react with pinacol boronate esters to form primary amines and anilines (Scheme 1c).13
Despite these recent advances in the synthesis of amines from alkenes, there are a number of limitations. Particularly relevant to the work we report, methods for the synthesis of secondary amines from alkenes are limited to isolated examples, and these reactions proceed in low yield (Scheme 1b).14-18 In addition, the utility of reactions to form primary amines is limited by the properties of the reagent used for amination. Hydrazoic acid has been use as the reagent to provide primary amines from alkyl boron reagents in high yields,6 but the explosion hazard of hydrazoic acid limits the utility of this method. Moreover, reactions conducted with the reagent formed from n-butyl lithium and a solution of MeONH2 require long times with boronic esters at elevated temperatures, and 3 equivalents of each are required for acceptable yields (Scheme 1).
To address these limitations, we followed an alternative strategy involving alkylzirconium intermediates. Srebnik and Zheng reported the stepwise addition of Schwartz's reagent (bis(cyclopentadienyl)zirconium chloride hydride), followed by O-mesitylsulfonyl hydroxylamine (MSH), to form primary alkylamine products from alkenes.19 Although this reaction provides a route to terminal primary amines, MSH decomposes over time and requires a multi-step synthesis from hydroxylamine.20 Thus, a method is needed to convert alkylzirconium compounds to primary and secondary alkylamines in high yield with an accessible reagent for the sequence of hydrozirconation and amination to be practical.
Herein we report a method for the synthesis of primary and secondary amines from alkenes by a one-pot procedure based on hydrozirconation that occurs in good to high yields without isolation of intermediates. This protocol is based on preliminary data for the conversion of N-allyl indoles to the corresponding amines21 and is sufficiently mild to tolerate a range of aprotic functional groups. This process includes commercially available and readily synthesized reagents and forms products that were isolated, in most cases, without column chromatography. These attributes make the reaction suitable for the one-pot, anti-Markovnikov hydroamination of complex molecules.
Our studies on the hydroamination of alkenes via hydrozirconation began with an evaluation of the conditions for the conversion of the alkylzirconium compounds to the corresponding amines. The reaction of 1-octylzirconocene chloride (generated from 1-octene and Schwartz reagent) with N-methylhydroxylamine-O-sulfonic acid was complete in 30 minutes at 50 °C, but the yields of the N- methyloctylamine product were variable. Higher yields were obtained consistently if the alkylzirconocene formed in the first step was used immediately.22 Simple evaporation of the solvent from the final solution and aqueous extraction provided pure amine products. Acid-sensitive substrates were subjected to a modified aqueous extraction and purified by column chromatography.
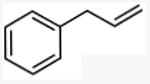
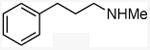
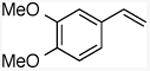
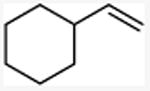
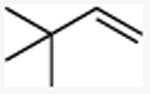
With these conditions in hand, the reactions of a variety of alkenes were evaluated (Table 1). The amination reaction was conducted on a 4 mmol scale without reduction in yield (entry 2), and, at 10 mmol scale, the yield remained synthetically useful (entry 3). The reaction in entry 14 shows that a Boc-protected secondary amine is stable to Schwartz reagent, and the reactions in entries 13, 7, and 8 show that the reaction occurs with strained and 1,1-disubstituted alkenes to form the corresponding amines cleanly. Amination of the sterically hindered vinyl cyclohexane (entry 11) and tert-butyl ethylene (entry 12) proceeded in good yield, although 1.1 equivalents of tert-butyl ethylene were necessary to ensure complete conversion of Schwartz's reagent. Vinylarenes (entries 9 and 10) were converted to substituted phenethylamines without competing amination at the benzylic position. Yields were lower for the reaction of 3-trifluoromethyl styrene than that of 3,4-dimethoxy styrene, and this lower yield is consistent with a higher equilibrium concentration of the benzylic zirconocene complex due to stabilization from the electron deficient arene.
Table 1. Reactions to form N-methylamines.
|
| |||
|---|---|---|---|
| entrya | alkene | product | yieldb |
| 1 |
|
|
89 |
| 2 |
|
|
92c |
| 3 |
|
|
79d |
| 4 |
|
|
83 |
| 5 |
|
|
89 |
| 6 |
|
|
84 |
| 7 |
|
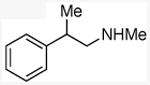
|
67 |
| 8 |
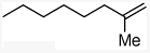
|
|
61e |
| 9 |
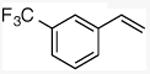
|
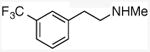
|
53 |
| 10 |
|
|
69 |
| 11 |
|
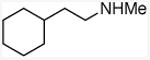
|
67 |
| 12 |
|
|
71f |
| 13 |
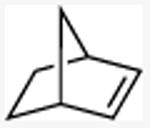
|
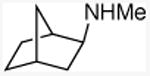
|
80 |
| 14 |
|
|
72e |
conditions: alkene (0.4 mmol), THF (1.5 mL), Cp2ZrHCl (0.4 mmol), MeNHOSO3H (0.6 mmol).
Isolated yield (%) without column chromatography, average of two runs.
4 mmol scale.
10 mmol scale.
isolated yield (%) after column chromatography.
1.1 equiv alkene, yield based on Cp2ZrHCl.
The synthesis of primary amines from commercially available hydroxylamine-O-sulfonic acid was also studied (Table 2). The amination step was complete in minutes at room temperature, and the products were isolated in high yields. In one case, the combination of hydrozirconation and conversion of the alkylzirconium intermediate to the primary amine (entry 4) gave a mixture of benzylic and terminal amines; the same sequence conducted with N-methylhydroxylamine-O-sulfonic acid formed only the terminal amine (Table 1, entry 6). The absence of the benzylic amine product from the sequence that generates the secondary amine suggests that the amination of the branched alkylzirconocene does not occur with the more hindered N-methyl aminating reagent and the remaining starting material is converted to the protonolysis product, propylbenzene.
Table 2.
Reactions to form primary amines.
conditions: alkene (0.4 mmol), THF (1.5 mL), Cp2ZrHCl (0.4 mmol); H2NOSO3H (0.6 mmol).
isolated yield (%) without column chromatography.
combined yield of linear and branched products.
It is well established that the hydrozirconation of internal alkenes forms terminal alkylzirconium products. To test if this feature of hydrozirconation would lead to a method for the one-pot conversion of internal alkenes to terminal amines, the hydrozirconation-amination process was conducted with a mixture of internal alkenes (Table 2, entry 5). 1-Octylamine was the only observed product, indicating complete conversion to the linear alkylzirconocene and amination of the linear alkylzirconocene intermediate.
The alkyl substituent on nitrogen was varied to investigate the potential of this route to form additional terminal secondary amines from alkenes (Table 3). Under the standard conditions, the combination of hydrozirconation of 1-octene, followed by amination of the alkylzirconium compound with N-(3-phenylpropyl)hydroxylamine-O-sulfonic acid, consistently provided 40-50% yields of alkylamine product (entry 1). However, when this sequence was conducted with the amination step at 80 °C, the N-(3-phenylpropyl)amine product was isolated in 72% yield (entry 2). N-heptyl hydroxylamine-O-sulfonic acid was synthesized from heptanal in 3 steps, and the combination of hydrozirconation of octene and reaction with this N-alkyl hydroxylamine-O-sulfonic acid afforded 79% yield of N-heptyl octylamine (entry 3) following the conditions from entry 2. The presence of additional substituents alpha to nitrogen was detrimental to the yield of the desired amine (entries 4 and 5); no product was observed in the case of N,N-dimethyl hydroxylamine-O-sulfonic acid to form tertiary amines at any temperature (entry 6).
Table 3.
Reactions to form secondary amines.

conditions: alkene (0.2 mmol), THF (0.75 mL), Cp2ZrHCl (0.2 mmol); aminating reagent (0.3 mmol), 50 °C, 30 min.
yield determined by NMR spectroscopy with CH2Br2 as an internal standard.
reaction performed at 80 °C, isolated yield (%) after column chromatography, average of two runs.
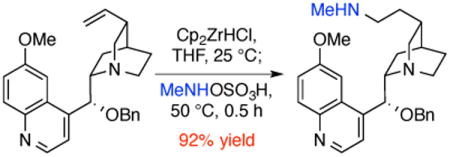
To investigate the ability of this method to be used with complex molecules containing basic nitrogens and oxygenated frameworks, the hydroamination of the cinchona alkaloid 9-O-benzyl quinine 1a was conducted. This substrate might be expected to quench the zirconium hydride by binding of the nitrogen in the quinoline or highly basic quinuclidine. This substrate also contains a doubly benzylic ether, which could be cleaved under reducing conditions. Despite these possible side reactions, the transformation of the alkene to the secondary alkylamine proceeded cleanly to afford the product 1b in 92% yield.
The reaction of mannose-derived 2a was conducted to assess the ability of the hydrozirconation-amination sequence to be conducted with a highly oxygenated substrate. Given the oxophilicity and Lewis acidity of zirconium, cleavage or epimerization at the anomeric position of 2a might be expected to occur. However, this reaction of the alkene formed the secondary alkylamine product 2b in 66% isolated yield. The sole byproduct observed from this reaction was formed by reduction of the alkene, presumably due to protonolysis of the intermediate alkyl zirconocene.
In summary, a one-pot hydroamination of unactivated alkenes has been developed. The synthesis of primary and secondary amines from alkenes containing a variety of functional groups occurs under the reported conditions, and this method was found to be applicable to the derivatization of complex molecules.
Experimental Section
General
Unless otherwise stated, reactions were performed using standard Schlenk and drybox techniques. Liquid alkene substrates were stored over 4 Å molecular sieves in the drybox. Bis(cyclopentadienyl) zirconium chloride hydride was purchased from Strem Chemicals and stored at room temperature in a drybox. Hydroxylamine-O-sulfonic acid was purchased from Sigma Aldrich and stored at 0 °C or in the drybox freezer at -30 °C. N-Boc-(2,2-diphenyl-hept-6-enyl)-methyl-amine,23 9O-benzyl quinine,24 2-methyl-1-octene, and 2-allylphenyl 2,3,4,6-tetra-O-benzyl-α-D-mannopyranoside25 were prepared by literature procedures. Thin-layer chromatography (TLC) plates were visualized by UV fluorescence quenching, potassium permanganate, or p-anisaldehyde staining. Chemical shifts are reported in ppm relative to the residual solvent peak (CDCl3 = 7.26 ppm for 1H and 77.0 ppm for 13C; (CD3)2SO = 2.50 ppm for 1H and 39.52 ppm for 13C) and are reported relative to Me4Si (δ 0.00). Mass spectrometry analyses (ESI–MS) were performed using an ion trap analyzer.
N-methylhydroxylamine-O-sulfonic acid
A 250 mL Schlenk flask was charged with N-methylhydroxylamine hydrochloride (5.00 g, 59.9 mmol) and a stir bar, dried under high vacuum for one hour, and backfilled with nitrogen. Under a constant flow of nitrogen, 10 mL of dry, degassed DCM was added, and the flask was cooled to -78 °C. Aliquots of ClSO3H4 (1 mL) were added every 15 min to total 8 portions (8.00 mL, 120. mmol). The reaction mixture was stirred at -78 °C for an additional 30 min and warmed to room temperature. Dry, degassed Et2O (20 mL) was added slowly, and the solid was filtered under nitrogen, washing with three 10 mL portions of Et2O to yield 5.7 g of white, fluffy solid (74% yield), which could be stored indefinitely in the drybox at -30 °C. 1H NMR (400 MHz, DMSO-d6) δ: 2.88 (s, 3H) 13C NMR (101 MHz, DMSO) δ 36.4. HRMS–ESI (m/z): [M – H]–1 calcd for CH4NO4S, 125.9867; found 125.9866.
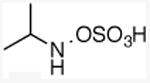
N-isopropylhydroxylamine-O-sulfonic acid
A 250 mL Schlenk flask was charged with N-isopropyllhydroxylamine hydrochloride (623 mg, 5.62 mmol) and a stir bar, dried under high vacuum for one hour, and backfilled with nitrogen. Under a constant flow of nitrogen, 20 mL of dry, degassed DCM was added, and the flask was cooled to -78°C. Aliquots of ClSO3H were added every 15 min over 2 hours, to total 1.50 equiv (563 μL, 8.43 mmol). The reaction mixture was stirred at -78 °C for an additional 15 min and warmed to room temperature over 30 min. Dry, degassed Et2O (10 mL) was added slowly, and the solid was filtered under nitrogen and washed with three 5 mL portions of Et2O to yield 640 mg of white, fluffy solid (74% yield), which could be stored indefinitely in the drybox at -30 °C. 1H NMR (500 MHz, DMSO) δ 3.54 (h, J = 6.5 Hz, 1H), 1.21 (d, J = 6.5 Hz, 6H). 13C NMR (126 MHz, DMSO) δ 52.6, 16.6. HRMS–ESI (m/z): [M – H]–1 calcd for C3H8NO4S, 154.0180; found 154.0180.
N-(3-phenylpropyl)hydroxylamine-O-sulfonic acid
3-phenylpropionaldehyde oxime
The title compound was prepared via a modified literature procedure.27 In a 50 mL flask, a solution of 3-phenylpropionaldehyde (1.98 mL, 15.0 mmol) in 10 mL of H2O was stirred, open to air. A solution of hydroxylamine hydrochloride (5.21 g, 75 mmol) in EtOH (10 mL) was added at room temperature, and the solution was cooled to 0 °C. NaOH (1.0 g, excess) in 5 mL of H2O was added slowly, and the solution was warmed to room temperature. The reaction was heated at 70 °C and stirred for 2 hours. The reaction was allowed to cool to room temperature, and the precipitate was filtered, washed with water, dried under high vacuum, and used without further purification.
N-(3-phenylpropyl)hydroxylamine
In a 100 mL flask under air, 3-phenylpropionaldehyde oxime (745 mg, 5.00 mmol) was stirred at 0 °C in MeOH (10 mL). A mixture of 6N HCl(aq) and MeOH (1:1 v/v) and a solution of NaBH3CN (408 mg, 6.50 mmol) in MeOH were alternatingly added dropwise over 30 min, maintaining pH <3. The solution was warmed to room temperature, maintaining pH <3, and stirred until no oxime remained, as determined by thin-layer chromatography (about 30 min). The reaction was quenched with brine (10 mL), basified with aqueous 6N NaOH to pH >9, and transferred to a separatory funnel. The solution was extracted with 2 × 20 mL Et2O, and the combined organic layers were washed with brine, dried with MgSO4, and concentrated in vacuo. The crude material was taken up in benzene (10 mL) and treated with conc HCl (1 equiv). The solvent was removed under high vacuum and the crude HCl salt was used in the next step.
N-(3-phenylpropyl)hydroxylamine-O-sulfonic acid
A 50 mL Schlenk flask was charged with solid N-(3-phenylpropyl)hydroxylamine hydrochloride (915 mg, 4.87 mmol) and a stir bar, dried under high vacuum for one hour, and backfilled with nitrogen. Under a constant flow of nitrogen, 8 mL of dry, degassed DCM was added, and the flask was cooled to -78 °C. Aliquots of ClSO3H were added every 15 min over 2 hours to total 1.50 equiv (488 μL, 7.31 mmol). The reaction mixture was stirred at -78 °C for an additional 15 min and warmed to room temperature over 30 min. Dry, degassed Et2O (10 mL) was added slowly, and the precipitate was filtered under nitrogen and washed with three 5 mL portions of Et2O to yield 890 mg of white, fluffy solid (79%), which could be stored indefinitely in the drybox at -30 °C. 1H NMR (600 MHz, DMSO) δ 11.24 – 11.00 (br s, 1H), 7.30 (m, 2H), 7.21 (m, 3H), 6.06 – 3.94 (br s, 1H), 3.18 (m, 2H), 2.65 (m, 2H), 1.90 (m, 2H). 13C NMR (151 MHz, DMSO) δ 140.7, 128.5, 128.3, 126.1, 49.2, 31.7, 25.0. HRMS–ESI (m/z): [M – H]–1 calcd for C9H12NO4S, 230.0493; found 230.0494.
N-(heptyl)hydroxylamine-O-sulfonic acid
1-heptanal oxime
The title compound was prepared via a modified literature procedure.27 In a 100 mL round bottom flask, a solution of 3-phenylpropionaldehyde (1.41 mL, 10.0 mmol) in 15 mL of H2O was stirred, open to the air. A solution of hydroxylamine hydrochloride (5.21 g, 75 mmol) in EtOH (5 mL) was added at room temperature, and the solution was cooled to 0 °C. NaOH (0.6 g, 15 mmol) in 5 mL of H2O was added slowly, and the solution was warmed to room temperature. The reaction was heated at 70 °C and stirred for 1 hour. The reaction was allowed to cool to room temperature, and the product was allowed to precipitate over night. The precipitate was filtered, washed with water, dried under high vacuum, and used without further purification (800 mg).
N-(heptyl)hydroxylamine
In a 50 mL round bottom flask under air, heptanaol oxime (794 mg, 6.14 mmol) was stirred at -60 °C in MeOH (10 mL). A mixture of 6N HCl(aq) and MeOH (1:1 v/v) and a solution of NaBH3CN (502 mg, 8.00 mmol) in MeOH (5 mL) were alternatingly added dropwise over 30 min, maintaining pH <3. The solution was warmed to room temperature, maintaining pH <3, and stirred until no oxime remained, as determined by thin-layer chromatography (about 30 min). The reaction was quenched with brine (10 mL), basified with aqueous 6N NaOH to pH >8, and transferred to a separatory funnel. The solution was extracted with 2 × 20 mL Et2O, and the combined organic layers were washed with brine, dried with MgSO4, and concentrated in vacuo. The crude material was taken up in benzene (10 mL) and treated with conc HCl (1 equiv). The solvent was removed under high vacuum and the crude HCl salt was used in the next step (736 mg).
N-(heptyl)hydroxylamine-O-sulfonic acid
A 50 mL Schlenk flask was charged with solid N-(3-phenylpropyl)hydroxylamine hydrochloride (736 mg, 5.61 mmol) and a stir bar, dried under high vacuum for one hour, and backfilled with nitrogen. Under a constant flow of nitrogen, 5 mL of dry, degassed DCM was added, and the flask was cooled to -78 °C. Aliquots of ClSO3H were added every 15 min over 1.5 hours to total 1.50 equiv (560 μL, 7.31 mmol). The reaction mixture was stirred at -78 °C for an additional 15 min and warmed to room temperature over 30 min. The reaction was cooled to -78 °C and dry, degassed Et2O (10 mL) was added slowly. The precipitate was filtered under nitrogen and washed with three 5 mL portions of Et2O to yield 1.04 of white, fluffy solid (88%), which could be stored indefinitely in the drybox at -30 °C. 1H NMR (600 MHz, DMSO) δ 5.00 – 3.40 (br s, 2H), 3.25 – 2.99 (m, 2H), 1.73 – 1.46 (m, 2H), 1.38 – 1.15 (m, 8H), 0.86 (t, J = 5.1 Hz, 3H). 13C NMR (151 MHz, DMSO) δ 49.6, 31.0, 28.2, 25.7, 23.1, 22.0, 134.0. [M – H]–1 calcd for C7H16NO4S, 210.0806; found 210.0804.
N-(tert-butoxy carbonyl)-(2,2-diphenyl-hept-6-enyl)-7-methylamine
A 20 mL vial was loaded with a stir bar, (2,2-diphenyl-hept-6-enyl)-methylamine (279 mg, 1.00 mmol), 3 mL THF, and triethylamine (420 mL, 3.00 mmol). Di-tert-butyl dicarbonate (275 mg, 1.10 mmol) was added, and the mixture was stirred at room temperature for one hour. The reaction was diluted with H2O (20 mL) and EtOAc (20 mL) in a separatory funnel, and the aqueous phase was extracted with 2× 20 mL EtOAc. The combined organic layers were washed with brine, dried with MgSO4, and concentrated by rotary evaporation. The product was lyophilized in benzene under high vacuum to yield 379 mg of white solid (quant.) as a mixture of rotamers, and used without further purification. NMR analysis was performed at 60 °C. 1H NMR (600 MHz, CDCl3) δ 7.30 – 7.24 (m, 4H), 7.24 – 7.17 (m, 6H), 5.78 – 5.63 (m, 1H), 4.93 (d, J = 17.1 Hz, 1H), 4.89 (d, J = 10.1 Hz, 1H), 4.05 (s, 2H), 2.22 – 2.04 (m, 5H), 2.04 – 1.90 (m, 2H), 1.45 (s, 9H), 1.24 – 1.11 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 156.8, 147.1, 138.9, 128.6, 127.9, 126.0, 114.4, 79.4, 57.4, 51.5, 36.5, 36.3, 34.4, 28.4, 23.9.
General Procedure A for the Hydrozircation/Amination of Alkenes
In a drybox, an oven-dried, screw-top, 1 dram vial was charged with a stir bar, and bis(cyclopentadienyl) zirconium chloride hydride (103 mg, 0.400 mmol, 1.00 equiv).6 THF (1.5 mL, 3.75 M) and alkene (0.400 mmol, 1.00 equiv) were added. The vial was sealed with a cap equipped with a PTFE-lined septum and stirred at room temperature. After 0.5 to 4 h the reaction mixture became homogeneous,7 and N-alkylhydroxylamine-O-sulfonic acid (0.600 mmol, 1.5 equiv) was added. The vial was re-sealed, removed from the drybox, and heated at 50 °C for 30 min with stirring. The reaction was cooled to room temperature, diluted with wet THF (5 mL), and concentrated in vacuo. The crude solids were dissolved in aqueous 1 M HCl (5 mL), transferred to a separatory funnel, and washed twice with Et2O. The combined washes were extracted once more with 5 mL of 1 M HCl, and the combined aqueous phases were basified to pH >8 with 1 M NaOH and extracted twice with Et2O. The organic phases from the second extraction were washed with brine, dried with Na2SO4, and concentrated in vacuo to yield the desired product.
General Procedure B for the Hydrozirconation/Amination of Alkenes
In a drybox, an oven dried, 1 dram vial was charged with a stir bar, and bis(cyclopentadienyl) zirconium chloride hydride (103 mg, 0.400 mmol, 1.00 equiv).6 THF (1.5 mL) and alkene (0.400 mmol, 1.00 equiv) were added. The vial was sealed with a cap equipped with a PTFE-lined septum and stirred at room temperature. After 0.5 to 4 h the reaction mixture became homogeneous, and hydroxylamine-O-sulfonic acid (67.9 mg, 0.600 mmol, 1.5 equiv) was added. The vial was re-sealed, stirred at room temperature for 30 min, and removed from the drybox. The reaction was diluted with wet THF (5 mL), and concentrated in vacuo. The crude solids were dissolved in aqueous 1 M HCl (5 mL), transferred to a separatory funnel, and washed twice with Et2O. The combined washes were extracted once more with 5 mL of 1 M HCl, and the combined aqueous phases were basified to pH >8 with 1 M NaOH and extracted twice with Et2O. The organic phases from the second extraction were washed with brine, dried with Na2SO4, and concentrated in vacuo to yield the desired product.
General Procedure C for the Hydrozirconation/Amination of Alkenes
General Procedure A was followed until the workup step. The crude solids were dissolved in aqueous 1 M NaOH (5 mL), transferred to a separatory funnel, diluted with H2O, and extracted twice with Et2O. The combined organic phases were washed with brine, dried with Na2SO4, and concentrated in vacuo. The crude residue was purified via column chromatography, eluting with 0.5% NH4OH, 1-10% MeOH in DCM (v/v).
N-(3-phenylpropyl)octylamine
In a drybox, an oven-dried, 1 dram vial was charged with a stir bar and bis(cyclopentadienyl) zirconium chloride hydride (51.6 mg, 0.200 mmol, 1.00 equiv),6 THF (0.75 mL) and 1-octene (31.4 mL, 0.200 mmol, 1.00 equiv) were added. The vial was sealed with a cap equipped with a PTFE-lined septum and stirred at room temperature. After 1 h the reaction mixture became homogeneous, and N-(3-phenylpropyl)hydroxylamine-O-sulfonic acid (69.6 mg, 0.300 mmol, 1.5 equiv) was added. The vial was re-sealed, removed from the drybox, and heated at 80 °C for 30 min with stirring. The reaction was cooled to room temperature, diluted with wet THF (2 mL), and concentrated in vacuo. The crude solids were dissolved in aqueous 1 M NaOH (5 mL), transferred to a separatory funnel, diluted with H2O, and extracted twice with Et2O. The combined organic phases were washed with brine, dried with Na2SO4, and concentrated in vacuo. The crude residue was purified via column chromatography eluting with 0.5% NH4OH, 1-10% MeOH in DCM (v/v) to yield 37 mg of N-(3-phenylpropyl)octylamine (clear oil, 74%). 1H NMR (600 MHz, CDCl3) δ 7.34 – 7.27 (m, 2H), 7.25 – 7.14 (m, 3H), 2.74 – 2.61 (m, 4H), 2.59 (t, J = 7.3 Hz, 2H), 1.89 – 1.75 (m, 2H), 1.47 (m, 2H), 1.36 – 1.18 (m, 10H), 0.88 (t, J = 6.9 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 142.2, 128.3, 128.3, 125.7, 50.0, 49.5, 33.7, 31.8, 31.7, 30.1, 29.5, 29.2, 27.4, 22.6, 14.1.
N-(tert-butoxy carbonyl)-(2,2-diphenyl-heptyl)-7-methylamino)-methylamine
General procedure C. 120 mg (mixture of rotamers, clear oil, 74%). 1H NMR (600 MHz, CDCl3) δ 7.34 – 7.20 (m, 5H), 7.18 – 7.13 (m, 5H), 4.09 – 3.88 (m, 2H), 2.54 – 2.42 (m, 2H), 2.38 (s, 3H), 2.18 – 1.87 (m, 6H), 1.41 (m, 9H), 1.34 – 1.17 (m, 2H), 1.11 – 0.94 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 186.5, (186.1), 147.5, (146.5), 128.5, (128.3), 127.8, 126.0, (125.8), 79.8, (79.1), 57.9, (56.6), 52.1, (51.8), 51.3, (51.0), 36.5, 36.4, (36.3), 29.8, (29.3), 28.4, 28.1, (27.8), 24.4, (24.1). HRMS–ESI (m/z): [M + H]+ calcd for C26H39N2O2, 411.3007; found 411.2999.
9-O-benzyl-10,11-dihydro-11-methylamino-quinine
In a drybox, an oven-dried, screw-top, 1 dram vial was charged with a stir bar, and bis(cyclopentadienyl) zirconium chloride hydride (51.6 mg, 0.200 mmol, 1.00 equiv). THF-d8 (1.0 mL) and 9-O-benzyl quinine (82.9 mg, 0.200 mmol, 1.00 equiv) were added, the vial was sealed with a cap equipped with a PTFE-lined septum, and the reaction was stirred at room temperature. After 30 min the reaction mixture became homogeneous, and the solution was analyzed by NMR spectroscopy. The amount of remaining olefin was determined to be approximately 0.2 equiv, and a further portion of bis(cyclopentadienyl) zirconium chloride hydride (16.6 mg, 0.0640 mmol, 0.322 equiv) was added. The reaction was stirred for 1 h, at which point the reaction mixture became homogeneous, and N-methylhydroxylamine-O-sulfonic acid (38.1 mg, 0.300 mmol, 1.50 equiv) was added. The vial was re-sealed, removed from the drybox, and heated at 50 °C for 30 min with stirring. The reaction was cooled to room temperature, diluted with wet THF (2 mL), and concentrated in vacuo. The crude solids were dissolved in aqueous 1 M HCl (5 mL), transferred to a separatory funnel, diluted with H2O, and washed twice with Et2O. The combined washes were extracted once more with 5 mL of 1 M HCl, and the combined aqueous phases were basified to pH >8 with aqueous 1 M NaOH and extracted twice with Et2O. The organic phases from the second extraction were washed with brine, dried with Na2SO4, and concentrated in vacuo to yield 82 mg of the desired product as a white solid (92%). NMR analysis was performed at 60 °C to minimize peak broadening, due to constrained rotation at C9. 1H NMR (600 MHz, CDCl3) δ 8.74 (d, J = 4.4 Hz 1H), 8.03 (d, J = 9.2 Hz, 1H), 7.55 (s, 1H), 7.47 (d, J = 4.2 Hz, 1H), 7.41 – 7.28 (m, 6H), 5.82 – 5.64 (m, 1H), 4.55 – 4.43 (m, 2H), 4.01 (s, 3H), 3.70 – 3.56 (m, 1H), 3.35 – 3.21 (m, 2H), 2.90 – 2.78 (m, 1H), 2.60 (m, 3H), 2.39 (s, 3H), 2.08 – 1.97 (m, 1H), 1.91 – 1.75 (m, 3H), 1.65 – 1.47 (m, 4H), 1.27 (br s, 1H). 13C NMR (151 MHz, CDCl3) δ 158.4, 150.4, 147.3(×2), 144.7, 137.4, 131.8, 128.5, 127.9, 127.7, 127.2, 122.4, 118.6, 101.2, 77.2, 71.3, 59.9, 57.5, 56.5, 48.6, 43.3, 34.8, 32.8, 32.4, 29.7, 25.7. HRMS: (m/z + 1) Calc'd 446.2802 Found 446.2799. HRMS–ESI (m/z): [M + H]+ calcd for C28H36N3O2, 446.2802; found 446.2799.
2-(3-methylaminopropyl)phenyl-2,3,4,6-tetra-O-benzyl-α-D-mannopyranoside
General Procedure C. 26 mg (colorless solid, 66%). 1H NMR (500 MHz, CDCl3) δ 7.45 – 7.26 (m, 18H), 7.24 – 7.08 (m, 5H), 6.94 (td, J =7.3 Hz, 1.2 Hz, 1H), 5.58 (d, J =1.8 Hz, 1H), 4.93 (d, J =10.8 Hz, 1H), 4.80 (s, 1H), 4.80 (s, 1H), 4.71 (m, 2H), 4.67 (d, J =12.0 Hz, 1H), 4.56 (d, J =10.8 Hz, 1H), 4.48 (d, J =11.9 Hz, 1H), 4.17 (t, J =9.4 Hz, 1H), 4.07 (dd, J =9.4 Hz, 3.0 Hz, 1H), 3.98 – 3.92 (m, 1H), 3.88 – 3.76 (m, 2H), 3.70 (d, J =9.5 Hz, 1H), 2.56 – 2.47 (m, 4H), 2.35 (s, 3H), 1.75 – 1.67 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 154.3, 138.5, 138.38, 138.34, 138.2, 131.0, 129.9, 128.4, 128.4, 128.3, 128.2, 127.9, 127.8, 127.8, 127.74, 127.71, 127.68, 127.6, 127.4, 127.2, 122.1, 114.5, 96.5, 79.9, 75.1, 74.9, 74.8, 73.3, 72.73, 72.67, 72.4, 69.1, 51.8, 36.5, 30.3, 28.0. HRMS–ESI (m/z): [M + H]+ calcd for C44H50N1O6, 688.3633; found 688.3616.
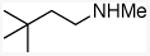
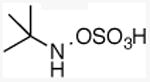
(3,3-dimethyl)butylmethylamine
General procedure A was followed, using 1.10 equivalents of 3,3-dimethyl-1-butene (56.7 μL, 0.400 mmol) to yield 42 mg (clear oil, 91%). 1H NMR (400 MHz, CDCl3) δ 2.63 – 2.52 (m, 2H), 2.44 (s, 3H), 2.21 (s, 2H), 1.45 – 1.32 (br s, 1H), 0.89 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 48.2, 43.7, 36.4, 29.8, 29.5.
(6-chloro)hexylmethylamine
General procedure A. 50 mg (clear oil, 83%). 1H NMR (600 MHz, CDCl3) δ 3.53 (t, J =6.7 Hz, 2H), 2.59 (t, J =7.2 Hz, 2H), 2.44 (s, 3H), 2.07 (br s, 1H), 1.81 – 1.71 (m, 2H), 1.58 – 1.47 (m, 2H), 1.47 – 1.40 (m, 2H), 1.40 – 1.28 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 51.8, 45.0, 36.3 32.4, 29.5, 26.7, 26.5. HRMS–ESI (m/z): [M + H]+ calcd for C7H17ClN, 150.1045; found 150.1044.
N-methyl octylamine
General procedure A. 51 mg (clear oil, 89%). 1H NMR (400 MHz, CDCl3) δ 2.54 (t, J =7.2 Hz, 2H), 2.41 (s, 3H), 1.62 – 1.53 (br s, 1H), 1.52 – 1.41 (m, 2H), 1.26 (m, 10H), 0.86 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 52.1, 36.4, 31.8, 29.8, 29.5, 29.2, 27.3, 22.6, 14.0.30
N-methyl(3-trimethylsilyl)propylamine
General procedure A. 48 mg (yellow oil, 83%). 1H NMR (500 MHz, CDCl3) δ 2.54 (t, J =7.1 Hz, 2H), 2.41 (s, 3H), 2.13 (br s, 1H), 1.52 – 1.37 (m, 2H), 0.49 – 0.42 (m, 2H), -0.04 (s, 9H).13C NMR (75 MHz, CDCl3) δ 55.5, 36.5, 24.3, 14.3, -1.5
N-methyl(3-phenyl)propylamine
General procedure A. 50 mg (yellow oil, 83%). 1H NMR (400 MHz, CDCl3) δ 7.37 – 7.29 (m, 2H), 7.28 – 7.20 (m, 3H), 2.78 – 2.60 (m, 4H), 2.49 (s, 3H), 1.88 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 142.1, 128.3, 128.3, 125.7, 51.5, 36.3, 33.6, 31.4.31
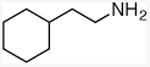
N-(2-cyclohexyl)ethyl methylamine
General procedure A. 50 mg (yellow oil, 89%). 1H NMR (400 MHz, CDCl3) δ 2.62 – 2.49 (m, 2H), 2.40 (s, 3H), 1.78 – 1.52 (m, 4H), 1.35 (m, 2H), 1.28 – 1.00 (m, 5H), 0.88 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 49.7, 37.5, 36.5, 35.6, 33.4, 26.6, 26.3.32
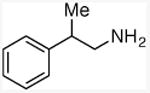
N-methyl(2-phenyl)propylamine
General procedure A. 41 mg (yellow oil, 69%). 1H NMR (500 MHz, CDCl3) δ 7.31 (t, J =7.6 Hz, 1H), 7.25 – 7.17 (m, 3H), 2.96 (tq, J =7.0 Hz, 7.0 Hz, 1H), 2.74 (d, J =7.2 Hz, 2H), 2.38 (s, 3H), 1.87 – 1.42 (m, 1H), 1.26 (d, J =7.0 Hz, 4H). 13C NMR (75 MHz, CDCl3) δ 145.2, 128.5, 127.1, 126.3, 59.2, 39.8, 36.3, 20.0.31
1-octylamine
General procedure B. 48 mg (clear oil, 92%). 1H NMR (500 MHz, CDCl3) δ 2.65 (t, J = 6.8 Hz, 2H), 1.69 (br s, 2H), 1.50 – 1.34 (m, 2H), 1.34 – 1.14 (m, 12H), 0.85 (t, J =6.8 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 42.1, 33.7, 31.8, 29.4, 29.3, 26.8, 22.6, 14.1.33
(2-phenyl)propylamine
General procedure B. 48 mg (yellow, 88%). 1H NMR (600 MHz, CDCl3) δ 7.31 (t, J =7.4 Hz, 2H), 7.24 – 7.17 (m, 3H), 2.90 – 2.80 (m, 2H), 2.79 – 2.70 (m, 1H), 1.56 (br s, 2H), 1.25 (d, J =6.9 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 144.9, 128.5, 127.3, 126.3, 49.4, 43.4, 19.2.34
N-methyl-exo-2-norbornylamine
General procedure A. 41 mg (yellow, 81%). 1H NMR (500 MHz, CDCl3) δ 2.44 (dd, J =7.4, 3.5 Hz, 1H), 2.36 (s, 3H), 2.16 (d, J =3.9 Hz, 1H), 1.55 (ddd, J =12.4, 7.5, 2.3 Hz, 1H), 1.50 – 1.36 (m, 3H), 1.13 – 1.00 (m, 4H), 1.23 (br s, 1H), 1.04 (d, J =2.1 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 63.8, 40.2, 39.6, 35.6, 34.8, 34.3, 28.5, 26.9.35
3,4-dimethoxy-N-methylphenethylamine
General procedure A. 55 mg (yellow oil, 71%). 1H NMR (400 MHz, CDCl3) δ 6.86 – 6.59 (m, 3H), 3.86 (s, 3H), 3.84 (s, 3H), 2.81 (t, J =6 Hz, 2H), 2.75 (t, J =6 Hz, 1H), 2.43 (s, 1H), 2.10 (br s, 1H). 13C NMR (151 MHz, CDCl3) δ 148.84, 148.82, 147.4, 132.3, 120.5, 111.86, 111.85, 111.84, 111.2, 111.2, 55.9, 55.8, 53.1, 36.1, 35.5.36
3-trifluoromethyl-N-methylphenethylamine
General procedure A. 63 mg (yellow oil, 78%). 1H NMR (600 MHz, CDCl3) δ 7.62 – 7.28 (m, 4H), 2.97 – 2.84 (m, 4H), 2.75 (br s, 1H), 2.46 (s, 3H).13C NMR (151 MHz, CDCl3) δ 140.6, 132.1, 130.8 (q, J =32.0 Hz), 128.9, 125.3 (q, J =3.7 Hz), 124.1 (q, J = 272.1 Hz), 123.1 (q, J =7.1, 3.3 Hz), 52.6, 35.9, 35.6. 19F NMR (376 MHz, CDCl3) δ -61.8.
(2-cyclohexyl)ethylamine
General procedure B. 36 mg (yellow oil, 70%). 1H NMR (500 MHz, CDCl3) δ 2.69 (t, J =7.0 Hz, 2H), 1.86 (br s, 2H), 1.74 – 1.56 (m, 5H), 1.43 – 1.29 (m, 3H), 1.27 – 1.01 (m, 3H), 0.99 – 0.76 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 41.4, 39.6, 35.2, 33.3, 26.5, 26.3.37
N,2-dimethyloctan-1-amine
General procedure C. 45 mg (yellow oil, 72%). 1H NMR (300 MHz, CDCl3) δ 2.47 (dd, J =11.5, 5.9 Hz, 1H), 2.41 (s, 3H), 2.33 (dd, J =11.5, 7.3 Hz, 1H), 1.58 (m, 1H), 1.48 (br s, 1H), 1.40 – 1.15 (m, 9H), 1.15 – 0.99 (m, 1H), 0.97 – 0.77 (m, 6H). 13C NMR (126 MHz, CDCl3) δ 58.8, 36.7, 35.0, 33.0, 31.9, 29.6, 26.9, 22.6, 18.1, 14.1. HRMS–ESI (m/z): [M + H]+ calcd for C10H24N, 158.1903; found 158.1902.
N-heptyloctan-1-amine
General procedure C, heating to 80 °C. 74 mg (yellow oil, 81%). 1H NMR (500 MHz, CDCl3) δ 2.78 – 2.42 (m, 4H), 1.52 – 1.34 (m, 4H), 1.34 – 1.00 (m, 19H), 0.86 (t, J =6.8 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 50.24 (2), 32.07 (2), 30.17 (2), 29.8, 29.52, 29.48, 27.7, 27.6, 22.90, 22.86, 14.3 (2).
Supplementary Material
Scheme 2. Hydroamination of complex substrates.

Acknowledgments
We thank the NIH-NIGMS (GM-055382) for support of this work.
Footnotes
Supporting Information Available. Spectroscopic data for all compounds. The Material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Muller TE, Hultzsch KC, Yus M, Foubelo F, Tada M. Chem Rev. 2008;108:3795–3892. doi: 10.1021/cr0306788. [DOI] [PubMed] [Google Scholar]
- 2.Muller TE, Beller M. Chem Rev. 1998;98:675–703. doi: 10.1021/cr960433d. [DOI] [PubMed] [Google Scholar]
- 3.Brown HC, Heydkamp WR, Breuer E, Murphy WS. J Am Chem Soc. 1964;86:3565–3566. [Google Scholar]
- 4.Barker TJ, Jarvo ER. Synthesis. 2011;24:3954–3964. [Google Scholar]
- 5.Jigajinni VB, Pelter A, Smith K. Tetrahedron Lett. 1978;19:181–182. [Google Scholar]
- 6.Kabalka GW, Henderson DA, Varma RS. Organometallics. 1987;6:1369–1370. [Google Scholar]
- 7.Kabalka GW, Wang Z, Goudgaon NH. Synthetic Comm. 1989;19:2409–2414. [Google Scholar]
- 8.Genêt JP, Hajicek J, Bischoff L, Greck C. Tetrahedron Lett. 1992;33:2677–2680. [Google Scholar]
- 9.Matsuda N, Hirano K, Satoh T, Miura M. Angew Chem Int Ed. 2012;51:3642–3645. doi: 10.1002/anie.201108773. [DOI] [PubMed] [Google Scholar]
- 10.Rucker RP, Whittaker AM, Dang H, Lalic G. J Am Chem Soc. 2012;134:6571–6574. doi: 10.1021/ja3023829. [DOI] [PubMed] [Google Scholar]
- 11.Rucker RP, Whittaker AM, Dang H, Lalic G. Angew Chem Int Ed. 2012;51:3953–3956. doi: 10.1002/anie.201200480. [DOI] [PubMed] [Google Scholar]
- 12.Zhu C, Li G, Ess DH, Falck JR, Kürti L. J Am Chem Soc. 2012;134:18253–18256. doi: 10.1021/ja309637r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mlynarski SN, Karns AS, Morken JP. J Am Chem Soc. 2012;134:16449–16451. doi: 10.1021/ja305448w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suzuki A, Sono S, Itoh M, Brown HC, Midland MM. J Am Chem Soc. 1971;93:4329–4330. [Google Scholar]
- 15.Brown HC, Midland MM, Levy AB. J Am Chem Soc. 1972;94:2114–2115. [Google Scholar]
- 16.Brown HC, Midland MM, Levy AB. J Am Chem Soc. 1973;95:2394–2396. [Google Scholar]
- 17.Akimoto I, Suzuki A. Synthetic Comm. 1981;11:475–480. [Google Scholar]
- 18.Kabalka GW, Wang Z. Synthetic Comm. 1990;20:231–237. [Google Scholar]
- 19.Zheng B, Srebnik M. J Org Chem. 1995;60:1912–1913. [Google Scholar]
- 20.Mendiola J, Rincón JA, Mateos C, Soriano JF, de Frutos Os, Niemeier JK, Davis EM. Org Process Res Dev. 2009;13:263–267. [Google Scholar]
- 21.Stanley LM, Hartwig JF. Angew Chem Int Ed. 2009;48:7841–7844. doi: 10.1002/anie.200904338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.See Supporting Information for details
- 23.Bexrud JA, Eisenberger P, Leitch DC, Payne PR, Schafer LL. J Am Chem Soc. 2009;131:2116–2118. doi: 10.1021/ja808862w. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Wang Y, Tang L, Deng L. J Am Chem Soc. 2004;126:9906–9907. doi: 10.1021/ja047281l. [DOI] [PubMed] [Google Scholar]
- 25.Luo SY, Tripathi A, Zulueta MML, Hung SC. Carbohydrate Research; Elsevier Ltd. 2012;352:197–201. doi: 10.1016/j.carres.2012.01.022. [DOI] [PubMed] [Google Scholar]
- 26.Plastic pipette tips affixed to an adjustable volume pipette were used to transfer chlorosulfonic acid
- 27.Biraboneye AC, Madonna S, Maher P, Kraus JL. ChemMedChem. 2010;5:79–85. doi: 10.1002/cmdc.200900418. [DOI] [PubMed] [Google Scholar]
- 28.Care was taken not to exceed 1 equivalent of Schwartz reagent
- 29.Allowing the hydrozirconation reaction to stir overnight before addition of aminating reagent resulted in reduced yields (61% for amination of 1-octene). Consumption of Schwartz reagent (insoluble in THF) was monitored visually
- 30.Stearns JF, Rapoport H. J Org Chem. 1977;42:3609–3614. [Google Scholar]
- 31.Hanada S, Ishida T, Motoyama Y, Nagashima H. J Org Chem. 2007;72:7551–7559. doi: 10.1021/jo070591c. [DOI] [PubMed] [Google Scholar]
- 32.Furstoss R, Tadayoni R, Esposito G, Lacrampe J, Heumann A, Waegell B. Can J Chem. 1976;54:3569–3577. [Google Scholar]
- 33.Card RJ, Schmitt JL. J Org Chem. 1981;46:754–757. [Google Scholar]
- 34.Bocchinfuso R, Robinson JB. Eur J Med Chem. 1999;34:293–300. [Google Scholar]
- 35.Pienemann T, Schaefer HJ. Synthesis. 1987;11:1005–1007. [Google Scholar]
- 36.Theodore LJ, Nelson WL. J Org Chem. 1987;52:1309–1315. [Google Scholar]
- 37.Kukula P, Koprivova K. J Catal. 2005;234:161–171. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.