

Abstract
We present a method of fabricating microneedles from polyvinylpyrrolidone (PVP) that enables delivery of intact proteins (or peptides) to the dermal layers of the skin. PVP is known to self-assemble into branched hollow fibers in aqueous and alcoholic solutions; we utilized this property to develop dissolvable patches of microneedles. Proteins were dissolved in concentrated PVP solution in both alcohol and water, poured into polydimethylsiloxane templates shaped as microneedles and, upon evaporation of solvent, formed into concentric, fibrous, layered structures. This approach of making PVP microneedles overcomes problems in dosage, uniform delivery and stability of protein formulation as compared to protein-coated metallic microneedles or photopolymerized PVP microneedles. Here we characterize the PVP microneedles and measure the delivery of proteins into skin. We show that our method of fabrication preserves the protein conformation. These microneedles can serve as a broadly useful platform for delivering protein antigens and therapeutic proteins to the skin, for example for allergen skin testing or immunotherapy.
Keywords: Allergy, Protein, Microneedle, Drug delivery, Diagnostics
1. Introduction
Human skin is equipped with networks of antigen-presenting cells that are the first cells to react in the innate immune response [1]. Delivering antigens and adjuvants to these cells in the skin can trigger innate and adaptive immune responses for both diagnostic and therapeutic purposes. Recent work has shown that such delivery could be achieved using polymers [2]: a patch of microneedles coated with inactivated virus and photopolymerized in situ was used to vaccinate mice to protect them from a lethal influenza challenge [3–5]. Here we present a method of preparing polyvinylpyrrolidone (PVP) microneedles that results in the intact delivery of proteins to the skin and is, in this regard, superior to the photopolymerization method.
Because of defects in peripheral tolerance in allergic patients, contact or ingestion of allergenic proteins triggers production of specific immunoglobulin E (IgE) antibodies, which are responsible for subsequent allergic reactions through binding and allergen-induced crosslinking of the IgE receptor (FcεRI) on mast cells. The prevalence of allergic diseases has increased in developed countries over the last few decades and these disorders now account for over $7 billion in healthcare expenditure annually in the USA. It is estimated that approximately 8% of children have true food allergies, but over 20% of children make alterations to their diets because of perceived adverse reactions to food [6–8]. Anaphylaxis due to food allergy results in over 20,000 hospital visits and 100– 200 deaths per year [9].
The gold-standard diagnostic test for food allergy is a double-blind placebo controlled food challenge (DBPCFC), but these challenges are complicated when multiple foods are considered as potential allergens, are difficult or impossible in young children, and are potentially dangerous because the readout is an allergic or anaphylactic response [10]. Hence, the skin prick testing (SPT) and serum IgE testing have been mainly used as surrogate tests to predict the risk of failure in a DBPCFC. In general, serum IgE testing is quantitative, but it is rife with false positives and negatives [11– 13], especially in patients with atopic dermatitis, who have high baseline IgE levels. On the other hand, qualitative skin testing is quite predictive of DBPCFC because it is a functional test of mast cell responses in the skin due to the allergen [14]. Unfortunately, because of the limitations of existing methodologies, only 30 or so allergens can be feasibly tested in a day.
SPT and conventional serum IgE testing use an ensemble of proteins (generally, solvent extracted hydrolysates of mechanically crushed foods), which, because of the massive numbers of epitopes probed, shows only the likelihood of allergic response, not the severity. Sampson and colleagues have shown, using microarrays comprising sequential, linear peptides, that the number of epitopes sensed by IgEs is proportional to the severity of reaction [15]. This finding appears to be general, as testing peptides from peanuts, milk, eggs, salmon and lentils showed similar results [16–19]. However, their method is not yet suitable for broad clinical application because of the need for a microarray slide scanner and the high false positives and negatives inherent in serum testing for allergens. A functional test like SPT that could measure biological responses to many thousands of allergenic peptides should be superior, but until now none has existed that could deliver and measure this many peptides in parallel. Furthermore, immunotherapy directed at the specific epitopes causing allergic responses could be useful for engendering tolerance, but it has not hitherto been practical to deliver potentially hundreds or thousands of specific peptides for immunotherapy.
Non-covalently aggregated polymers such as PVP hold high promise for the development of smart drug delivery materials because of their biocompatibility and low manufacturing cost. Here we use the self-assembling properties of PVP in aqueous and alcoholic solutions [20] to make microneedles. We demonstrate that these protein-encapsulated polymer microneedles are able to deliver proteins to the human intradermal space effectively. These microneedles dissolve after insertion, eliminating the need for disposal of hazardous, sharp needles after administration. By integrating allergen proteins within the microneedles in an array, it could be possible to test a whole library of allergenic proteins or their component peptides in a single assay, and this idea is demonstrated in a proof-of-concept, multi-allergen patch.
2. Materials and methods
2.1. Preparation of labeled proteins
Peanut flour was obtained from the Golden Peanut Company (Alpharetta, GA). The peanut flour was mixed with water and dissolved by sonication in a water bath for 15 min. To remove insoluble components and separate out the protein, the mixture was centrifuged at 3000g for 10 min. The middle layer of the supernatant containing soluble peanut proteins was collected with a syringe, while the top layer of the supernatant was discarded since it comprised fats and oil. The middle layer of the supernatant was further centrifuged at 18,000 xg for 10 min and the pellet, comprising insoluble components, was discarded [21]. The protein content in the supernatant was measured by bicinchoninic acid assay. The protein was then lyophilized and labeled with rhodamine using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide(EDC) and N-hydroxysulfosuccinimide (Sulfo-NHS) as the crosslinkers [22]. Briefly, 3.25 mg of 5(6)carboxytetramethylrhodamine (Sigma–Aldrich, Steinheim, Germany), 3.3 mg of EDC (Thermo Scientific, Rockford, IL) and 2.9 mg of Sulfo-NHS (ChemPep, Wellington, FL) were mixed and stirred for 1 h in 1 mL of 2-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 6.0). To this was added 20 mg of peanut protein in 5 mL of MES, then the mixture was stirred at 4 °C in the dark overnight. It was then purified using either a Slide-A-Lyzer dialysis cassette G2 (3000 MWCO, Thermo Scientific, Rockford, IL) in MES buffer or centrifugal filter devices (3000 MWCO, Millipore, Billerica, MA). The completeness of the dialysis was confirmed by checking the fluorescence of the wash. The labeled peanut protein was lyophilized and stored at −80 °C until use.
To label casein protein, 43 mg of casein from bovine milk (Sigma–Aldrich, Steinheim, Germany) was dissolved in 2.85 mL of 0.1 M NaHCO3 (pH 9.5). Then 1.8 mg of rhodamin B isothiocyanate (RBITC) (Sigma#x02013;Aldrich, Steinheim, Germany) dissolved in 150 µL of anhydrous dimethyl sulfoxide was added dropwise into the casein solution while stirring. The mixture was stirred for a further 2 h at room temperature in the dark. Labeled casein (RBITC#x02013;casein) was purified on a PD-10 desalting column (GE Healthcare Life Sciences, Pittsburgh, PA) equilibrated with H2O (pH adjusted to 8.5 using 1 N NaOH). The purified RBITC#x02013;casein was stored at 4 °C or used immediately to prepare microneedles. DQ ovalbumin was obtained from Invitrogen.
Preparation of streptavidin (SA)#x02013;PVP microneedles followed a similar scheme to other microneedles. To begin, 5 mg SA lyophilized powder (ImmunoPure Streptavidin, Pierce/Thermo Scientific) was reconstituted by dissolving in 0.5 mL of H2O. To prepare the SA#x02013;PVP microneedles, 100 mg of PVP (10 kDa) was dissolved in 1.9 mL of H2O and mixed with 7.5 µL of PEG400 and 0.1 mL of 10 mg mL−1 reconstituted SA. This mixture was cast in polydimethylsiloxane (PDMS) molds and placed in a vacuum chamber to remove air from the needle channels. The needles were left to dry on the bench top and peeled free from the mold after 24–36 h.
Photochemically crosslinked PVP microneedles were prepared by mixing 1-vinyl-2-pyrrolidone and 2,2′-azobis(2-methylpropio-nitrile) (10:1 w/w) with and without streptavidin (1 wt.%). The mixture was used to cast the PDMS molds. A vacuum was applied to remove the air from the needle channels. The needles were irradiated with UV light overnight and peeled from the molds.
2.2. Preparation of PDMS mold
To fabricate the mold for our microneedles, a silicon wafer with oxide mask was patterned using standard contact lithographic techniques with thick photoresist and subjected to deep reactive ion etching. The residual photoresist was removed using oxygen plasma and the wafers were washed in sulfuric acid. To facilitate easy removal of molded materials, all wafers were silanized overnight in a vacuum chamber prior to use.
To prepare PDMS molds, PDMS monomer and curing agent (10:1 w/w, Dow Corning, Midland, MI) were mixed and poured onto Si wafers in a sterile Petri dish. To remove bubbles of trapped air, a vacuum was applied for 20–30 min and the Petri dishes were gently rapped. To cure the PDMS, the Petri dish was incubated at 37 °C overnight.
2.3. Preparation of allergen microneedles
To form PVP-based biodegradable microneedles incorporating labeled-proteins, we dissolved 200 mg of PVP (MW 10 kDa, Sigma#x02013;Aldrich, USA) in 3.4 mL of ethanol (EtOH) and 15 µL of plasticizer (poly ethylene glycol−400). Labeled protein (2 mg, 1 wt.% of PVP) was dissolved and sonicated in 100 µL of water to obtain a clear solution, then added to the PVP#x02013;EtOH mixture. We adjusted the pH to 8–8.5 (for casein) or 9–10 (for peanut protein) by titrating 0.1 N NaOH until the mixture was clear. For the DQ ovalbumin microneedles, EtOH was replaced by deionized water to avoid precipitation of the protein.
The PVP#x02013;protein mixture was poured onto PDMS molds. A vacuum was applied to force out trapped air in the needle channels of the molds. The molds were left in a fume hood to evaporate the solvent (EtOH or water). The dried microneedles were carefully peeled from the mold and stored in a desiccator.
Photochemically crosslinked PVP microneedles were prepared by mixing 1-vinyl-2-pyrrolidone and 2,2′;-azobis(2-methylpropio-nitrile) (with and without proteins), poured onto PDMS molds. Trapped air was removed under a vacuum and then irradiated with ultraviolet (UV) light overnight. The crosslinked microneedles were peeled and stored in a desiccator.
2.4. Streptavidin binding activity using biotin quantitation assay
The binding activity of SA in the microneedles was analyzed by measuring the unbound biotin present after incubating it with biotin and horseradish peroxidase (biotin#x02013;HRP). The SA#x02013;PVP microneedles were dissolved in H2O to the final concentration of 3.33 mg mL−1 of SA and 330 mg mL−1 of PVP. Dissolved SA–PVP microneedles or a mixture of PVP + SA (weight ratio same as in microneedles) were treated with 5 mg mL−1 biotin–HRP and incubated at room temperature for 2 h. Free biotin was measured using a biotin quantitation kit (Thermo Scientific, Rockford, IL) according to the manufacturer’s directions. Briefly, 10 µL of 4’-hydroxyazo-benzene-2-carboxylic acid/avidin (HABA) was mixed with 80 µL of phosphate-buffered saline. Then 10 µL of the pre-mixture of SA#x02013;biotin–HRP was added to the solution and incubated at room temperature for 30 min. Absorbance at 500 nm, reflecting displacement of HABA from the SA, was measured using a NanoDrop 2000 (Thermo Scientific, Rockford, IL). To determine the total biotin input, a control solution contained only biotin–HRP. PVP or SA alone was also tested using the same method as control samples.
For sodium dodecyl sulfate#x02013;polyacrylamide gel electrophoresis (SDS#x02013;PAGE) experiments, the SA–PVP microneedle solution was mixed within sample buffer (NuPage, Life Technologies, Carlsbad, CA). The mixture (containing ~10 µg of SA) was loaded into the well and fractionated by using 10% Bis#x02013;Tris SDS#x02013;PAGE gels (1.0 mm × 10 well) (NuPage), then stained using a Coomassie staining kit (NuPage).
2.5. Skin penetration and microscopic examination of fluorescence
Human foreskin was obtained from discarded neonatal foreskins after elective circumcision. Institutional Review Board approval was obtained prior to tissue collection. The sample was cut into an ~1 cm × 1 cm square and pinned onto a Styrofoam board, with the epidermis facing up. A small patch of fluorescently labeled microneedles (~0.5 cm × 0.5 cm) was applied onto the skin and secured in place with a Tegaderm (3 M). The microneedles were pressed down using a small Petri dish for 30 s. After 5 min, the Tegaderm was removed and the skin was gently stripped with another clean Tegaderm to remove any residues on the surface. Still pinned on the sytrofoam, the skin was immersed in 4% paraformaldehyde and fixed overnight. Fixed skin was embedded in the optimum cutting temperature formulation on dry ice and cryosectioned. Tissue slides were preserved in mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; Vector Labs) and examined by widefield fluorescence microscopy. Fluorescence intensity was quantified using ImageJ (http://rsbweb.nih.gov/ij/download/).
2.6 Scanning electron microscopy (SEM) characterization
The morphology of the microneedles was characterized using SEM. The microneedles along with their bases were attached to an SEM sample stub using a double-stick carbon tab. The samples were coated with a 7 nm thick gold–palladium layer using a Den-ton Desk II vacuum sputter coater. Imaging was carried out on a Hitachi S-3400N VP scanning electron microscope at the Cell Sciences Imaging Facility, Stanford University.
2.7. Coating steel microneedles
AdminPatch array 1500 steel microneedles (AdminMed) were dip-coated using a standard technique [5]. Briefly, a coating solution formulated with 1% (w/v) carboxymethylcellulose (Aquacide II, EMD Millipore), 0.5% (w/v) Poloxamer 188, 15% (w/v) sucrose (Sigma) and 0.5% (w/v) rhodamine B labeled casein. Skin penetration tests were performed immediately after coating using the procedure described for polymer microneedles.
3. Results and discussion
3.1. Physical characteristics of fluorescently labeled allergen microneedles
Microneedles were prepared as described containing various proteins that were labeled with a fluorophor. After the microneedles were peeled from the mold, we imaged them using wide-field microscopy (Fig. 1A). The microneedles were of consistent shape and spacing, regardless of the protein constituent, and reflected the dimensions of the mold. The measured dimensions showed a height of approximately 400 µm and spacing of 500 µm, center to center (Fig. 1B).
Fig. 1.

Microscopic images of labeled allergen microneedles. (A) Microneedles incorporated with rhodamine-labeled peanut protein (left), fluorescein isothiocyanate-labeled casein protein (middle) and DQ ovalbumin (right). (B) The dimensions of microneedles are shown, consistent with the PDMS mold.
To further characterize the microneedles, the structures were examined using SEM (Fig. 2A). The PVP/casein microneedles had a rugged texture, in contrast to the smoother texture of the PVP/ peanut protein and control PVP microneedles. These features could reflect different arrangements of the incorporated proteins within the matrix of PVP fibers. To study the structure of the interior of the microneedles, they were broken and visualized by SEM. We found that the interior of the needles was uniform, with some hints of concentric circular features (Fig. 2B middle, also seen in Fig. 3), indicating the orderly self-assembly of molecules, as shown in the schematic representation (Fig. 2C). These results demonstrate the surface and interior textures of PVP microneedles incorporating allergen proteins and control PVP microneedles.
Fig. 2.
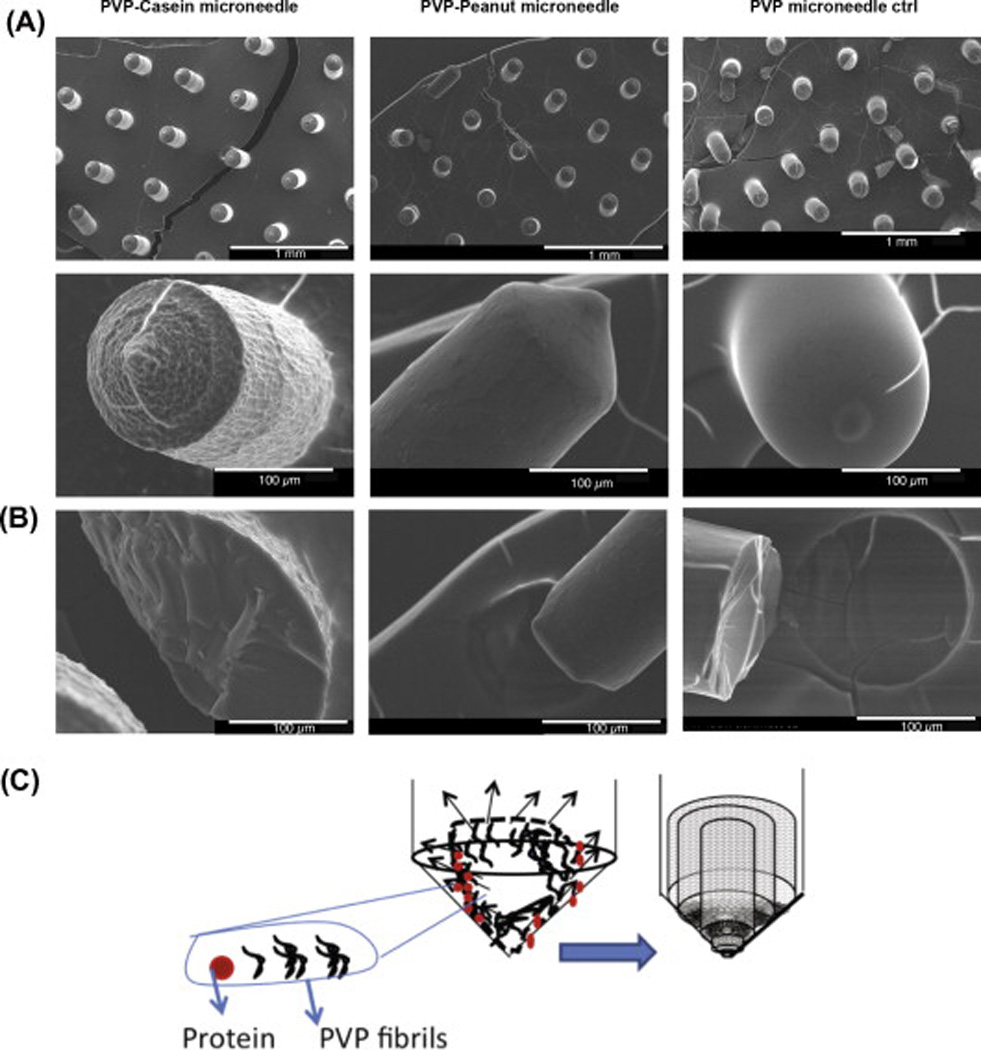
SEM images of microneedles. (A) (Row 1) Microneedle array of PVP-casein, PVP-peanut proteins and PVP alone (Control). (Row 2) Magnified single microneedle showing the surface morphology. (B) Morphology of the interior of the broken microneedle. (C) Schematic representation of the aggregate separation and packed arrangement of the PVP fibers.
Fig. 3.
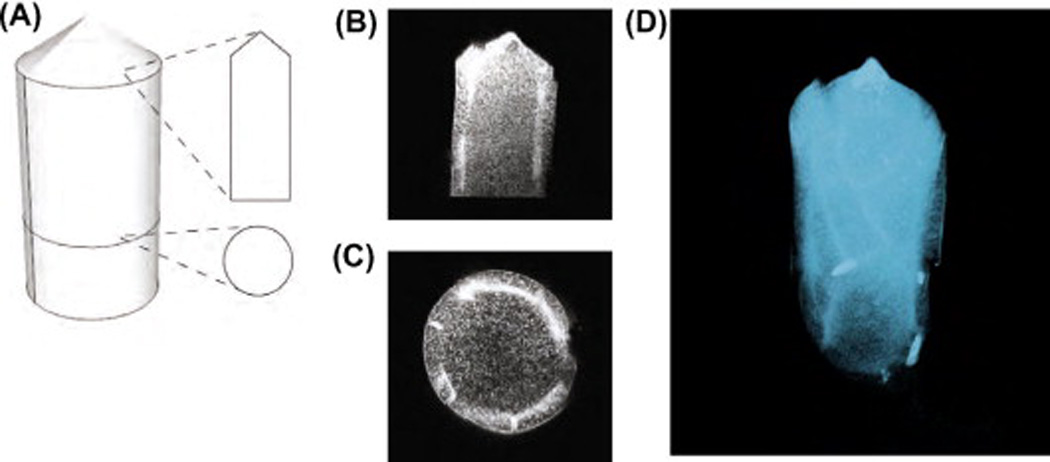
Confocal images of PVP-protein microneedle. (A) Schematic representation showing the vertical and horizontal cross-section of a rhodamine B-labeled casein microneedle. (B) Vertical optical cross-section. (C) Transverse optical cross-section. (D) Rendered view of a three-dimensional reconstructed microneedle. The protein is concentrated in concentric layers beneath the surface.
Upon drying, the concentrated PVP solution formed concentric fibrous PVP structures. This is due to the ‘‘coffee drying effect’’ originally demonstrated by Deegan and colleagues in explaining a polymer solution drying on a planar surface [23,24]. We found this effect during the drying process after the polymer solution had been poured into the PDMS templates. As the polymer solution dried, PVP fibrils were deposited on the periphery of the drying solution, and were next studied by fluorescence microscopy.
To study the distribution of allergen proteins inside the microneedles, labeled microneedles were secured in embedding resin and imaged by fluorescence confocal microscopy. Optical sections of the microneedles showed that fluorescently labeled allergen proteins were distributed throughout the microneedle (schematically illustrated in Fig. 3A). Notably, the distribution was not uniform. A concentrated subsurface layer of protein was found in the needles (Fig. 3B), with a partial ring-like structure observed in the horizontal cross-section (Fig. 3C). Areas of concentrated proteins were also found along the cylindrical axis of the needle (Fig. 3D). This finding is consistent with our interpretation of the coffee ring phenomenon, which noted separation of heteromeric aggregates during the drying process [25]. Presumably PVP#x02013;pro-teins formed smaller fibers or aggregates as compared to plain PVP fibers. The cone angle of the microneedle was measured to be ~49°, which is consistent with the sloping, <111>-oriented side-walls of the anisotropically wet-etched silicon master (~55°) used to shape the PDMS mold.
3.2. Microneedle fabrication process does not disrupt the biotin binding activity of SA
To test whether the binding activity of a protein is preserved after the process of forming a PVP#x02013;protein microneedle, we prepared microneedles that contained SA. SA is tetrameric (MW ~66 kDa) and binds four biotins with high affinity (KD ~10~14 M). The binding sites for biotin lie at the interfaces of the subunits of SA, so if formation of the microneedles disrupted the quaternary or tertiary structure of ‘SA, we would expect poor binding of biotin. We prepared PVP microneedles with SA and evaluated the biotin binding activity of SA recovered from the microneedles. The SA microneedles were prepared using the same method, with a 100/1 ratio (w/w) of PVP to SA. The SA microneedles were dissolved in H2O and mixed with biotin–HRP. The level of unbound biotin was then measured with a biotin quantitation kit. The level of decrease in free biotin indicates the level of biotin binding activity of SA in the microneedles. To evaluate the function of recovered SA, we compared the redissolved SA–PVP microneedles with microneedles prepared by photochemical crosslinking [5]. As a reference, we used an unpolymerized mixture of SA and PVP (mixed in the same weight ratios). We found a similar amount of decrease in free biotin among the samples, corresponding to 90.8% and 81.6% of SA still active in the microneedles, respectively (Fig. 4A). This result indicates that, at least for SA, its function and likely its conformation are preserved after the fabrication of microneedles.
Fig. 4.
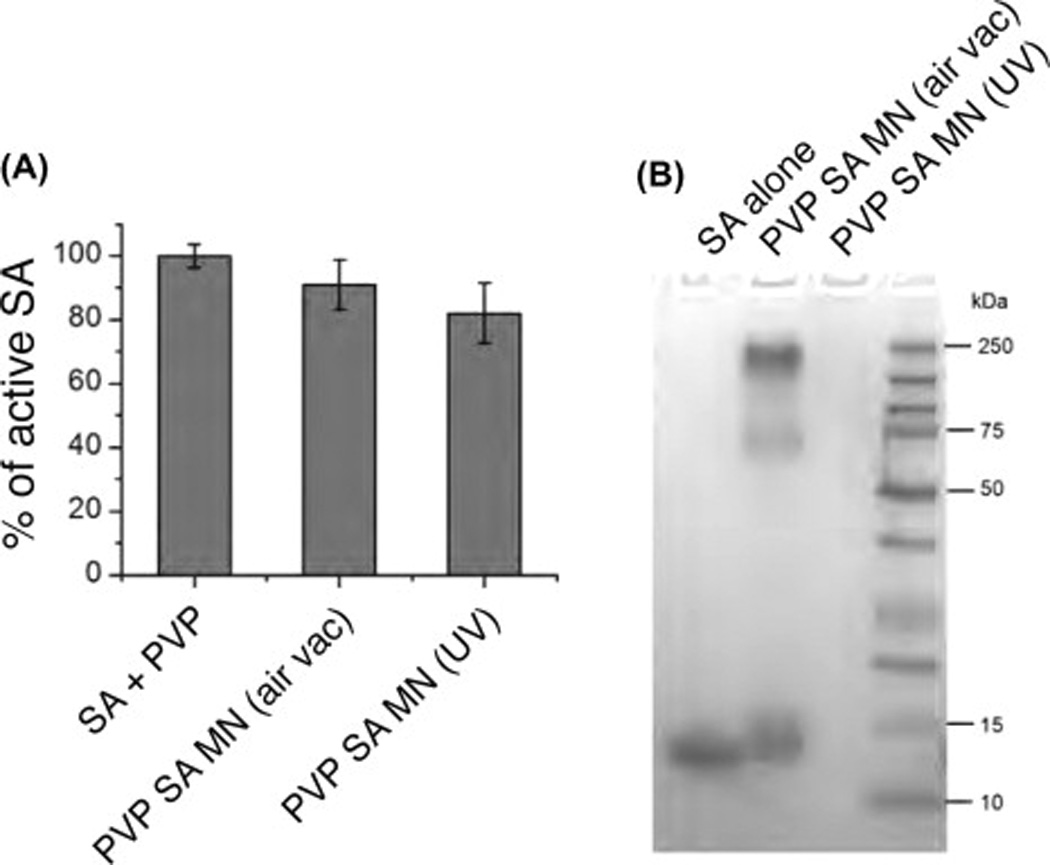
Characterization of SA recovered from microneedles. (A) SA recovered from SA–PVP microneedles retained its biotin binding activity. The intactness of the SA was evaluated by measuring the amount of unbound biotin using a biotin quantitation assay (n = 3, mean ± SD). (B) SDS–PAGE of SA recovered from microneedles. Lane 1: control SA; Lane 2: SA–PVP microneedles prepared using the air–vacuum method; Lane 3: SA–PVP microneedles prepared using UV-photocrosslinking method.
To further analyze the SA after formation into SA–PVP microneedles, we dissolved microneedles prepared as above and analyzed them using SDS–PAGE (Fig. 4B). We found, as expected that monomeric SA ran at about ~16 kDa (Lane 1). In the SA obtained from dissolving SA–PVP microneedles, we found bands at ~16 and 66 kDa, corresponding to monomeric and tetrameric (intact) SA (Lane 2). We also saw a high molecular weight, probably multimeric, complex. In dissolving SA–PVP microneedles that were fabricated by the photocrosslinking process, we saw no bands, as the very high molecular weight SA–PVP created by photocrosslinking could not enter into the gel (Lane 3). These results show that in our formulation of PVP microneedles, streptavidin can form a non-covalent complex with PVP while at the same time retaining its quaternary structure and binding activity. Taken together, these results show that the method of fabrication presented here results in intact, active proteins.
3.3. In vitro human foreskin test
To test the capability of these microneedles to penetrate into human skin, we applied the fluorescently labeled allergen microneedles onto human foreskin. Since human foreskin is soft and pliant, we first pinned it to a Styrofoam board (Fig. 5A). To characterize the kinetics of microneedle dissolution in the foreskin, we inserted the microneedles into the foreskin and monitored them over time by removing them at a set time and imaging them by wide-field microscopy. Significant dissolution was observed after 2.5 min, and after 5 min the microneedles were completely dissolved (Fig. 5B). To evaluate the delivery of proteins into the skin, we imaged the skin by fluorescence microscopy after application of microneedles comprising rhodamine B-labeled casein. Fluorescence from the rhodamine was observed up to 187 µm beneath the epidermis (Fig. 6). Taken together, these results show that the PVP microneedles can rapidly dissolve and successfully deliver proteins into the dermal portions of the skin.
Fig. 5.

Penetration of casein microneedles into human foreskin. (A) Schematic representation showing the experimental setup. (B) Microneedles dissolve in human foreskin. Images are microneedles prior to insertion or remaining on the Tegaderm 1, 2.5 and 5 min after insertion into human foreskin.
Fig. 6.
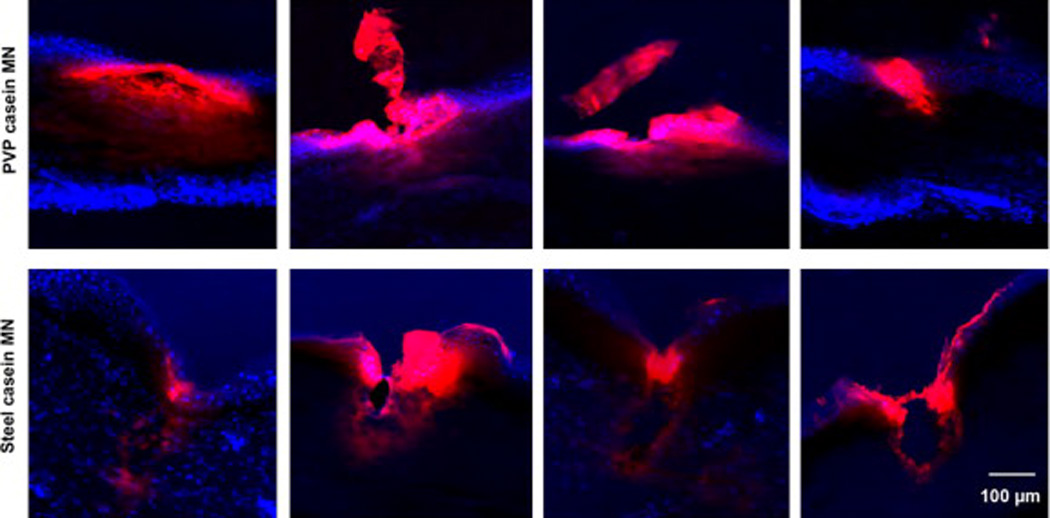
Penetration of microneedles into skin. Fluorescent images showing a comparison of the penetration of PVP-rhodamine B labeled casein microneedles and casein-coated AdminPatch array 1200 steel needles into human foreskin. Blue, DAPI staining; red, rhodamine B. Four representative penetration sites are shown for each type of microneedle.
3.4. Comparison of penetration of steel needles and polymer microneedles
Because metal needles are so commonly used for delivering antigens in the skin (e.g. skin prick testing), we compared the capability of our PVP microneedles to deliver antigenic proteins with metal needles. We coated Adminpatch array 1200 steel needles with rhodamine B-labeled casein, and compared with our PVP microneedles containing the same labeled casein protein. The steel needles showed clear penetration and, upon removal, tearing of the skin tissue (Fig. 6). Fluorescence imaging showed that most of the casein was pushed off the metal needles just at the surface of the skin, with poor delivery below the superficial epidermis. On the other hand, the polymer microneedles showed good penetration, and casein fluorescence was seen spread widely to about 100 µm deep. The results show that the PVP microneedles result in less apparent tissue trauma and deliver antigen more deeply than conventional steel microneedles.
3.5. Preparation of microneedle arrays
In the clinical setting, skin prick testing often requires multiple allergens to be evaluated in one encounter. We therefore tested the capacity of the PVP microneedles platform to incorporate multiple allergens. A six-patch microneedle array was developed as a proof-of-concept model. A PDMS mold was first prepared that has six wells for six separate microneedle patches (Fig. 7A). PVP microneedles labeled with fluorescein or rhodamine B were cast into the mold in a checkerboard pattern and mounted onto a single Tegaderm patch (Fig. 7B). The needles in each patch were well formed (Fig. 7C). Fluorescence from each domain of the multi-patch array was detected after excitation with a UV lamp (Fig. 7D).
Fig. 7.

Array of microneedles. (A) PDMS mold with six patches used to prepare the array. (B) An array of six PVP microneedles labeled with alternating fluorescent dyes (fluorescein and rhodamine B) peeled off the mold by a Tegaderm patch. (C) Images of individual microneedle patches in the array. (D) Image of the microneedles array under UV illumination.
4. Discussion
Microneedles made from various biodegradable or dissolving polymers have been reported recently, including carboxymethyl-cellulose (CMC) [26], maltose [27], poly(lactic-co-glycolic acid) (PLGA) [28] and chitosan [29], in addition to PVP. Compared to these formulations, our microneedles offer a few advantages. First, the conditions used during manufacturing are mild and the process is simple, inexpensive and easily scaled. Allergen proteins are dissolved in aqueous solution and the micromolding is completed in one step at room temperature. The process works with peptides, full proteins and non-protein components. In contrast, high temperature is needed to melt PLGA polymers and maltose, which may damage peptides and proteins [27,28]. As we showed in Fig. 4B, PVP microneedles prepared by photopolymerization caused extensive crosslinking of encapsulated proteins. A more recent report described a separable arrowhead PVP microneedle prepared without using photopolymerization [30]. However, in this method, polymer microneedle tips need to be mounted on a steel shaft to enhance penetration, which increases the complexity and cost of the fabrication process [30].
Second, the needle dissolution is rapid, which is especially favorable in clinical practice. As shown in the human foreskin test (Fig. 5), complete dissolution was observed after 5 min. This speed contrasts with the published CMC microneedles, which showed incomplete dissolution 24 h after insertion into rat skin [26]. The addition of trehalose increased the water solubility. Nevertheless, complete needle dissolution still required several hours [26]. PLGA and chitosan microneedles showed even slower release of encapsulated proteins, lasting for days [28,29]. In another report, human growth hormone (hGH) was encapsulated in CMC to prepare microneedles using a simple and mild fabrication method which did not affect the functional activity of the hGH [26]. However, the kinetics of dissolution of these microneedles was not suitable for use in allergen skin testing. These other microneedles may be useful in continuous delivery of therapeutics over a long period of time, but are not suitable for conducting an allergy skin test, which requires fast delivery.
Although dissolvable polymeric microneedles have gained considerable attention in recent years as a method for delivering drugs and vaccine transdermally, few reports have explored their utility in allergy skin testing. To the best of our knowledge, only one patent described polymeric microneedle devices for diagnosis of allergy [31]. However, in this patent, proteins were loaded by coating the surface of the needles, which is cumbersome and inefficient; furthermore, PVP was not able to be used successfully.
5. Conclusion
In summary, we present a technique for fabricating microneedles with PVP that offers the capability of delivering multiple, intact proteins or peptides to the skin for diagnostic or therapeutic applications. When applied to testing linear peptide epitopes of food allergens, this approach offers advantages for easy, low-cost and functional testing of thousands of epitopes in parallel.
Acknowledgements
We acknowledge Si Wan Kim for technical assistance and Jon Mulholland of the Stanford Cell Sciences Imaging Facility. We acknowledge funding from the NIH (K08 AI079268) to M.J.B., from the Gold Family Trust in the form of an unrestricted Grant to C.M., from the Epidermolysis Bullosa Medical Research Foundation and Jackson Gabriel Silver Foundation to A.T.L. and M.P.M., from the Stanford University School of Medicine SPARK and Spectrum (Stanford’s Clinical Translational Science Award from NIH) to A.T.L., J.A.R and M.P.M. and from the Office of Research and Development, Palo Alto VA Medical Center to M.P.M.
Footnotes
References
- 1.Kupper TS, Fuhlbrigge RC. Immune surveillance in the skin: mechanisms and clinical consequences. Nat Rev Immunol. 2004;4:211–222. doi: 10.1038/nri1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee JW, Park JH, Prausnitz MR. Dissolving microneedles for transdermal drug delivery. Biomaterials. 2008;29:2113–2124. doi: 10.1016/j.biomaterials.2007.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu Q, Zarnitsyn VG, Ye L, Wen Z, Gao Y, Pan L, et al. Immunization by vaccine-coated microneedle arrays protects against lethal influenza virus challenge. Proc Natl Acad Sci USA. 2009;106:7968–7973. doi: 10.1073/pnas.0812652106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi HJ, Yoo DG, Bondy BJ, Quan FS, Compans RW, Kang SM, et al. Stability of influenza vaccine coated onto microneedles. Biomaterials. 2012;33:3756–3769. doi: 10.1016/j.biomaterials.2012.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sullivan SP, Koutsonanos DG, Del Pilar Martin M, Lee JW, Zarnitsyn V, Choi SO, et al. Dissolving polymer microneedle patches for influenza vaccination. Nat Med. 2010;16:915–920. doi: 10.1038/nm.2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Branum AM, Lukacs SL. Food allergy among U.S. children: trends in prevalence and hospitalizations. NCHS Data Brief. 2008;124:1–8. [PubMed] [Google Scholar]
- 7.Branum AM, Lukacs SL. Food allergy among children in the United States. Pediatrics. 2009;124:1549–1555. doi: 10.1542/peds.2009-1210. [DOI] [PubMed] [Google Scholar]
- 8.Patel DA, Holdford DA, Edwards E, Carroll NV. Estimating the economic burden of food-induced allergic reactions and anaphylaxis in the United States. J Allergy Clin Immunol. 2011;128:110. doi: 10.1016/j.jaci.2011.03.013. U87. [DOI] [PubMed] [Google Scholar]
- 9.Sicherer SH. Epidemiology of food allergy. J Allergy Clin Immunol. 2011;127:594–602. doi: 10.1016/j.jaci.2010.11.044. [DOI] [PubMed] [Google Scholar]
- 10.Asero R, Fernandez-Rivas M, Knulst AC, Bruijnzeel-Koomen CA. Double-blind, placebo-controlled food challenge in adults in everyday clinical practice: a reappraisal of their limitations and real indications. Curr Opin Allergy Clin Immunol. 2009;9:379–385. doi: 10.1097/ACI.0b013e32832d9049. [DOI] [PubMed] [Google Scholar]
- 11.Niggemann B, Beyer K. Diagnostic pitfalls in food allergy in children. Allergy. 2005;60:104–107. doi: 10.1111/j.1398-9995.2004.00591.x. [DOI] [PubMed] [Google Scholar]
- 12.Norgaard A, Bindslev-Jensen C, Skov PS, Poulsen LK. Specific serum IgE in the diagnosis of egg and milk allergy in adults. Allergy. 1995;50:636–647. doi: 10.1111/j.1398-9995.1995.tb02580.x. [DOI] [PubMed] [Google Scholar]
- 13.Sicherer SH. Clinical implications of cross-reactive food allergens. J Allergy Clin Immunol. 2001;108:881–890. doi: 10.1067/mai.2001.118515. [DOI] [PubMed] [Google Scholar]
- 14.Williams LW, Bock SA. Skin testing and food challenges in allergy and immunology practice. Clin Rev Allergy Immunol. 1999;17:323–338. doi: 10.1007/BF02737614. [DOI] [PubMed] [Google Scholar]
- 15.Shreffler WG, Beyer K, Chu TH, Burks AW, Sampson HA. Microarray immunoassay: association of clinical history, in vitro IgE function, and heterogeneity of allergenic peanut epitopes. J Allergy Clin Immunol. 2004;113:776–782. doi: 10.1016/j.jaci.2003.12.588. [DOI] [PubMed] [Google Scholar]
- 16.Cerecedo I, Zamora J, Shreffler WG, Lin J, Bardina L, Dieguez MC, et al. Mapping of the IgE and IgG4 sequential epitopes of milk allergens with a peptide microarray-based immunoassay. J Allergy Clin Immunol. 2008;122:589–594. doi: 10.1016/j.jaci.2008.06.040. [DOI] [PubMed] [Google Scholar]
- 17.Perez-Gordo M, Lin J, Bardina L, Pastor-Vargas C, Cases B, Vivanco F, et al. Epitope mapping of atlantic salmon major allergen by peptide microarray immunoassay. Int Arch Allergy Immunol. 2011;157:31–40. doi: 10.1159/000324677. [DOI] [PubMed] [Google Scholar]
- 18.Shreffler WG, Lencer DA, Bardina L, Sampson HA. IgE and IgG4 epitope mapping by microarray immunoassay reveals the diversity of immune response to the peanut allergen, Ara h 2. J Allergy Clin Immunol. 2005;116:893–899. doi: 10.1016/j.jaci.2005.06.033. [DOI] [PubMed] [Google Scholar]
- 19.Vereda A, Andreae DA, Lin J, Shreffler WG, Ibanez MD, Cuesta-Herranz J. Identification of IgE sequential epitopes of lentil (Len c 1) by means of peptide microarray immunoassay. J Allergy Clin Immunol. 2010;126(596–601):e1. doi: 10.1016/j.jaci.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiu P, Mao C. Biomimetic branched hollow fibers templated by self-assembled fibrous polyvinylpyrrolidone structures in aqueous solution. ACS Nano. 2010;4:1573–1579. doi: 10.1021/nn9009196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Juliana KR, Zhengxing C. Effects of processing methods on the physic-functional properties of peanut flour (Arachis hypogaea L.) Biotechnology. 2008;7:168–174. [Google Scholar]
- 22.Nakajima N, Ikada Y. Mechanism of amide formation by carbodiimide for bioconjugation in aqueous media. Bioconjug Chem. 1995;6:123–130. doi: 10.1021/bc00031a015. [DOI] [PubMed] [Google Scholar]
- 23.Deegan RD, Bakajin O, Dupont TF, Huber G, Nagel SR, Witten TA. Capillary flow as the cause of ring stains from dried liquid drops. Nature. 1997;389:827–829. [Google Scholar]
- 24.Kuncicky DM, Velev OD. Surface-guided templating of particle assemblies inside drying sessile droplets. Langmuir. 2007;24:1371–1380. doi: 10.1021/la702129b. [DOI] [PubMed] [Google Scholar]
- 25.Wong TS, Chen TH, Shen X, Ho CM. Nanochromatography driven by the coffee ring effect. Anal Chem. 2011;83:1871–1873. doi: 10.1021/ac102963x. [DOI] [PubMed] [Google Scholar]
- 26.Lee JW, Choi SO, Felner EI, Prausnitz MR. Dissolving microneedle patch for transdermal delivery of human growth hormone. Small. 2011;7:531–539. doi: 10.1002/smll.201001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee K, Lee CY, Jung H. Dissolving microneedles for transdermal drug administration prepared by stepwise controlled drawing of maltose. Biomaterials. 2011;32:3134–3140. doi: 10.1016/j.biomaterials.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 28.Park JH, Allen MG, Prausnitz MR. Polymer microneedles for controlled-release drug delivery. Pharm Res. 2006;23:1008–1019. doi: 10.1007/s11095-006-0028-9. [DOI] [PubMed] [Google Scholar]
- 29.Chen MC, Ling MH, Lai KY, Pramudityo E. Chitosan microneedle patches for sustained transdermal delivery of macromolecules. Biomacromolecules. 2012;13:4022–4031. doi: 10.1021/bm301293d. [DOI] [PubMed] [Google Scholar]
- 30.Chu LY, Prausnitz MR. Separable arrowhead microneedles. J Control Release. 2011;149:242–229. doi: 10.1016/j.jconrel.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuwahara T, Matsudo T, Tokumoto S. Microneedle device for diagnosis of allergy. United States Patent US 20100030100A1. 2010 [Google Scholar]