

Abstract
Arsenic is a well-documented human carcinogen associated with skin carcinogenesis. Our previous work reveals that arsenite exposure is able to induce cell transformation in mouse epidermal cell JB6 Cl41 through the activation of ERK, rather than JNK pathway. Our current studies further evaluate downstream pathway in low dose arsenite-induced cell transformation in JB6 Cl41 cells. Our results showed that treatment of cells with low dose arsenite induced activation of c-Jun/AP-1 pathway, and ectopic expression of dominant negative mutant of c-Jun (TAM67) blocked arsenite-induced transformation. Furthermore, our data indicated that cyclin D1 was an important downstream molecule involved in c-Jun/AP-1-mediated cell transformation upon low dose arsenite exposure, because inhibition of cyclin D1 expression by its specific siRNA in the JB6 Cl41 cells resulted in impairment of anchorage-independent growth of cells induced by low dose arsenite. Collectively, our results demonstrate that c-Jun/AP-1-mediated cyclin D1 expression is at least one of the key events implicated in cell transformation upon low dose arsenite exposure.
Keywords: c-Jun, AP-1, Cyclin D1, Arsenite, Transformation
Introduction
Arsenic is a widespread pollutant and its toxic properties at high doses have been recognized for years (Bernstam and Nriagu, 2000; Evans, 1977). Acute exposure to arsenite at relatively high doses has been shown to result in cell apoptosis, which contributes to the pathogenic effects such as inflammation, hyperplasia and fibrosis (Lage et al., 2006; Pedlar et al., 2002). Epidemiologic investigations also indicate that long-term low dose arsenic exposure associates with carcinogenic effects in different tissues, especially in lung and skin via inhalation and ingestion (IARC, 1980). Arsenite induces cell transformation of various types of cells to a more malignant phenotype in several cultured cell models at relatively low doses that represent the exposure concentrations in natural environment (Huang et al., 1996; Ouyang et al., 2008; Pi et al., 2008). It is known that cell transformation is a complex process which involves many transcription factors and their downstream genes (Huang et al., 1996; Landolph, 1994; Pi et al., 2008). Nuclear factor kappa B (NFκB) (Ouyang et al., 2008) and nuclear factor E2-related factor 2 (Nrf2) (Pi et al., 2008) are reported to be involved in arsenite-induced cell transformation. AP-1 is another well-characterized transcription factor composed of homodimers and/or heterodimers of the Jun and Fos gene families. Overexpression of c-Jun greatly enhances the tumorigenic properties of the MCF7 human mammary carcinoma cell line, inducing elevated motility, and increased tumor formation in nude mice (Doucas et al., 1991; Smith et al., 1999; Vogt, 2001). In current studies, by applying dominant negative mutant of c-Jun (TAM67) to block AP-1’s function, we exploited the role of c-Jun/AP-1 pathway in the transformation potential of low dose arsenite in mouse epidermal JB6 Cl41 cells.
The mechanism of c-Jun/AP-1-induced oncogenic transformation is due to its abnormal regulation of its specific target genes. Cyclin D proteins are regulators of G1 to S phase transitions, and they bind to cyclin-dependent kinase 4 (CDK4) and increase its kinase activity, thereby causing the phosphorylation and inactivation of the retino-blastoma tumor suppressor protein (Shaulian and Karin, 2001; Sherr, 1996). Aberrant cyclin D1 expression has been observed in carcinogenesis (Barnes and Gillett, 1998; Fusenig and Boukamp, 1998; Weinstein, 2000), and overexpression of cyclin D1 was reported in several human cancers, including skin cancer (Rodriguez-Puebla et al., 1999). Previous studies have shown that the human cyclin D1 gene regulatory sequences contain two AP-1 binding sites (Albanese et al., 1995; Herber et al., 1994; Shaulian and Karin, 2001). c-Jun is shown to bind these sites and suggested to regulate cyclin D1 transcription (Bakiri et al., 2000; Beier et al., 1999; Shaulian and Karin, 2001). Our current study further reveals that this AP-1/c-Jun-dependent cyclin D1 induction played an important role in transformation potential of low dose arsenite in JB6 Cl41 cells.
Materials and methods
Cell culture and reagents
Mouse epidermal cell, JB6 Cl41, and its stable transfectants, were cultured as monolayer at 37 °C, 5% CO2 using MEM containing 5% fetal bovine serum (FBS), 2 mM L-glutamine, and 25 μg of gentamicin/ml. JB6 Cl41 cells were neither able to cause spontaneous transformation in soft agar, nor to form tumors in nude mice. The cultures were detached with trypsin and transferred to new 75-cm2 culture flasks (Fisher, Pittsburgh, PA) twice a week. FBS was purchased from Life Technologies, Inc.; MEM was from Calbiochem (San Diego, CA); and sodium arsenite was purchased from Aldrich (Milwaukee, WI).
Plasmids and cyclin D1 small interference RNA construction
The AP-1-luciferase reporter plasmid (pAP-1-Luc, Cat#219074) was purchased from Stratagene (La Jolla, CA), and used to determine AP-1-dependent transcriptional activity. In this plasmid, the expression of the Photinus pyralis (firefly) luciferase gene is controlled by a synthetic promoter that contains direct repeats of the transcription recognition sequences for AP-1. The cyclin D1 promoter-driven luciferase reporter was constructed by inserting a 1.23 kb EcoR I-Pvu II fragment of the cyclin D1 gene promoter, which contains the cyclin D1 promoter sequence from −1095 to +135 relative to the translation initiation site, into the pA3LUC vector as described previously (Ouyang et al., 2005; Yan et al., 1997). pcDNA3.1 plasmid containing c-Jun dominant negative mutant (pcDNA3.1/His-TAM67) was kindly provided by Dr. Tim G. Bowden (College of Pharmacy, University of Arizona, Tucson, AZ) and Dr. Matthew Young (Center for Cancer Research, National Cancer Institute, Frederick, MD). The cyclin D1 siRNA expression plasmid was made by using GeneSuppressor ™ System (Imgenex Co. San Diego, CA). The siRNA target sequences for cyclin D1 were: 5′-GUU GUG CAU CUA CAC UGA CTT-3′, and 5′-GUC AGU GUA GAU GCA CAA CTT-3′(NM_007631). Constructs containing the reversed target sequences were used as negative siRNA control.
Stable and transient transfection
JB6 Cl41 cells were transfected with 1 μg AP-1 luciferase reporter plasmid or cyclin D1 promoter-driven luciferase reporter plasmid with or without 4 μg TAM67 by LipofectAMINE transfection kit (Gibco BRL, Rockville, MD) according to the manufacturer’s instructions. 5 μg of siRNA cyclin D1 construct was stably transfected into JB6 Cl41 cells using the same kit. The stable transfectants were established by G418 selection (400 μg/ml). Stable transfectants were cultured in G418-free MEM for at least two passages before experimentation. After chronic exposure to low dose arsenite, the JB6 Cl41 parental cells and TAM67 transfectants were transiently transfected with AP-1-luciferase reporter and cyclin D1 promoter-driven luciferase reporter. 48 h post transfection, the above cells were subjected to reporter assay.
Gene reporter assay
Confluent monolayer of AP-1-, or cyclin D1-luciferase reporter stable transfectants were trypsinized, and 8×103 viable cells suspended in 100 μl of medium were seeded to each well of 96-well plates. After the cell density reached 80–90% confluence, the cells were treated with arsenite for time periods as indicated in the figure legends. The cells were then lysed with 50 μl lysis buffer, and the luciferase activity was measured using Promega Luciferase assay reagent with a luminometer (Wallac 1420 Victor2 multipliable counter system). The results were expressed as AP-1 activity relative to the medium control or cyclin D1 induction relative to the medium control.
Western blot
JB6 Cl41 cells and their transfectants (2×105) were cultured in each well of 6-well plates to 70–80% confluence with normal culture medium, and then treated with arsenite for different time frames. The cells were washed once with ice-cold PBS and extracted with SDS-sample buffer. The cell extracts were separated on polyacrylamide-SDS gels, transferred, and probed with the antibodies against phosphor-specific c-Jun (Ser73), c-Jun, cyclin D1 (Cell Signaling, Danvers, MA) and β-Actin (Sigma, St. Louis, MO). The protein bands specifically bound to the primary antibodies were detected using an alkaline phosphatase-linked secondary antibody and an ECF Western blot system (Amersham, Piscataway, NJ).
Reverse transcription polymerase chain reaction (RT-PCR)
After exposure to arsenite for indicated time periods, total RNA was extracted from the cells using Trizol reagent (Invitrogen, Carlsbad, California). Total cDNAs were synthesized by ThermoScript™ RT-PCR system (Invitrogen). Cyclin D1 mRNA amount presented in the cells was measured by semiquantitative RT-PCR according to the previous reports (Klein et al., 2003; Li et al., 2004). The PCR products were separated on 2% agarose gels, stained with EB, and scanned the images from a UV light.
Anchorage-independent growth
JB6 Cl41 cells and their transfectants (1×105) were cultured in each well of 6-well plates to 50–60% confluence with normal culture medium. The cells were treated with 2.5–10 μM arsenite for 3 days and then recovered in normal medium for another 3 days. After the repeated treatment with arsenite for 6 months, the cells were used for anchorage-independent growth assay which was performed as described previously (Huang et al., 1999a; Yan et al., 2006). Briefly, 2.5 ml of 0.5% agar in BMEM supplemented with 10% fetal bovine serum was laid onto each well of 6-well tissue culture plates. 2×104 of JB6 Cl41 cells were mixed with 2 ml of 0.5% agar BMEM and layered on top of the 0.5% agar layer. The plates were incubated at 37 °C in 5% CO2 for 3 weeks. The colonies with more than 16 cells were then scored.
Statistical analysis
The significance of the differences between the treated and untreated groups, or parental cells and stable transfectants was determined with the Student’s t test. The results are expressed as mean±S.D of three independent experiments.
Results
c-Jun/AP-1 activation participated in the transformation potential of low dose arsenite in mouse epidermal cells JB6 Cl41
Skin is a major target of environmental carcinogen arsenite. The role of AP-1 in the epidermis of the skin has been suggested for differentiation, carcinogenesis, UV-response, photo-aging and wound repair (Angel et al., 2001; Zenz and Wagner, 2006). To determine the potential involvement of the AP-1 pathway in the transformation potential of low dose arsenite in skin cells, we tested the AP-1 activity in response to low dose arsenite in mouse epidermal cell JB6 Cl41. We found that exposure to low dose arsenite at 2.5–10 μM for 2 days, which was not toxic to JB6 Cl41 cells, did increase AP-1 activity in JB6 Cl41 cells (Fig. 1A). c-Jun phosphorylation, which is essential for AP-1 activity, was also observed upon low dose arsenite treatment (Fig. 1B). Consistently, when JB6 Cl41 cells were repeatedly treated with low dose arsenite ranging from 10 to 2.5 μM for 4, 5 and 6 months, AP-1 activity was also readily increased detected by luciferase reporter assay (Fig. 1C) and c-Jun phsophorylation (Fig. 1D). More importantly, compared with the medium control, repeated arsenite exposure at low doses resulted in the increased anchorage-independent growth capacity of JB6 Cl41 cells in soft agar assay (Figs. 1E and F), indicating that repeated exposure of JB6 Cl41 cells to low dose arsenite could cause malignant transformation. However, the transformation colonies were smaller than those induced by TPA as we previously reported (Huang et al., 1999b).
Fig. 1.

Exposure to low dose arsenite triggered c-Jun/AP-1 pathway activation and transformation in mouse epidermal cells JB6 Cl41 cells. (A) JB6 Cl41 cells stably transfected with AP-1 luciferase reporter (1×103) were seeded into a 96-well plate and cultured in MEM containing 5% FBS at 37 °C overnight. After the cells were treated with the indicated doses of arsenite for 48 h, the treated cells were extracted with lysis buffer, and the luciferase activity was measured using the Promega luciferase assay kit as described in Materials and methods. AP-1 transcription activity was presented as the induction fold of luciferase post arsenite treatment compared with the basal luciferase without arsenite treatment (relative AP-1 activity). The symbol (*) indicates a significant increase as compared with that of medium control (p<0.05). (B) JB6 Cl41 cells were treated with arsenite in different doses as indicated for 48 h. Western blotting assay was carried out to determine c-Jun activation using antibodies against phosphorylated c-Jun at Ser73 or total c-Jun. (C–F) 1×105 JB6 Cl41 cells were seeded into each well of 6-well plates and cultured till 50–60% confluent. The cells were then repeatedly treated with or without arsenite (ranging from 10 to 2.5 μM) every 3 days for 4, 5 and 6 months as described in the section of Materials and methods. Then the cells were subjected to the following tests. (C) The cells were transiently transfected with AP-1 luciferase to detect its activity. The symbol (*) indicates a significant increase as compared with that of medium control (p<0.05). (D) The cells were extracted for Western blotting assay to determine c-Jun activation. (E and F) The cell transformation was detected by soft agar assay. The colony formation was observed under inverted microscope and photographed (E), and numbers of colonies were scored, and presented as colonies per 10,000 seeded cells (F). The symbol (*) indicates a significant increase as compared with that of medium control (p<0.05). Each bar indicates the mean and standard deviation of three independent experiments.
To elucidate the role of AP-1 pathway in the transformation potential of low dose arsenite, we overexpressed the dominant negative mutant form of c-Jun (TAM67) in JB6 Cl41 cells to abolish c-Jun/AP-1 activity induced by low dose arsenite in short term exposure (Figs. 2A and B) as well as in long term exposure (Figs. 2C and D). Consistent with the inhibition of AP-1 activity, ectopic expression of TAM67 consequently blocked cell transformation upon chronic low dose arsenite exposure in JB6 Cl41 cells (Figs. 2E and F). These results demonstrate that the c-Jun/AP-1 pathway is involved in low dose arsenite-induced transformation in JB6 Cl41 cells.
Fig. 2.

c-Jun/AP-1 pathway activation was responsible for low dose arsenite-induced transformation in JB6 Cl41 cells. (A) JB6 Cl41 AP-1-luciferase stable transfectant (Vector) and JB6 Cl41/TAM67 AP-1-luciferase stable transfectant (TAM67) were treated with 5 μM of arsenite for the indicated time periods and AP-1 activity was assayed. The symbol (*) indicates a significant increase between the arsenite-treated and non-treated parental cells (p<0.05). (B) Vector and TAM67 stable transfectants were treated with 5 μM of arsenite for the indicated time periods and c-Jun activation was tested by Western blotting. (C–F) Vector and TAM67 stable transfectants were repeatedly treated with or without arsenite for up to 6 months. The cells were then transiently transfected with AP-1 luciferase to detect its activity (C). The cells were also extracted for Western blotting assay to determine c-Jun activation (D). The cell transformation was detected by soft agar assay (E and F). The symbol (*) indicates a significant increase as compared with that of medium control (p<0.05). Each bar indicates the mean and standard deviation of three independent experiments.
Cyclin D1 was a key downstream target of c-Jun/AP-1 pathway in response to low dose arsenite in JB6 Cl41 cells
It has been thought that the contribution of the AP-1 pathway to tumorigenesis could be associated with its regulation of cell apoptosis or cell growth. Our previous studies have shown that arsenite exposure was able to up-regulate cyclin D1 protein expression in HaCat cells, which further mediates cell cycle alternation (Ouyang et al., 2005). Therefore, we further tested whether there was a link between low dose arsenite-induced c-Jun/AP-1 activation and cyclin D1 protein expression. As we expected, low dose arsenite treatment resulted in a marked increase in cyclin D1 protein expression in acute and chronic exposure responses in JB6 Cl41 cells (Figs. 3A and D). Cyclin D1 mRNA and transcription were elevated by low dose arsenite treatment as well (Figs. 3B and C). More interestingly, this cyclin D1 induction was dramatically impaired in TAM67 stable transfectants (Figs. 4A and D), probably due to the reductions of cyclin D1 transcription and mRNA (Figs. 4B and C). These results demonstrate that c-Jun/AP-1 activation is critical for cyclin D1 protein induction by low dose arsenite exposure.
Fig. 3.
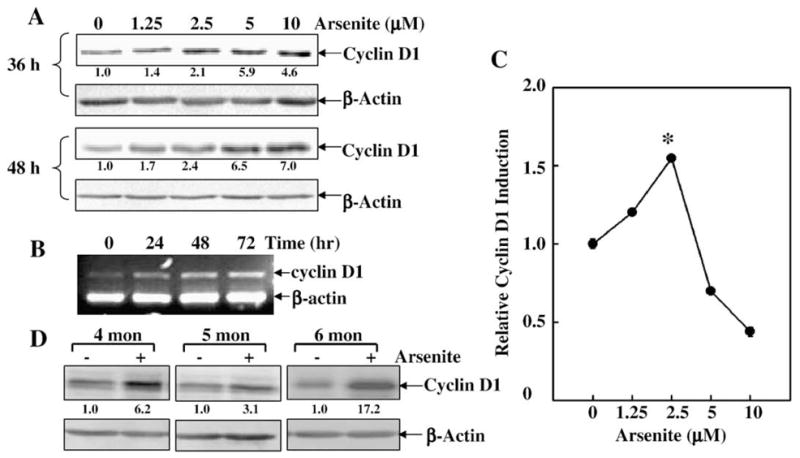
Cyclin D1 was induced by low dose arsenite exposure in JB6 Cl41 cells. (A) JB6 Cl41 cells were treated with arsenite in different doses as indicated for 36 h and 48 h. Western blotting assay was carried out to determine cyclin D1 protein expression. (B) JB6 Cl41 cells were exposed to 2.5 μM of arsenite for the indicated time points, and then extracted with Trizol reagent for the total RNA isolation. Cyclin D1 was amplified with their specific primers by RT-PCR for 30 cycles. β-actin was used as an internal control. (C) JB6 Cl41 cells stably transfected with cyclin D1 promoter-driven luciferase reporter were treated with arsenite for 48 h to detect cyclin D1 transcription. Cyclin D1 induction was presented as the induction fold of luciferase post arsenite treatment compared with the basal luciferase without arsenite treatment (relative cyclin D1 induction). The symbol (*) indicates a significant increase as compared with that of medium control (p<0.05). (D) After JB6 Cl41 cells were exposed to chronic arsenite treatment (4, 5 and 6 months), cyclin D1 protein levels were determined by Western blotting. Each bar indicates the mean and standard deviation of three independent experiments.
Fig. 4.
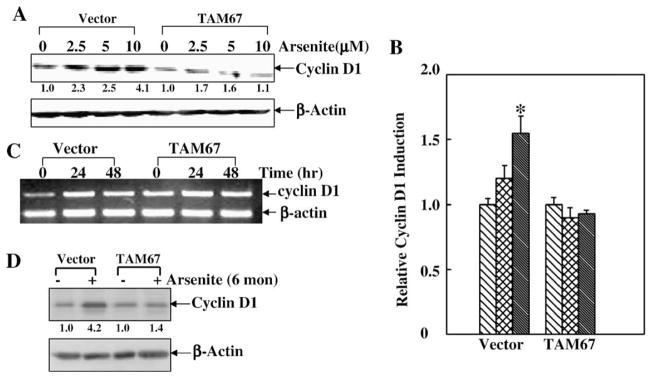
Cyclin D1 induction by low dose arsenite was mediated by c-Jun/AP-1 pathway. (A) JB6 Cl41 vector and TAM67 stable transfectants were treated with different doses of arsenite for 48 h and cyclin D1 protein induction was tested by Western blotting. (B) The above cells were transfected with cyclin D1 promoter-driven luciferase, and treated with different doses of arsenite for 48 h and cyclin D1 transcription was assayed. The symbol (*) indicates a significant increase as compared with that of medium control (p<0.05). Each bar indicates the mean and standard deviation of three repeated experiments. (C) The cells as indicated were exposed to 5 μM of arsenite for 24 h and 48 h, and the mRNA level of cyclin D1 was detected by RT-PCR. β-actin was used as an internal control. (D) After the cells were exposed to chronic arsenite treatment, cyclin D1 protein level was determined by Western blotting.
Cyclin D1 was involved in low dose arsenite-induced transformation of JB6 Cl41 cells
To evaluate the contribution of cyclin D1 to low dose arsenite-induced cell transformation, we employed JB6 Cl41 cells that were stably transfected with siRNA cyclin D1 (Ouyang et al., 2005). As shown in Fig. 5A, introduction of siRNA cyclin D1 dramatically reduced the basal as well as induced levels of cyclin D1 protein expression, verifying the efficiency of cyclin D1 siRNA. Knockdown of cyclin D1 expression by its siRNA abrogated the JB6 Cl41 cell transformation induced by low dose arsenite (Figs. 5B and C). Collectively, these results indicate that cyclin D1 is not only induced by low dose arsenite exposure through the c-Jun/AP-1 pathway, but also is at least one of the key events responsible for low dose arsenite-induced transformation.
Fig. 5.
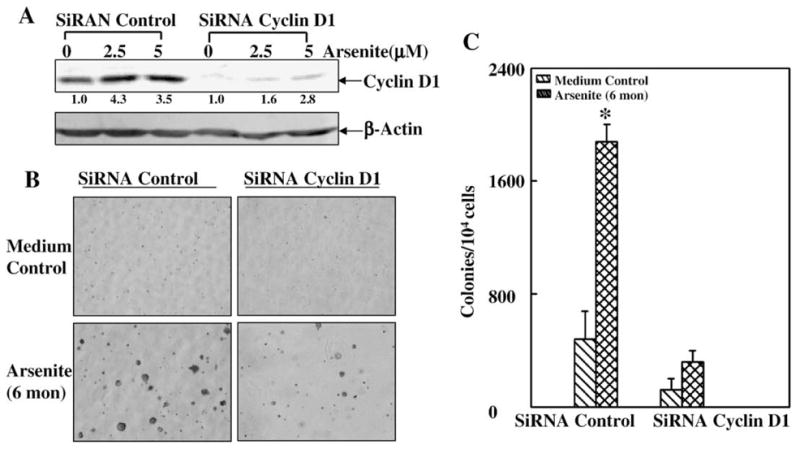
Cyclin D1 was involved in low dose arsenite-induced cell transformation in JB6 Cl41 cells. (A) JB6 Cl41 parental cells and SiRNA cyclin D1 stable transfectants were treated with different doses of arsenite for 48 h and cyclin D1 protein induction was tested by Western blotting. (B and C) After the above cells were exposed to chronic arsenite treatment, the anchorage-independent cell growth was assayed in soft agar. The symbol (*) indicates a significant increase as compared with that of medium control (p<0.05). Each bar indicates the mean and standard deviation of three repeated experiments.
Discussion
To better represent the oncogenic effect of low concentration of arsenite in environmental contamination, we repeatedly treated the mouse epidermal cell JB6 Cl41, with low dose arsenite in normal medium ranging from 2.5 to 10 μM for up to 6 months, and detected the cell anchorage-independent growth in soft agar. We found that treatment of cells with low dose arsenite caused cell transformation through induction of cyclin D1 expression mediated by c-Jun/AP-1 pathway.
Arsenite is a well documented human carcinogen (IARC, 1980; Lansdown, 1995). Arsenic contamination at low dose from both natural and manmade sources poses a risk to humans. It causes cancers of the skin and bladder; and also of the kidney and liver (Tseng, 1977; Tseng et al., 1968; Chen et al., 1992; Smith et al., 1992; Hopenhayn-Rich et al., 1998; Hwang et al., 2006). However, attempts to induce tumors in experimental animals with arsenic compound as a single agent have mostly failed in the past (Wang et al., 2002; Waalkes et al., 2004; 2007; Benbrahim-Tallaa and Waalkes, 2008). Therefore, in vitro studies using cultured cell lines such as keratinocytes and prostate epithelial cells, which may represent in vivo targets of arsenic, provide a relevant and reasonable approach to study the molecular events in arsenic carcinogenesis (Benbrahim-Tallaa and Waalkes, 2008). The findings from our lab and others have revealed that arsenic oncogenic properties were associated with epigenetic mechanisms including modulation of the activities of signaling pathways and gene expression that is responsible for cell growth and cellular transformation (Simeonova et al., 2000; Ouyang et al., 2005; 2006; 2007b; 2008; Hwang et al., 2006). In these works, we found that JB6 mouse epidermal cell is one of the optimal models for the study of genetic susceptibility to transformation promotion and progression at the molecular level. The transformed variants that grow under anchorage-independent conditions are tumorigenic in nude or BALB/c mice in the absence of tumor promoting conditions (Bernstein and Colburn, 1989; Dong et al., 1997b; Ouyang et al., 2007a). JB6 Cl41 cells also showed quite similar cellular responses with human keratinoyte HaCat cells like the activation of signaling pathways of PI-3K/Akt/IKK/NFκB/cyclin D1 leading to arsenite-induced malignant transformation, which was further confirmed to be tumorigenic in nude mice (Ouyang et al., 2006; 2008). Therefore, we took advantage of the normal mouse epidermal JB6 Cl41 cell model to further exploit the mechanisms of arsenite transformation potentials (Barrandon et al., 1989). In the present studies, we demonstrate that c-Jun/AP-1 pathway is involved in the cyclin D1-mediated arsenite transformation. Combined with our previous works (Ouyang et al., 2008), it indicates that for the regulation of important cellular molecules, such as cyclin D1, which is responsible for cell growth and consequently oncogenic transformation, there may be several parallel key signaling pathways, like c-Jun/AP-1 and PI-3K/Akt/IKK/NFκB pathways, involved. Thus it is possible that each pathway is partially, but not entirely, responsible for cyclin D1 regulation in response to arsenite, and that they might even crosstalk to each other and form the complicated signaling transduction network. The final outcome of these pathways will determine the fate of the cells in response to arsenite eventually.
The transcription factor AP-1 regulates many cellular processes, including proliferation, inflammation, differentiation and apoptosis. Elevated AP-1 activity has been detected in multiple human cancers and tumor cell lines (Young et al., 2003), suggesting a role for AP-1 in tumor promotion and progression (Matthews et al., 2007). The involvement of AP-1 in EGF and TPA-induced cell transformation in JB6 Cl41 cells was demonstrated by Nancy Colburn et al. using expression of a transactivation minus mutant of c-Jun (TAM67) (Brown et al., 1993; Dong et al., 1994; 1997a). Our current studies demonstrate that low dose arsenite exposure is able to activate c-Jun/AP-1 pathway and induce cell transformation in JB6 Cl41 cells, while inhibition of c-Jun/AP-1 pathway by TAM67 impaired low dose arsenite-induced cell transformation, suggesting that the c-Jun/AP-1 pathway might contribute to arsenite skin carcinogenic effects. Similar with our findings, Chiu et al. found that the activation of AP-1 transcription factors may play a regulatory role in lung transformation. Significant elevations in c-fos and c-Jun mRNA levels were observed within 30 min after exposure to arsenic and by enhancement of AP-1 DNA binding activity and transactivation activity (Li et al., 2002).
It is likely that the phenotype of the transformed cell results from the combined action of several positively and negatively misregulated targets (Vogt, 2001). The cyclin D1 gene is rearranged and over-expressed in a major fraction of human tumors (Hwang et al., 2006). Thus, increased expression of cyclin D1 can play a critical role in tumor development and in maintenance of the malignant phenotype. Current studies also show that low dose arsenite exposure results in a marked cyclin D1 induction, and knockdown of cyclin D1 by its siRNA effectively abolished arsenite-induced cell transformation. This clearly reveals the involvement of cyclin D1 expression in low dose arsenite-induced cell transformation. It is interesting to note that over time, cells transfected with TAM67 or siRNA for cyclin D1 lose the ability to form colonies in soft agar without exposure to arsenite, which might be due to the decreased cyclin D1 basal expression in these transfectants. It further indicates the importance of cyclin D1 for cellular transformation dependent or independent of promotion factors. RT-PCR and luciferase assay results indicated that low dose arsenite induced cyclin D1 expression through regulating its transcription. The cyclin D1 gene promoter region contains binding sites for a number of transcription factors including AP-1 (Hwang et al., 2006). When blocking c-Jun/AP-1 pathway activation by overexpres-sing TAM67, we found the induction of cyclin D1 at protein and transcription levels were both dramatically inhibited. Thus, our results provide the direct evidence that cyclin D1 is a downstream target of the c-Jun/AP-1 signal cascade, and is critical for the cell transformation upon arsenite exposure in JB6 Cl41 cells.
In summary, our studies demonstrate that the c-Jun/AP-1 pathway plays a role in the arsenite-induced transformation of JB6 Cl41 cells through the induction of cyclin D1. These results not only provide information for understanding the molecular mechanisms underlying the carcinogenic effect of arsenite on its major target tissue of skin, but also suggest that the c-Jun/AP-1/cyclin D1 pathway might be a target for chemoprevention of arsenite-induced skin cancer.
Acknowledgments
Grant Support: NIH/NCI/NIEHS (CA094964, CA112557, ES012451 and ES000260).
References
- Albanese C, Johnson J, Watanabe G, Eklund N, Vu D, Arnold A, Pestell RG. Transforming p21[IMAGE] mutants and c-Ets-2 activate the Cyclin D1 promoter through distinguishable regions. J Biol Chem. 1995;270:23589–23597. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- Angel P, Szabowski A, Schorpp-Kistner M. Function and regulation of AP-1 subunits in skin physiology and pathology. Oncogene. 2001;20:2413–2423. doi: 10.1038/sj.onc.1204380. [DOI] [PubMed] [Google Scholar]
- Bakiri L, Lallemand D, Bossy-Wetzel E, Yaniv M. Cell cycle-dependent variations in c-Jun and JunB phosphorylation: a role in the control of cyclin D1 expression. EMBO J. 2000;19:2056–2068. doi: 10.1093/emboj/19.9.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes DM, Gillett CE. Cyclin D1 in breast cancer. Breast Cancer Res Treat. 1998;52:1–15. doi: 10.1023/a:1006103831990. [DOI] [PubMed] [Google Scholar]
- Barrandon Y, Morgan JR, Mulligan RC, Green H. Restoration of growth potential in paraclones of human keratinocytes by a viral oncogene. Proc Natl Acad Sci USA. 1989;86:4102–4106. doi: 10.1073/pnas.86.11.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier F, Lee RJ, Taylor AC, Pestell RG, LuValle P. Identification of the cyclin D1 gene as a target of activating transcription factor 2 in chondrocytes. Proc Natl Acad Sci. 1999;96:1433–1438. doi: 10.1073/pnas.96.4.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benbrahim-Tallaa L, Waalkes M. Inorganic arsenic and human prostate cancer. Environ Health Perspect. 2008;116:158–164. doi: 10.1289/ehp.10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstam L, Nriagu J. Molecular aspects of arsenic stress. J Toxicol Environ Health, B Crit Rev. 2000;3:293–322. doi: 10.1080/109374000436355. [DOI] [PubMed] [Google Scholar]
- Bernstein LR, Colburn NH. AP1/jun function is differentially induced in promotion-sensitive and resistant JB6 cells. Science. 1989;244:566–569. doi: 10.1126/science.2541502. [DOI] [PubMed] [Google Scholar]
- Brown PH, Alani R, Preis LH, Szabo E, Birrer MJ. Suppression of oncogene-induced transformation by a deletion mutant of c-jun. Oncogene. 1993;8:877–886. [PubMed] [Google Scholar]
- Chen CJ, Chen CW, Wu MM, Kuo TL. Cancer potential in liver, lung, bladder and kidney due to ingested inorganic arsenic in drinking water. Br J Cancer. 1992;66:888–892. doi: 10.1038/bjc.1992.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Birrer MJ, Watts RG, Matrisian LM, Colburn NH. Blocking of tumor promoter-induced AP-1 activity inhibits induced transformation in JB6 mouse epidermal cells. Proc Natl Acad Sci. 1994;91:609–613. doi: 10.1073/pnas.91.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Z, Crawford HC, Lavrovsky V, Taub D, Watts R, Matrisian LM, Colburn NH. A dominant negative mutant of jun blocking 12-O-tetradecanoylphorbol-13-acetate-induced invasion in mouse keratinocytes. Mol Carcinog. 1997a;19:204–212. doi: 10.1002/(sici)1098-2744(199707)19:3<204::aid-mc8>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Dong Z, Huang C, Brown RE, Ma WY. Inhibition of activator protein 1activity and neoplastic transformation by aspirin. J Biol Chem. 1997b;272:9962–9970. doi: 10.1074/jbc.272.15.9962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucas V, Spyrou G, Yaniv M. Unregulated expression of c-Jun or c-Fos proteins but not Jun D inhibits oestrogen receptor activity in human breast cancer derived cells. EMBO J. 1991;10:2237–2245. doi: 10.1002/j.1460-2075.1991.tb07760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans S. Arsenic and cancer. Br J Dermatol. 1977;97:13–14. [Google Scholar]
- Fusenig NE, Boukamp P. Multiple stages and genetic alterations in immortalization, malignant transformation, and tumor progression of human skin keratinocytes. Mol Carcinog. 1998;23:144–158. doi: 10.1002/(sici)1098-2744(199811)23:3<144::aid-mc3>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Herber B, Truss M, Beato M, Müller R. Inducible regulatory elements in the human cyclin D1 promoter. Oncogene. 1994;9:1295–1304. [PubMed] [Google Scholar]
- Hopenhayn-Rich C, Biggs ML, Smith AH. Lung and kidney cancer mortality associated with arsenic in drinking water in Córdoba. Int J Epidemiol. 1998;27:561–569. doi: 10.1093/ije/27.4.561. [DOI] [PubMed] [Google Scholar]
- Huang C, Ma WY, Dong Z. Requirement for phosphatidylinositol 3-kinase in epidermal growth factor-induced AP-1 transactivation and transformation in JB6 P+ cells. Mol Cell Biol. 1996;16:6427–6435. doi: 10.1128/mcb.16.11.6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Li J, Ma WY, Dong Z. JNK activation is required for JB6 cell transformation induced by tumor necrosis factor-alpha but not by 12-O-tetradecanoylphorbol-13-acetate. J Biol Chem. 1999a;274:29672–29676. doi: 10.1074/jbc.274.42.29672. [DOI] [PubMed] [Google Scholar]
- Huang C, Ma WY, Li J, Goranson A, Dong Z. Requirement of Erk, but not JNK, for arsenite-induced cell transformation. J Biol Chem. 1999b;274:14595–14601. doi: 10.1074/jbc.274.21.14595. [DOI] [PubMed] [Google Scholar]
- Hwang BJ, Utti C, Steinberg M. Induction of cyclin D1 by submicromolar concentrations of arsenite in human epidermal keratinocytes. Toxicol Appl Pharmacol. 2006;217:161. doi: 10.1016/j.taap.2006.08.006. [DOI] [PubMed] [Google Scholar]
- IARC. International Agency for Research on Cancer. Monogr Eval Carcinog Risk Hum. 1980;23:37–141. [Google Scholar]
- Klein A, Guhl E, Tzeng YJ, Fuhrhop J, Levrero M, Graessmann M, Graessmann A. HBX causes cyclin D1 overexpression and development of breast cancer in transgenic animals that are heterozygous for p53. Oncogene. 2003;22:2910–2919. doi: 10.1038/sj.onc.1206539. [DOI] [PubMed] [Google Scholar]
- Lage CR, Nayak A, Kim CH. Arsenic ecotoxicology and innate immunity. Integr Comp Biol. 2006;46:1040–1054. doi: 10.1093/icb/icl048. [DOI] [PubMed] [Google Scholar]
- Landolph JR. Molecular mechanisms of transformation of C3H/10T1/2 C1 8 mouse embryo cells and diploid human fibroblasts by carcinogenic metal compounds. Environ Health Perspect. 1994;102:119–125. doi: 10.1289/ehp.94102s3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdown A. Physiological and toxicological changes in the skin resulting from the action and interaction of metal ions. Crit Rev Toxicol. 1995;25:397–462. doi: 10.3109/10408449509049339. [DOI] [PubMed] [Google Scholar]
- Li M, Cai JF, Chiu JF. Arsenic induces oxidative stress and activates stress gene expressions in cultured lung epithelial cells. J Cell Biochem. 2002;87:29–38. doi: 10.1002/jcb.10269. [DOI] [PubMed] [Google Scholar]
- Li J, Chen H, Tang MS, Shi X, Amin S, Desai D, Costa M, Huang C. PI-3K and Akt are mediators of AP-1 induction by 5-MCDE in mouse epidermal Cl41 cells. J Cell Biol. 2004;165:77–86. doi: 10.1083/jcb.200401004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews CP, Colburn NH, Young MR. AP-1 a target for cancer prevention. Curr Cancer Drug Targets. 2007;7:317–324. doi: 10.2174/156800907780809723. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Ma Q, Li J, Zhang D, Liu Z-g, Rustgi AK, Huang C. Cyclin D1 induction through I{kappa}B kinase {beta}/nuclear factor-{kappa}B pathway is responsible for arsenite-induced increased cell cycle G1-S phase transition in human keratinocytes. Cancer Res. 2005;65:9287–9293. doi: 10.1158/0008-5472.CAN-05-0469. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Li J, Ma Q, Huang C. Essential roles of PI-3K/Akt/IKK{beta}/NF{kappa}B pathway in cyclin D1 induction by arsenite in JB6 Cl41 cells. Carcinogenesis. 2006;27:864–873. doi: 10.1093/carcin/bgi321. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Hu Y, Li J, Ding M, Lu Y, Zhang D, Yan Y, Song L, Qu Q, Desai D, Amin S, Huang C. Direct evidence for the critical role of NFAT3 in benzo[a]pyrene diol-epoxide-induced cell transformation through mediation of inflammatory cytokine TNF induction in mouse epidermal Cl41 cells. Carcinogenesis. 2007a;28:2218–2226. doi: 10.1093/carcin/bgm115. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Li J, Zhang D, Jiang BH, Huang C. PI-3K/Akt signal pathway plays a crucial role in arsenite-induced cell proliferation of human keratinocytes through induction of cyclin D1. J Cell Biochem. 2007b;101:969–978. doi: 10.1002/jcb.21279. [DOI] [PubMed] [Google Scholar]
- Ouyang W, Luo W, Zhang D, Jian J, Ma Q, Li J, Shi X, Chen J, Gao J, Huang C. PI-3K/Akt pathway-dependent cyclin D1 expression is responsible for arsenite-induced human keratinocyte transformation. Environ Health Perspect. 2008;116:1–6. doi: 10.1289/ehp.10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedlar R, Ptashynski M, Evans R, Klaverkamp J. Toxicological effects of dietary arsenic exposure in lake whitefish (Coregonus clupeaformis) Aquat Toxicol. 2002;57:167–189. doi: 10.1016/s0166-445x(01)00198-9. [DOI] [PubMed] [Google Scholar]
- Pi J, Diwan BA, Sun Y, Liu J, Qu W, He Y, Styblo M, Waalkes MP. Arsenic-induced malignant transformation of human keratinocytes: involvement of Nrf2. Free Radic Biol Med. 2008;45:651–658. doi: 10.1016/j.freeradbiomed.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Puebla ML, Robles AI, Conti CJ. ras activity and cyclin D1 expression: an essential mechanism of mouse skin tumor development. Mol Carcinog. 1999;24:1–6. [PubMed] [Google Scholar]
- Shaulian E, Karin M. AP-1 in cell proliferation and survival. Oncogene. 2001;20:2390–2400. doi: 10.1038/sj.onc.1204383. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- Simeonova PP, Wang S, Toriuma W, Kommineni V, Matheson J, Unimye N, Kayama F, Harki D, Ding M, Vallyathan V, Luster MI. Arsenic mediates cell proliferation and gene expression in the bladder epithelium: association with activating protein-1 transactivation. Cancer Res. 2000;60:3445–3453. [PubMed] [Google Scholar]
- Smith AH, Hopenhayn-Rich C, Bates MN, Goeden HM, Hertz-Picciotto I, Duggan HM, Wood RS, Kosnett MJ, Smith MTS, Vallyathan V, Luster MI. Cancer risks from arsenic in drinking water. Environ Health Perspect. 1992;97:259–267. doi: 10.1289/ehp.9297259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L, Wise S, Hendricks D, Sabichi A, Bos T, Reddy P, Brown P, Birrer M. cJun overexpression in MCF-7 breast cancer cells produces a tumorigenic, invasive and hormone resistant phenotype. Oncogene. 1999;18:6063–6070. doi: 10.1038/sj.onc.1202989. [DOI] [PubMed] [Google Scholar]
- Tseng WP. Effects and dose response relationships of skin cancer and blackfoot disease with arsenic. Environ Health Perspect. 1977;19:109–119. doi: 10.1289/ehp.7719109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng WP, Chu HM, How SW, Fong JM, Lin CS, Yeh S. Prevalence of skin cancer in an endemic area of chronic arsenicism in Taiwan. J Natl Cancer Inst. 1968;40:453–463. [PubMed] [Google Scholar]
- Vogt PK. Jun, the oncoprotein. Oncogene. 2001;20:2365–2377. doi: 10.1038/sj.onc.1204443. [DOI] [PubMed] [Google Scholar]
- Wang JP, Qi L, Moore MR, Ng JC. A review of animal models for the study of arsenic carcinogenesis. Toxicol Lett. 2002;133:17–31. doi: 10.1016/s0378-4274(02)00086-3. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Liu J, Ward JM, Diwan BA. Animal models for arsenic carcinogenesis: inorganic arsenic is a transplacental carcinogen in mice. Toxicol Appl Pharmacol. 2004;198:377–384. doi: 10.1016/j.taap.2003.10.028. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Liu J, Diwan BA. Transplacental arsenic carcinogenesis in mice. Toxicol Appl Pharmacol. 2007;222:271–280. doi: 10.1016/j.taap.2006.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein IB. Disorders in cell circuitry during multistage carcinogenesis: the role of homeostasis. Carcinogenesis. 2000;21:857–864. doi: 10.1093/carcin/21.5.857. [DOI] [PubMed] [Google Scholar]
- Yan YX, Nakagawa H, Lee MH, Rustgi AK. Transforming growth factor-alpha enhances Cyclin D1 transcription through the binding of early growth response protein to a cis-regulatory element in the Cyclin D1 promoter. J Biol Chem. 1997;272:33181–33190. doi: 10.1074/jbc.272.52.33181. [DOI] [PubMed] [Google Scholar]
- Yan Y, Li J, Ouyang W, Ma Q, Hu Y, Zhang D, Ding J, Qu Q, Subbaramaiah K, Huang C. NFAT3 is specifically required for TNF-{alpha}-induced cyclooxygenase-2 (COX-2) expression and transformation of Cl41 cells. J Cell Sci. 2006;119:2985–2994. doi: 10.1242/jcs.03014. [DOI] [PubMed] [Google Scholar]
- Young MR, Yang HS, Colburn NH. Promising molecular targets for cancer prevention: AP-1, NF-[kappa]B and Pdcd4. Trends Mol Med. 2003;9:36–41. doi: 10.1016/s1471-4914(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Zenz R, Wagner EF. Jun signalling in the epidermis: from developmental defects to psoriasis and skin tumors. Int J Biochem Cell Biol. 2006;38:1043–1049. doi: 10.1016/j.biocel.2005.11.011. [DOI] [PubMed] [Google Scholar]