

Abstract
Gastrointestinal disorders such as chronic or acute diarrhea, malabsorption, abdominal pain, and inflammatory bowel diseases can indicate immune deficiency. The gastrointestinal tract is the largest lymphoid organ in the body, so it is not surprising that intestinal diseases are common among immunodeficient patients. Gastroenterologists therefore must be able to diagnose and treat patients with primary immunodeficiency. Immune-related gastrointestinal diseases can be classified as those that develop primarily via autoimmunity, infection, an inflammatory response, or malignancy. Immunodeficient and immunocompetent patients with gastrointestinal diseases present with similar symptoms. However, intestinal biopsy specimens from immunodeficient patients often have distinct histologic features, and these patients often fail to respond to conventional therapies. Therefore, early recognition of symptoms and referral to an immunologist for a basic immune evaluation is required to select appropriate treatments. Therapies for primary immunodeficiency comprise immunoglobulin replacement, antibiotics, and, in severe cases, bone marrow transplantation. Treatment of immunodeficient patients with concomitant gastrointestinal disease can be challenging, and therapy with immunomodulators often is required for severe disease. This review aims to guide gastroenterologists in the diagnosis and treatment of patients with primary immunodeficiency.
Keywords: Immune System, Hypogammaglobulinemia, IBD, Inflammatory Intestinal Disease
Primary immunodeficiencies are a group of more than 150 disorders, often inherited, that are caused by intrinsic defects in the immune system. The immune defects can affect the humoral (B cell) immune system, such as in Bruton's agammaglobulinemia; the cellular (T cell) immune system, such as in DiGeorge syndrome; and both T- and B-cell immunity, such as in severe combined immunodeficiency (SCID)1,2 and in innate defects.
Gastrointestinal (GI) disorders present in 5% to 50% of patients with primary immunodeficiencies. This is in part because the gut is the largest lymphoid organ in the body, containing the majority of lymphocytes and producing large amounts of immunoglobulin (Ig). The mucosal immune system is uniquely regulated to manage its constant exposure to viruses, parasites, and bacterial antigens, all of which are in close proximity to a large reservoir of lymphocytes, macrophages, and dendritic cells. Its response is one of suppression or tolerance, unlike the systemic immune system. Dysfunction of the regulatory mechanisms maintaining this balance between active immunity and tolerance in the gut may lead to mucosal inflammation and damage and GI diseases. Therefore, it is not surprising that GI disorders are common manifestations, and often the initial presenting symptom, in patients with dysfunction in humoral immunity or cell-mediated immunity (Table 1).
Table 1.
Immunodeficiency and Associated GI Disorders
| Immunodeficiency | Molecular defect | Laboratory findings | GI manifestation | Other clinical findings |
|---|---|---|---|---|
| Selective IgA deficiency | Gene defect unknown; defective maturation of B cells into IgA-secreting plasma cells | Serum IgA absent or near absent, usually <10 mg/dL; normal IgG and IgM levels although IgG2 subclass deficiency may be present; impaired specific antibody response in some patients | Diarrhea, celiac sprue, NLH | Usually asymptomatic; some with recurrent bacterial infections, atopy, and autoimmunity |
| Agammaglobulinemia, X-linked or AR | X-linked (BTK), autosomal-recessive (μ, heavy chain, λ5, Igα, Igβ, BLNK) | Absent IgM, IgG, and IgA; B cells <1% of lymphocytes; absent specific antibody response | GI disorders rare, chronic diarrhea, malabsorption | Recurrent and severe bacterial infections, enteroviral infections, absent lymphoid tissue, autoimmunity |
| Hyper-IgM syndrome | Mutations in CD40L, CD40, AICDA, UNG | Low IgG and IgA; normal or increased IgM; normal or increased B-cell numbers; impaired specific antibody response; decreased T-cell responses in CD40L/CD40 deficiency | Diarrhea, progressive liver disease, sclerosing cholangitis | Recurrent bacterial infections, opportunistic infections, neutropenia, autoimmune disease |
| CVID | Mutations in ICOS, CD19, CD20, CD81, TNFRSF13B; TNFRSF13C; mostly unknown | Low IgG and IgA and/or IgM; absent specific antibody response; normal or decreased B-cell numbers; variably decreased T-cell responses | Diarrhea, NLH, flat villous lesions, IBD-like disease, pernicious anemia, hepatitis | Variable clinical phenotype: recurrent bacterial infections, autoimmune disease, lymphoproliferative ann/or granulomatous disease |
| SCID | Multiple defects: RAG1/2, JAK3, CD45, CD3 chain, ZAP70, Artemis, ligase 4, Cernunnos, IL-2RG, IL-7Rα, ADA → defects in T and B cells | Decreased serum immunoglobulins; marked diminished/absent T-cell, B-cell, and NK cell numbers depending on functional deficiency; diminished response to mitogens PHA, ConA, PWM | Chronic diarrhea, oral candidiasis, IBD | Failure to thrive, recurrent and severe bacterial, viral, and/or fungal infections early in life |
| CGD | Multiple defects: X-linked owing to defects in CYBB encoding the gp91 phox component of NADPH oxidase autosomal recessive owing to defects in NCF1, NCF2, or CYBA defects in components of NADPH oxidase | Defective oxidative burst in neutrophils by DHR or NBT equivalent | Granulomatous colitis, perianal fistulae, hepatic abscess, gastric outlet obstruction, small-bowel obstruction, granulomatous stomatitis, oral ulcers, esophageal dysmotility, hepatomegaly splenomegaly | Recurrent abscess, skin infections, recurrent bacterial and fungal infections (particularly S aureus and Aspergillus) |
| WAS | Mutations in WAS; cytoskeletal defect affecting hematopoietic stem cell derivatives | Immunoglobulins variable in concentration secondary to accelerated synthesis and catabolism (decreased IgM; normal or slightly low IgG; often increased IgA and IgE); antibody response to polysaccharides decreased; normal B-cell numbers; progressive decrease in T-cell numbers with abnormal lymphocyte responses to anti-CD3; platelet numbers are reduced and small in size | Colitis, bloody diarrhea, malabsorption | Thrombocytopenia, eczema, autoimmune disease, bacterial and viral infections, lymphoreticular malignancy |
| IPEX | Defects in F0XP3, encoding a T-cell transcription factor | Increased IgA and IgE; normal B-cell numbers; lack of CD4+CD25+F0XP3+ regulatory T cells; eosinophilia | Severe enteropathy with watery, often bloody, diarrhea associated with eosinophilic inflammation | Early onset diabetes, thyroiditis, hemolytic anemia, thrombocytopenia, eczema |
| X-linked anhidrotic ectodermal dysplasia with immunodeficiency | Mutations of NEMO (IKBKG), a modulator of NF-κB activation | Impaired response to polysaccharides in most cases; high serum levels of IgM, and low serum levels of IgG, IgA, or IgG2 in several cases | Failure to thrive, recurrent diarrhea, colitis | Severe, recurrent infections (mycobacteria and pyogenic bacteria) with an early onset; ectodermal dysplasia, hypohydrosis, widely spaced cone- or peg-shaped teeth, and hypotrichosis; some with osteopetrosis and lymphedema |
| DiGeorge syndrome (chromosome 22q11.2 deletion syndrome) | Contiguous gene defect in 90% affecting thymic development; evidence that point mutations in the TBX1 gene are involved | Immunoglobulins usually normal although occasionally IgE increased and IgA reduced; normal B-cell numbers; low to absent T-cell numbers in complete forms; varying degrees of T-cell function according to thymic deficiency | Mucocutaneous candidiasis | Conotruncal malformation; hypoparathryoidism; abnormal facies; absent thymus in complete forms |
| Hermansky–Pudlak syndrome, type 1 | Mutation in the HPS1 gene on chromosome 10q23 that forms part of BLOC-3 | Normal platelet count; prolonged bleeding time, with abnormal platelet function assays | Granulomatous colitis | Oculocutaneous albinism, abnormal platelet aggregation with bleeding diathesis, and pulmonary fibrosis |
AICDA, activation-induced cytidine deaminase; AR, autosomal recessive; BLNK, B-cell linker protein; BLOC-3, biogenesis of lysosome-related organelles complex-3; BTK, Bruton tyrosine kinase; CYBA, cytochrome b α subunit; CYBB, cytochrome b β subunit; DHR, dihydrorhodamine; ICOS, inducible costimulator; JAK3, Janus activating kinase 3; NADPH, nicotinamide adenine dinucleotide phosphate; NF-κB, nuclear factor-kB; PHA, phytohemagglutinin; PWM, pokeweed mitogen; RAG, recombinase activating gene; TBX1, T-box 1; TNFRSF, TNF-receptor superfamily; UNG, uracil DNA glycosylase.
GI manifestations can be broadly classified into 4 groups: infection, inflammation, malignancy, and autoimmune. The diversity of disorders involving the GI tract speaks to the differing forms of immune regulation along the length of the intestine, and the varying nature of the challenge (ie, food antigens in the small bowel and commensal flora in the colon). Many of these disorders mimic classic forms of disease (in the absence of immunodeficiency) such as celiac sprue, inflammatory bowel disease (IBD), and pernicious anemia but differ in pathogenesis and are often unresponsive to conventional therapies.
This review highlights the GI manifestations of the more common primary immunodeficiency disorders, focusing on the recognition of these diseases, appropriate diagnostic testing, and therapy.
Predominantly Antibody Deficiency
Selective Immunoglobulin A Deficiency
The most common primary immunodeficiency, estimated at 1 in 300 to 700 in Caucasians, is selective IgA deficiency (IgA < 7 mg/dL with normal or increased levels of other immunoglobulins).3,4 IgA deficiency is not well defined from a genetic aspect; the decreased IgA production may be the result of immune dysfunction in the regulation of terminal maturation of B cells into IgA-secreting plasma cells.5–7 The majority of patients are asymptomatic, although the absence of IgA has been associated with recurrent upper respiratory infections (frequently in those with concomitant IgG2 subclass deficiency), autoimmune disorders, and allergic diseases.4,8 T-cell immunity as well as natural killer activity appears to be normal in most patients. In addition, certain HLA haplotypes, including B8 and DR3, are associated with selective IgA deficiency.9–11
Although secretory IgA is the major antibody in the intestinal mucosa, the prevalence of GI disorders in patients with IgA deficiency is not as high as one would expect. It is thought that the transportation of IgM from the mucosa into the intestinal lumen by the polymeric immunoglobulin receptor can compensate for the lack of IgA in the intestine and mucosal surfaces, although several studies have challenged this belief.12–16 There is, however, a demonstrated link between IgA deficiency and giardiasis, celiac disease, nodular lymphoid hyperplasia (NLH), ulcerative colitis, Crohn's disease, pernicious anemia, and gastric and colonic adenocarcinoma17; these disorders, however, are associated much more commonly with common variable immunodeficiency (CVID).
GI infections related to Giardia lamblia have been reported with an increased frequency in primary immunodeficiency patients.18 Once ingested, G lamblia cysts release trophozoites, which colonize the small intestine and cause bloating, cramping, excessive flatus, and watery diarrhea. Steatorrhea and villus flattening can occur with chronic infection owing to effacement of the mucosa and the subsequent disruption of the absorption of lipids and carbohydrates. The degree of mucosal damage appears to be associated with the duration of the infection; some epithelial damage may be irreversible. Diagnosis is made by examining the stool for cysts or trophozoites of G lamblia, or by examination of duodenal aspirates, which can yield more determinate results. The parasitic load can be unremitting in IgA-deficient patients, despite treatment with metronidazole. Luminal IgA may be involved in clearing this parasite, as shown in studies on jejunal biopsy specimens from infected patients who stained positive for anti-human IgA. Presumably the lack of secretory IgA in these patients allows for attachment and proliferation of the organism on the intestinal epithelium; however, mouse models have suggested that clearance is T-cell mediated.19
The association between celiac disease and IgA deficiency frequently is reported as having a causal relationship, although the association of these 2 diseases may have a genetic basis, given shared HLA haplotypes. Genetic studies have shown that an important susceptibility locus between celiac disease and IgA deficiency is haplotype HLA-A1, Cw7, B8, DR3, and DQ2,20–23 and celiac disease is associated with HLA-DQ2 and DQ8.20,21,23 However, given the high incidence of both disorders, between 1 in 100 and 1 in 300 in the population, it is conceivable that the presence of one disease in the setting of the other is purely coincidental.
Secretory IgA can bind to wheat gluten and gliadin, and the absence of IgA may lead to abnormal processing of these antigens. Symptoms of celiac disease are similar in patients with or without IgA deficiency. The histopathology of celiac disease in IgA-deficient patients is indistinguishable from the pathology seen in patients with conventional celiac disease: increased numbers of intraepithelial lymphocytes, villous shortening or flattening, crypt hyperplasia, and infiltration of the lamina propria with lymphoid cells. A distinguishing feature is the absence of IgA-secreting plasma cells in intestinal biopsy specimens in IgA-deficient patients.24–26 Antigliadin IgA, antitissue transglutaminase IgA, and antiendomysial IgA antibodies cannot be used as screening tests for this population, which is why some celiac panels come with a measurement of serum IgA to rule out the possibility of IgA deficiency.26 Tissue transglutaminase IgG may be a better screening test.27–29 Celiac disease associated with IgA deficiency is responsive to gluten withdrawal, unlike the flat villous lesion seen in CVID.30–32 Failure to respond to a gluten-free diet should lead one to consider CVID.
NLH also is reported in IgA deficiency. These nodules often appear in multiples and are commonly 5 mm or greater in diameter. They are found in the lamina propria, superficial submucosa of the small intestine, or both, and occasionally can occur in the stomach, large intestine, or rectum. The lesions can be associated with mucosal flattening, causing malabsorption and even obstruction when large. Diagnosis is made by small-bowel enteroscopy or contrast barium studies.33 Immunohisto-chemical staining demonstrates that these nodules contain large amounts of IgM-bearing cells, possibly as compensation for the absent IgA.34 These nodules are very sensitive to oral steroids.35 When presented with IgA deficiency, NLH also has been associated with lymphomas (usually B-cell origin)36 and gastric carcinomas.37
Other GI manifestations reported in patients with IgA deficiency include pernicious anemia,38–40 Crohn's disease, and ulcerative colitis.41–44
Asymptomatic patients with IgA deficiency are generally not treated. Patients with GI manifestations should be treated in the same way as patients without this immunodeficiency. Current preparations of Ig do not contain sufficient amounts of IgA; therefore, patients with IgA deficiency should receive treatment for specific complications and should be monitored over time because some may progress to CVID. Because patients with IgA deficiency may have IgG or IgE antibodies to IgA (although rare), serious reactions may occur if these patients receive blood products. In cases in which blood transfusion is required, the patient should be screened for anti-IgA antibodies, and blood product should be prepared from an IgA-deficient individual or saline-washed.
X-Linked Agammaglobulinemia
X-linked agammaglobulinemia (XLA) results from a defect in Bruton's tyrosine kinase, an intracellular kinase, leading to the maturation arrest of pre-B cells and subsequent failure of the generation of mature B cells.45 The incidence is approximately 1 in 100,000 live births. The diagnosis of XLA typically is made in a male infant with recurrent sinopulmonary infections beginning at 4 months of age, when maternal antibodies are depleted. Laboratory findings include a profound reduction in all classes of immunoglobulin caused by the failure of B cells to differentiate into plasma cells, and humoral responses to specific antigens are markedly depressed or absent. Peripheral CD19+ B cells are usually less than 0.1%.46 T-cell numbers and function are normal.
In contrast to other antibody-deficiency syndromes, GI manifestations are less common in XLA, presumably because the T-cell dysfunction present in other immunodeficiency syndromes is what drives intestinal disease. Chronic diarrhea is the most common GI symptom reported in XLA patients.47–49 Infectious diarrhea, most commonly related to G lamblia, Salmonella, Campylobacter, and Cryptosporidium, likely is secondary to bacterial overgrowth and lack of antibody.49,50 GI infections are treated accordingly based on culture results but may require longer antibiotic courses. Enteroviral infections such as coxsackievirus and echovirus can occur in the gut and can lead to severe neurologic defects.51–53
Rare cases of gastric adenocarcinoma and colorectal cancer also have been described in XLA.54–56 Small-bowel strictures and transmural intestinal fissures resembling Crohn's disease have been seen on pathology.57 In contrast to Crohn's disease, no granulomas or plasma cells are identified when the strictures are resected in patients with XLA. There are no germinal centers in the gut-associated lymphoid tissues, and NLH does not develop.
XLA is treated with replacement IgG therapy, either intravenous or subcutaneously. Passive protection to many viruses and bacteria is obtained through immunoglobulin replacement therapy; however, treatment with antibiotics for documented or suspected infections is necessary because commercial preparations of IgG may not have adequate titers against uncommon organisms that patients with XLA may encounter.
X-Linked Hyper-IgM Syndrome
X-linked hyper-IgM syndrome results from a mutation in the gene that encodes the CD40 ligand.58 Because these patients lack CD40 ligand on T cells, there is no interaction with CD40 on B cells, which is required for immunoglobulin class switching of IgM to IgG or IgA. The X-linked form of hyper-IgM is the most common; however, autosomal-recessive forms also exist. Boys with hyper-IgM syndrome present early in life with recurrent bacterial or opportunistic infections. They are susceptible to a variety of intracellular pathogens such as mycobacterial species, fungi, and viruses. Pneumonia caused by Pneumocystis is a presenting feature of this syndrome in approximately 40% of cases.59 On laboratory evaluation, these patients have significantly low or absent levels of IgG and IgA, and normal or increased levels of IgM. Antibody (IgG) responses to vaccinations are poor or nonprotective. T-lymphocyte numbers are usually normal, and B-cell numbers are normal or slightly reduced.
Patients may present with oral ulcers, gingivitis, and rectal ulcers, which all may be secondary to neutropenia. Diarrhea occurs in about half of these patients and is secondary to Cryptosporidium parvum, G lamblia, Salmonella, or Entamoeba histolytica infection.59–61 In many cases the diarrhea is protracted or recurrent, causing failure to thrive and weight loss; Cryptosporidium is the most frequently isolated pathogen.62,63 Cholangiopathy with Cryptosporidium in the biliary tree is a common complication of both clinical and subclinical infection. It can result in disturbed liver function tests with increased γ-glutamyl transferase levels and can lead to the development of sclerosing cholangitis progressing to cirrhosis with a risk of cholangiocarcinoma.64–66 Hepatitis B, C, and cytomegalovirus infections also have been documented to possibly progress to hepatocellular carcinoma.66–68 NLH involving the GI tract also has been reported. Lymphoid hyperplasia may result in lymphadenopathy, hepatosplenomegaly, and tonsillar enlargement.
Treatment for hyper-IgM is with monthly replacement of Ig and antibiotics for specific infectious complications. Careful monitoring is especially essential in those with Cryptosporidium infection, given the complications described earlier, and prophylaxis against pneumocystis can be considered. To reduce the risk of Cryptosporidium infection, it is recommended that patients boil drinking water or filter it through a professionally fitted filter with less than a 1-μm pore size. The granulocyte colony-stimulating factor filgrastim may be used as a daily subcutaneous injection to treat neutropenia, although some patients may not respond. Hematopoietic cell transplantation alone or combined with liver transplantation also has been used to correct this disease.
Common Variable Immunodeficiency
CVID is the most common symptomatic primary immunodeficiency; its prevalence is estimated at 1 in 25,000 to 50,000.2,3 The pathogenesis of CVID has not been delineated clearly; however, mutations in several genes associated with B-cell development, including autosomal-recessive mutations in BAFF-R, CD20, CD19, CD81, CD21, and inducible costimulator, have been found in a small subset of patients.69–72 Affected patients typically present with recurrent bacterial infections of the upper and lower respiratory tracts, which may lead to bronchiectasis. In addition to chronic infections, CVID patients have a wide range of clinical manifestations, including autoimmune disease (mostly immune thrombocytopenic purpura and autoimmune hemolytic anemia), granulomatous/lymphoid infiltrative disease, and increased incidence of malignancy.73–76 The diagnosis is based on significantly reduced levels of IgG, IgA, and/or IgM, with poor or absent antibody production to protein and carbohydrate vaccines, such as tetanus or diphtheria toxoids; Haemophilus influenzae type b conjugate; measles, mumps, and rubella vaccines; and pneumococcal polysaccharide vaccines, with exclusion of other causes of hypogammaglobulinemia.77,78 Most patients are diagnosed with CVID between the ages of 20 and 40 years; however, the diagnosis commonly is delayed by 6 to 8 years, even after the onset of characteristic symptoms.77 Patients are treated with monthly infusions of IgG; this treatment alone may not be effective in treating other manifestations of this disorder.
Reviews from the 1970s to the present have noted a high incidence of GI manifestations in CVID, ranging from 20% to 60% of patients.47,57,59,79–84 Many of these GI manifestations mimic disease seen in the absence of immunodeficiency (eg, pernicious anemia, sprue-like lesions, and IBD), but the pathology is not comparable (there are no plasma cells) and they often do not respond to conventional therapies. The most frequently reported GI symptom is transient or persistent diarrhea.85,86 Similar to other antibody deficiency disorders, G lamblia is the most common organism found, although other pathogens such as C parvum, cytomegalovirus, Salmonella species, and Campylobacter jejuni have been reported.82,85 Small-bowel mucosal abnormalities in giardiasis include villous blunting, increased intraepithelial lymphocytes, and NLH. The trophozoite form can be identified on small-bowel biopsy. G lamblia is treated with metronidazole, which usually is effective but may require several courses in patients with CVID. Despite their exposure to the wide range of antibiotics used to treat systemic infections, these patients have no higher incidence rate of Clostridium difficile infections. This is likely owing to the high titers of anti–C difficile antibodies administered in monthly Ig treatments, which may leak into the gut.87,88
In the stomach, Helicobacter pylori infection has been associated with gastritis.89–91 Atrophic gastritis, which resembles autoimmune gastritis developing into pernicious anemia, may occur in the absence of demonstrable antiparietal cell antibodies in CVID patients. Treatment of pernicious anemia in CVID patients is similar to that of patients without CVID: monthly replacement of vitamin B12 and careful monitoring of the gastric mucosa for changes associated with malignancy. Gastric adenocarcinoma, in the setting of atrophic gastritis or de novo, has an increased frequency in CVID patients.
In the small bowel, sprue-like manifestations with villous atrophy have been reported in CVID patients and can be associated with severe malabsorption. Patients report symptoms of bloating, diarrhea, and weight loss. Small-bowel pathology is similar to what is seen in classic celiac sprue, with short villi, crypt hyperplasia, intraepithelial lymphocytosis, and, in some cases, an increase in apoptotic bodies in crypt epithelial cells (Figure 1).57,82,92–94 Unlike the diagnosis of classic sprue, the diagnosis of CVID should be suspected when the number of plasma cells are reduced or absent in the lamina propria, and the affected patients do not make antibodies to tissue transglutaminase, endomysium, or gliadin. Affected CVID patients typically do not respond to gluten withdrawal and do not express the genes associated with classic celiac disease, HLA-DQ2 and HLA-DQ8. Although the inflammatory process responds to steroid therapy, prolonged therapy with steroids is not advisable in CVID patients. Other immune modulators such as 6-mercaptopurine (6-MP) or azathioprine (AZA) can be used in addition to Ig replacement therapy. Careful use of budesonide may be required if unresponsive to 6-MP or AZA until the symptoms are controlled (eg, reversal of weight loss and dehydration). In severe cases of malabsorption, when significant loss of essential nutrients (eg, calcium, zinc, and vitamins A, E, and D) lead to bone loss and neurologic deficits, limited use of total parenteral nutrition may be required78,95; an indwelling catheter can serve as a source of infection and should be monitored.
Figure 1.
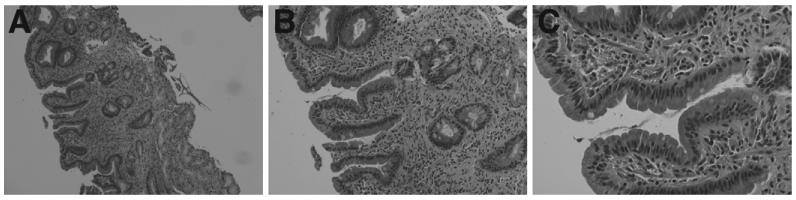
H&E-stained duodenum from a patient with CVID showing mild to moderate villous flattening, active and chronic inflammation, and intraepithelial lymphocytosis with decreased numbers of plasma cells.
Liver disease, including primary biliary cirrhosis and autoimmune hepatitis, which may lead to persistently increased liver enzyme levels, also occurs in CVID. The cause remains unknown, with liver biopsy specimens showing mild periportal changes or granulomas. In one cohort study, 43% of patients had abnormal liver function tests, predominantly increased alkaline phosphatase. Nodular regenerative hyperplasia leading to portal hypertension and cholestasis was found in 14 of 40 subjects in a cohort of subjects who had these abnormalities in liver function tests.76,83,96,97 Hepatitis C was an older complication of intravenous immunoglobulin infusions and is not seen in the new formulations.
GI malignancy is also more common in CVID patients, in comparison with patients with IgA deficiency or XLA.76,82,98–100 Those with a history of chronic atrophic gastritis with achlorhydria, intestinal metaplasia, and pernicious anemia have an increased risk of developing gastric adenocarcinoma. Benign lymphoproliferative disorders such as NLH also have been observed; the cause is still unknown, but it has been suggested that the lymphoid hyperplasia is a compensatory response for the antibody deficiency, although treatment with replacement Ig has not been shown to correct this. As mentioned earlier in the IgA deficiency section, NLH may be observed on endoscopy distributed diffusely throughout the stomach, ileum, and colon. Massive hyperplasia can result in intestinal obstruction and intussusception, as well as malabsorption mediated by the effacement of mucosal folds. It is controversial as to whether NLH can lead to the development of lymphoma.
An inflammatory bowel-like disease resembling Crohn's or ulcerative colitis also has been described in patients with CVID.47,57,80–82,101 Unlike XLA and IgA deficiency, IBD-like disease is reported more commonly in patients with CVID, suggesting that this tendency toward inflammation is driven by T cells or, more likely, T-cell dysregulation; the exact pathogenesis, however, is still unknown. Distinguishing inflammatory disease from infectious disease can be challenging because both can have symptoms and signs of diarrhea, weight loss, and malabsorption. In CVID patients with IBD-like disease, colonic biopsy specimens show an absence of plasma cells in the lamina propria, intraepithelial or subepithelial lymphocytosis (microscopic or lymphocytic colitis), prominent apoptosis, granulomas, and crypt distortion (Figure 2).
Figure 2.
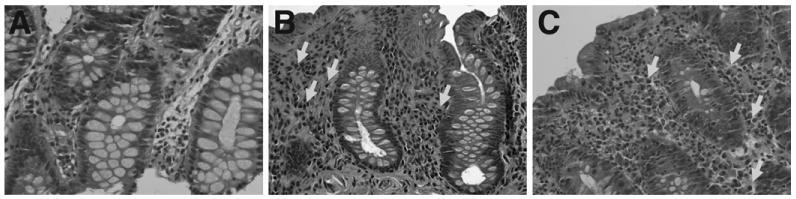
H&E (original magnification, ×40) from a patient with (A) CVID, (B) IBD, and (C) hypogammaglobulinemia. Patients with CVID lack plasma cells in the intestinal lamina propria in contrast to patients with classic IBD or hypogammaglobulinemia. Arrows represent plasma cells. Reprinted from Agarwal et al.80
Treatment of colitis for affected CVID patients is the same as for patients with classic IBD, including corticosteroids, 5-aminosalicyclic acid, 6-MP, and AZA. The later do not compromise immune function to a significant degree, and monthly Ig treatments also protect patients from infectious complications. Corticosteroids at any dose can lead to a significant risk of infections. Infliximab has been used with some benefit in severe enteropathy; however, patients with significant T-cell defects should be monitored for fungal infections.102,103
Combined T- and B-Cell Immunodeficiency
Severe Combined Immunodeficiency
SCID is a heterogeneous group of congenital disorders characterized by molecular defects in both T- and B-cell function and are classified as T-B+NK+, T-B+NK−, T-B−NK+, or T-B−NK−.3 SCID is estimated to be in 1 of 50,000 to 500,000 live births. Infants with SCID present in the first year of life with severe recurrent bacterial or viral infections, eczematoid rashes, failure to thrive, and overwhelming sepsis. The genetic mutations that result in the SCID phenotype are deficiencies in adenosine deaminase, purine nucleoside, ZAP70, JAK3, and defects in the interleukin (IL)-7 receptor104; the X-linked form, which is more common, results from a defect in the common gamma chain. The diagnosis of SCID is established when the absolute lymphocyte count is less than 2500 cells/mm3, T cells make up less than 20% of the total lymphocytes, and the response to mitogens is less than 10% of the control. Serum levels of immunoglobulins are usually very low, and specific antibody responses are impaired. Diagnosis of infants often is delayed by several months as a result of the protection of maternal antibodies. When there is a prior SCID-affected child with a known specific molecular defect, a prenatal diagnosis can be made through genetic testing. Early recognition and diagnosis is crucial to these patients because bone marrow transplantation is necessary for survival.105 A newborn screening program is already in place in several states.106,107 Patients are often kept in isolation while awaiting the diagnosis and before treatment. Replacement Ig therapy is given, as well as prophylaxis for Pneumocystis jiroveci pneumonia. Live vaccinations should not be given to the patient or the patient's caregivers because they can cause life-threatening infections. Blood products are irradiated before administration to prevent graft-versus-host disease (GVHD).
Because SCID phenotypes involve both T- and B-cell dysfunction, GI disorders often occur in SCID patients.108 Oral, esophageal, and perianal candidiasis is common and can affect oral intake. Affected children develop severe diarrhea and malabsorption early in life, and stool should be sent to a laboratory to check for viral and opportunistic infections, especially rotavirus. Chronic infection with rotavirus has been reported in patients who have received the live vaccination. Cytomegalovirus and adenovirus infections also have been identified in GI biopsy specimens. GI biopsy specimens show hypocellular lamina propria, without plasma cells or lymphocytes. Villous atrophy may occur in some infants owing to damage in the intestine after viral or bacterial infections.57 Patients with SCID who receive blood transfusions or allogeneic bone marrow transplantation are susceptible to GVHD and a GVHD-like process affecting the colon and small intestine.
Disorder of Phagocyte Function
Chronic Granulomatous Disease
Chronic granulomatous disease (CGD) is an immunodeficiency syndrome caused by an inability of the phagocytes to produce adequate reactive oxygen metabolites to kill ingested microorganisms, affecting 1 in 250,000 births in the United States. The most common form is X-linked (gp91phox); others are autosomal recessive.109 Genetically heterogeneous CGD is the result of a mutation in the components of nicotinamide adenine dinucleotide phosphate oxidase. Consequently, affected individuals suffer from recurrent infections caused by catalase-positive organisms (eg, Staphylococcus aureus, Burkholderia cepacia, Serratia marcescens, Aspergillus species, Chromobacterium violaceum, and Nocardia species) at epithelial surfaces (eg, skin, gut, lungs), as well as in organs with a large number of phagocytes such as the liver. The diagnosis is established by showing defective uptake of dihydrorhodamine by neutrophils using flow cytometry.
GI involvement occurs in approximately 50% of patients with CGD, ranging from noncaseating granulomatous colitis, protein-losing enteropathy, and inflammatory disease mimicking Crohn's.110–112 The rate of GI involvement has been reported to be higher in the X-linked gp91 phagocyte oxidase (phox) deficiency compared with the autosomal-recessive forms of the disease.113 Patients with CGD may present with granulomatous lesions either at diagnosis or later, and throughout the GI tract, from the oral cavity to the colon, causing dysphagia, dysmotility, or obstruction. This may resolve with a combination of corticosteroids and antibiotics but in many cases also requires surgery. Granulomas, giant cells, and macrophages laden with brown-yellow fine pigment are commonly present in gastric biopsy specimens. It is speculated that chronic antigenic stimulation from organisms not killed by phagocytes results in granuloma formation and bowel wall thickening.114
Gut inflammation is chronic and relapsing and also more common in the X-linked form. It has a phenotype that overlaps those of Crohn's disease and ulcerative colitis but without the extraintestinal manifestations often associated with IBD.115 Patients may present with perirectal abscesses, GI tract obstruction, and recurrent diarrhea with anal fissures. Endoscopy of the colon reveals features similar to idiopathic inflammatory bowel disease, such as colonic narrowing, cobblestone pattern, thickened bowel wall, fistulization, pancolitis, patchy friability, pseudopolyps, and hemorrhage.111,116 At the microscopic level are large granulomas (usually in the muscularis), submucosal edema, crypt abscesses, as well as lipid-laden histiocytes in the colonic mucosa and submucosa, suggestive of CGD colitis. Interestingly, in a study performed by our group, CGD patients with or without colitis had considerably higher levels of anti- Saccharomyces cerevisiae antibodies to IgA, anti-Saccharomyces cerevisiae antibodies to IgG, antibodies directed against the outer membrane porin protein C of Escherichia coli and Pseudomonas fluorescens (anti-I2), and anti-CBir1, but low to absent perinuclear antineutrophil cytoplasmic antibodies compared with IBD-predictive cut-off levels. Higher antibody levels were not associated with a history of colitis, suggesting that the increased levels of antimicrobial antibodies in CGD may reflect a specific defect in innate immunity in the face of chronic antigenic stimulation.115
CGD-associated colitis typically responds rapidly to treatment with glucocorticoids; oral prednisone usually is initiated after biopsy confirmation of the granulomata.112 Doses can be tapered gradually over a period of 3 to 6 months; however, relapses upon discontinuation are common. Other treatment options include interferon-γ, antitumor necrosis factor (anti-TNF), and cyclosporine. These options are based on established IBD therapies117; reports of infectious complications may preclude their use in CGD patients. We have reported a case of successful therapy with granulocyte-macrophage colony-stimulating factor.118
Hepatic abscess is also common in CGD and may be recurrent and prolonged.119–123S aureus and Pseudomonas aeruginosa are the most common organisms identified in patients with hepatic abscess—any patient presenting with these organisms should be evaluated for CGD. Patients may present with increased transaminase levels, fever, abdominal pain, fatigue, weight loss, and night sweats. Abscesses are treated with appropriate antimicrobial agents and surgical drainage.
Management of CGD includes antibiotic prophylaxis with trimethoprim-sulfamethoxazole and itraconazole to reduce the incidence of bacterial and fungal infections, although break-through infections do occur.124,125 Prophylactic administration of subcutaneous interferon-γ also has been used. Hematopoietic stem cell transplantation from an HLA-identical donor can be curative for CGD. Since the introduction of nonmyeloablative regimens, the risks surrounding transplant have decreased, allowing for transplantation in patients with ongoing infections. More transplants also are being performed using unrelated donors.126 Because it results from single-gene defects, CGD is well suited for gene therapy; however, this type of therapy currently remains under investigation.127,128
Disease of Immune Dysregulation
Immune Dysfunction, Polyendocrinopathy, Enteropathy, X-Linked
Mutations in forkhead box P3 (FOXP3) gene, a transcriptional regulator required for regulatory T cells and maintenance of peripheral tolerance, leads to immune dysfunction, polyendocrinopathy, enteropathy, X-linked (IPEX).129–132 IPEX typically presents during the first few months of life with diabetes mellitus, intractable diarrhea, failure to thrive, eczema, and hemolytic anemia.133 Other autoimmune manifestations, including thyroiditis, thrombocytopenia, and neutropenia, also have been reported. B-cell subsets and CD3+ T cells, including CD4+ and CD8+ subsets, are present in normal numbers and proliferate to mitogens and antigens, although affected patients typically have decreased numbers of regulatory T cells and are unable to suppress T-cell proliferation.134 Serum IgG, IgA, and IgM levels are usually normal but can be slightly reduced from enteric protein loss. IgE usually is increased and eosinophilia may be present as well. The diagnosis is made by showing decreased FOXP3 protein expression and a reduction in the number of regulatory T cells. The diagnosis also can be confirmed by mutation testing of the FOXP3 gene; identification of a FOXP3 mutation allows carrier detection and prenatal diagnosis in male fetuses of known carrier females.
The most consistent feature is severe enteropathy, including watery diarrhea (at times mucoid or bloody) that is intractable despite gut rest, usually occurring before 3 months of age. Clinical presentation can be similar to that of severe food allergies, although in this case the patient is unresponsive to an elemental formula. Failure to thrive usually follows as a result of enteropathy and malabsorption. Histopathology includes loss of normal small-bowel mucosa as a result of total or partial villous atrophy. Involvement of the large intestine is common with lymphocytic and plasma cell infiltration in the lamina propria; eosinophils also may be present. There is mucosal and submucosal destruction, although the muscular layer is preserved. Crohn's disease, ulcerative colitis, celiac disease, and GVHD are often on the differential diagnosis, based on histology.135
Patients are often severely ill by the time a diagnosis of IPEX is made; and, in the absence of aggressive therapy, children usually die before 2 years of age as a result of malnutrition, electrolyte imbalance, or infection.136,137 Symptomatic treatment includes bowel rest with total parenteral nutrition and, if necessary, insulin injections and red blood cell and platelet transfusions. Although immunosuppression can be used in the interim, hematopoietic stem cell transplant is curative.138,139 Immunosuppressive agents such as cyclosporine A, tacrolimus, sirolimus, and corticosteroids have been used with some success to improve diarrhea137,140–144; however, these agents do not maintain long-term remission. Chronic therapy may be toxic and may facilitate opportunistic infections.145,146
Interleukin-10–Receptor Deficiency
IL-10 is secreted by a wide variety of cells and has pleiotropic effects on many cell types, including T cells, B cells, and myeloid cells. IL-10 plays a critical role in maintaining the balance of the immune system by limiting the secretion of proinflammatory cytokines, such as TNF-α and IL-12. This mediation of the signaling that controls inflam mation initially was described in a murine model; mice deficient in either IL-10 or IL-10 –receptor 2 (IL-10R2) developed severe enterocolitis.147–149
Loss-of-function mutations in the IL-10Rα (IL-10RA) or IL-10Rβ (IL-10RB) encoding the IL-10R1 and IL-10R2 has been described in children with severe enterocolitis.150–152 These patients presented within the first year of life with enterocolitis and perianal disease, as well as the formation of multiple abscesses and enterocutaneous fistula, requiring several surgical interventions. Histopathology showed ulcerations of the intestinal mucosa with inflammatory infiltrates of the epithelium and the formation of abscesses extending to the muscularis propria. The patients also suffered from chronic folliculitis and recurrent respiratory infections. They were treated with various anti-inflammatory drugs, including steroids, methotrexate, thalidomide, and anti-TNFα monoclonal antibodies, without success of remission or long-term improvement. The mutations found in these patients abrogate IL-10–induced signaling, shown by deficient phosphorylation of signal transducer and activator of transcription-3 (STAT3) upon stimulation with IL-10. After a successful allogeneic stem cell transplantation, a cohort of 66 patients with early onset IBD was analyzed for mutations in the genes encoding IL-10, IL-10R1, and IL-10R2. Sixteen patients with IL-10 or IL-10R deficiency were identified: 3 patients had mutations in IL-10, 5 patients had mutations in IL-10R1, and 8 patients had mutations in IL-10R2.150 Refractory colitis was seen in all patients within the first 3 months of life and was associated with perianal disease and, in some patients, folliculitis and arthritis. Allogeneic stem cell transplantation was performed in 5 patients and was reported to induce sustained clinical remission. Patients had a median follow-up time of 2 years. More recently, 2 other patients with Crohn's-like symptoms presented with perianal and rectovaginal fistulae.151 Endoscopy and histopathology showed extensive ulceration of the ileum and focal active colitis with neutrophils infiltrating the surface epithelium. Both patients carried homozygous loss-of-function mutations in IL-10 itself, leading to an amino acid exchange at codon 113 (Gly113Arg), which likely interfered with the dimerization of IL-10.
These findings in patients with either IL-10 or IL-10R mutations highlight the importance of IL-10 in controlling the intestinal immune system and show that the loss of IL-10 signaling cannot be compensated by any other pathway. Although rare, infantile IBD patients with perianal disease should be screened for IL-10 and IL-10R deficiency. Defects in the IL-10R may be evaluated by functional assays.152 Peripheral blood mononuclear cells from healthy individuals show strong phosphorylation of STAT3 upon stimulation with IL-10, whereas peripheral blood mononuclear cells from IL-10R–deficient patients do not. Functional abnormalities can be confirmed by sequencing IL-10RA and IL-10RB and subsequent functional testing using STAT3 and/or TNF-α assays.
Well-Defined Syndrome With Gastrointestinal Immunodeficiency
Wiskott–Aldrich Syndrome
Wiskott–Aldrich syndrome (WAS) is an X-linked disorder caused by mutations in the WAS protein (WASp). This protein is involved in the regulation of actin polymerization in hematopoietic cells and has domains involved in signaling, cell locomotion, and immune synapse formation.153 The classic clinical manifestation of the complete absence of WASp is described as the triad of thrombocytopenia, eczema, and recurrent infections, although missense mutations, resulting in expression of defective WASp, lead to X-linked thrombocytopenia with minimal immunodeficiency. Laboratory evaluation reveals normal or slightly low IgG levels, high IgA and IgE levels, and low levels of IgM that are secondary to accelerated synthesis and catabolism.154–156 Antibody responses are variable and may be adequate to some antigens and abnormal to others, including polysaccharide antigens and bacteriophage ΦX174.153 Complete blood counts show reduced platelet numbers (20,000 – 50,000 per mm3) and small platelet size. Diminished lymphocyte proliferation in response to mitogens occurs in approximately 50% of patients. The number and phagocytic activity of neutrophils are normal, although chemotactic responses are defective.157 Screening for WASp mutations is performed by flow cytometry; however, this does not identify carriers of, or those patients with, X-linked thrombocytopenia. Sequence analysis of the WAS gene is essential to confirm the diagnosis.
Affected patients often present with bleeding diathesis manifested by purpura, hematemesis, melena, epistaxis, or hematuria, which can be life-threatening. Inflammatory bowel disease resembling ulcerative colitis has been described in up to 10% of patients158–160 and WAS should be considered in the differential diagnosis in early onset IBD.
Curative therapy for WAS is hematopoietic cell transplantation; however, gene therapy is under investigation.161–163 Conventional management includes antibiotic prophylaxis as well as platelet transfusions, to treat major bleeding episodes. Immunosuppressive treatments may be required for autoimmune manifestations. Splenectomy in patients with WAS increases the risk of septicemia and, if performed, requires lifelong antibiotic prophylaxis. To avoid splenectomy, autoimmune cytopenias can be treated with the monoclonal antibody targeting CD20 (rituximab). Patients with significant antibody deficiency may receive replacement IgG either intravenously or subcutaneously.
Early Evaluation of Primary Immunodeficiency
Because GI disease may be the first presentation of an underlying immunodeficiency, it is imperative to consider immunodeficiency in any patient with intractable diarrhea, malabsorption, and failure to thrive that is resistant to conventional treatments. Opportunistic or unusual infections in the setting of presumed IBD should prompt clinical suspicion and immune evaluation. A gastroenterologist can initiate an evaluation with laboratory tests, radiographic imaging, and intestinal biopsy specimens; however, a referral to an immunologist is essential. It is often helpful to ask the pathologist to review slides when a question of immunodeficiency exists, given some of the unique pathologic findings or lack thereof (eg, plasma cells). It is important to take notice of immunodeficiency secondary to defects in T and B cells because there appears to be an increased prevalence of GI disease when there are defects in both humoral and cellular immunity.
The evaluation of the immune system is best approached in stages, with the performance of the basic screening tests before continuing on to more advanced testing as necessary. The measurement of total serum protein, comprising albumin and globulin, may indicate a possible reduction of immunoglobulins. Normal globulin levels are generally more than 2 g/dL; if the globulin fraction is low (total protein minus albumin is <2 g/dL), quantitative levels of serum immunoglobulins should be obtained. Hypogammaglobulinemia can result from protein loss and is excluded by measuring serum albumin and urinary protein levels; enteral loss of protein can be excluded by measurement of stool α-1-antitrypsin.
Because many of the primary immune disorders involve defects in antibody production, evaluation of quantitative levels of immunoglobulins (IgG, IgA, IgM, and IgE) may be sufficient for diagnosis. This is especially important if treatment with immunoglobulin is being considered and must be performed before its administration. Quantification of IgG subclasses may be helpful in the assessment of an immunodeficiency, especially in IgA deficiency. Immunoglobulin values increase through adolescence, so comparison with age-matched controls is necessary for correct interpretation.
Further evaluation of a humoral defect includes the qualitative aspect of the antibody response, as measured by the ability to make antibodies to antigenic challenge in protective amounts. For example, if the IgG titer to measles, mumps, and rubella; tetanus; diphtheria; Haemophilus influenzae type b; pneumococcal; varicella; or meningococcal is low, vaccinations may be administered, followed by evaluation of postvaccination titers 4 to 6 weeks later. A patient who is receiving chronic treatment with corticosteroids and certain antiseizure medications also may have reduced serum immunoglobulin levels; however, the antibody response would be preserved in this case.
A complete blood count and lymphocyte panel to assess the number of lymphocytes and subpopulations (T cells, B cells, CD4+ T cells, and CD8+ T cells) should be obtained because lymphopenia may occur secondary to excessive loss of the cells into the lumen or through the trapping of cells in the inflamed bowel wall. Severe lymphopenia in an infant (<2000/mm3) is a critical finding that, if confirmed on a repeat test, should prompt an immediate immune evaluation for SCID. Thrombocytopenia and small platelet size suggest a diagnosis of WAS. Human immunodeficiency virus testing should be performed if there is clinical suspicion based on history or results of the lymphocyte panel.
Depending on the results of these initial tests, further evaluation or a more detailed study of cellular immune function can be performed. A lymphocyte proliferation in response to mitogens and antigens can provide more definitive insight into a patient's T-cell functional capability. The lymphocyte proliferation assay is an in vitro assay measuring responses to mitogens and antigens. Mitogens such as phytohemagglutinin, concanavalin A, and pokeweed mitogen are plant-derived lectins that bind to selected carbohydrates on lymphocyte surface glycoproteins. They activate intracellular signaling pathways, leading to increased metabolic activity and cell division, shown by incorporation of a radioactive marker. Mitogens activate cells without antigenic specificity; as such, more than 80% of T cells will respond to this form of activation. Failure of lymphocytes to respond to mitogens usually signals severely impaired T-cell function, as in the case of SCID. In contrast to mitogens, Candida antigens and recall antigens such as tetanus and diphtheria toxoids are entirely processed and presented by accessory cells; they stimulate only antigen-specific T cells. A stimulation index compares the results of the stimulated with unstimulated cells, using proliferation data from healthy controls as the normal reference range. These tests may not be possible in patients with significant T-cell lymphopenia. Corticosteroid therapy may lead to a reduced number of T cells, making interpretation of these tests difficult during therapy; however, response reappears rapidly with cessation of therapy.
Advanced testing can be performed to investigate specific disorders and is available mainly at academic laboratories investigating these disorders. For example, suspicion of CGD should lead to investigation of neutrophil function; this is accomplished with a dihydrorhodamine assay to determine a reduction or absence of phagocytic respiratory burst. Flow cytometric analysis of expression of cell surface and intracellular proteins can help in diagnosing X-linked hyper-IgM (through examination of CD40 ligand), Wiskott–Aldrich (through examination of WASp), and IPEX (through examination of FOXP3). Quantitative, real-time polymerase chain reaction for T-cell–receptor excision circles is used in neonatal screening assay for SCID. Genetic testing to identify carrier states or specific mutations can be performed for XLA, SCID, and DiGeorge.
Routine evaluation of the gut is beneficial for patients with immunodeficiency, given the high incidence of GI disease in these patients. Primary immune disorders are relatively common but likely underdiagnosed. In addition to the immunodeficiencies mentioned in this article, DiGeorge syndrome, chronic mucocutaneous candidiasis, and Hermansky–Pudlak syndrome also present with GI disease. Early evaluation and diagnosis can prevent potentially irreversible tissue damage. The type of immunodeficiency often is surmised by the nature of infections: bacterial infections indicate B-cell deficiencies, fungal or viral infections indicate T-cell deficiencies, and T- and B-cell deficiencies together suggest combined immunodeficiencies. Treatment with replacement Ig in most cases does not reverse or prevent the development of GI disease; additional therapies are necessary. A clinical history of recurrent infection, histologic features that do not fit the usual pattern of disease, and a poor response to conventional therapy should prompt the physician to pursue further immunologic evaluation.
Acknowledgments
Editorial support was provided by BSG Communications, through an educational grant from Baxter.
Abbreviations used in this paper
- 6-MP
mercaptopurine
- AZA
azathioprine
- CGD
chronic granulomatous disease
- CVID
common variable immunodeficiency
- FOXP3
forkhead box P3
- GI
gastrointestinal
- GVHD
graft-versus-host disease
- IBD
inflammatory bowel disese
- Ig
immunoglobulin
- IL
interleukin
- IPEX
immune dysfunction, polyendocrinopathy, enteropathy, X-linked
- NLH
nodular lymphoid hyperplasia
- SCID
severe combined immunodeficiency
- STAT
signal transducer and activator of transcription
- TNF
tumor necrosis factor
- WAS
Wiskott–Aldrich syndrome
- WASp
Wiskott-Aldrich syndrome protein
- XLA
X-linked agammaglobulinemia
Footnotes
Conflicts of interest The authors disclose no conflicts.
References
- 1.Chapel H. Classification of primary immunodeficiency diseases by the International Union of Immunological Societies (IUIS). Expert Committee on Primary Immunodeficiency, 2011. Clin Exp Immunol. 2012;168(58) doi: 10.1111/j.1365-2249.2012.04561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al-Herz W, Bousfiha A, Casanova JL, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2011;2:54. doi: 10.3389/fimmu.2011.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.International Union of Immunological Societies Expert Committee on Primary Immunodeficiencies. Notarangelo LD, Fischer A, et al. Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol. 2009;124(1161) doi: 10.1016/j.jaci.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yel L. Selective IgA deficiency. J Clin Immunol. 2010;30(10) doi: 10.1007/s10875-009-9357-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conley ME, Cooper MD. Immature IgA B cells in IgA-deficient patients. N Engl J Med. 1981;305(495) doi: 10.1056/NEJM198108273050905. [DOI] [PubMed] [Google Scholar]
- 6.Castigli E, Wilson SA, Garibyan L, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. 2005;37(829) doi: 10.1038/ng1601. [DOI] [PubMed] [Google Scholar]
- 7.Rachid R, Castigli E, Geha RS, et al. TACI mutation in common variable immunodeficiency and IgA deficiency. Curr Allergy Asthma Rep. 2006;6(357) doi: 10.1007/s11882-996-0004-9. [DOI] [PubMed] [Google Scholar]
- 8.Hammarström L, Vorechovsky I, Webster D. Selective IgA deficiency (SIgAD) and common variable immunodeficiency (CVID) Clin Exp Immunol. 2000;120(225) doi: 10.1046/j.1365-2249.2000.01131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cunningham-Rundles C. Physiology of IgA and IgA deficiency. J Clin Immunol. 2001;21(303) doi: 10.1023/a:1012241117984. [DOI] [PubMed] [Google Scholar]
- 10.De la Concha EG, Fernandez-Arquero M, Gual L, et al. MHC susceptibility genes to IgA deficiency are located in different regions on different HLA haplotypes. J Immunol. 2002;169:4637–4643. doi: 10.4049/jimmunol.169.8.4637. [DOI] [PubMed] [Google Scholar]
- 11.Mohammadi J, Ramanujam R, Jarefors S, et al. IgA deficiency and the MHC: assessment of relative risk and microheterogeneity within the HLA A1 B8, DR3 (8.1) haplotype. J Clin Immunol. 2010;30(138) doi: 10.1007/s10875-009-9336-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bienenstock J. The local immune response. Am J Vet Res. 1975;36(488) [PubMed] [Google Scholar]
- 13.Mellander L, Björkander J, Carlsson B, et al. Secretory antibodies in IgA-deficient and immunosuppressed individuals. J Clin Immunol. 1986;6(284) doi: 10.1007/BF00917328. [DOI] [PubMed] [Google Scholar]
- 14.Richman LK, Brown WR. Immunochemical characterization of IgM in human intestinal fluids. J Immunol. 1977;119(1515) [PubMed] [Google Scholar]
- 15.Brandtzaeg P, Karlsson G, Hansson G, et al. The clinical condition of IgA-deficient patients is related to the proportion of IgD-and IgM-producing cells in their nasal mucosa. Clin Exp Immunol. 1987;67(626) [PMC free article] [PubMed] [Google Scholar]
- 16.Brandtzaeg P, Bjerke K, Kett K, et al. Production and secretion of immunoglobulins in the gastrointestinal tract. Ann Allergy. 1987;59(21) [PubMed] [Google Scholar]
- 17.Brandtzaeg P. Update on mucosal immunoglobulin A in gastrointestinal disease. Curr Opin Gastroenterol. 2010;26(554) doi: 10.1097/MOG.0b013e32833dccf8. [DOI] [PubMed] [Google Scholar]
- 18.Eren M, Saltik-Temizel IN, Yüce A, et al. Duodenal appearance of giardiasis in a child with selective immunoglobulin A deficiency. Pediatr Int. 2007;49(409) doi: 10.1111/j.1442-200X.2007.02357.x. [DOI] [PubMed] [Google Scholar]
- 19.Heyworth MF, Carlson JR, Ermak TH. Clearance of Giardia muris infection requires helper/inducer T lymphocytes. J Exp Med. 1987;165(1743) doi: 10.1084/jem.165.6.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collin P, Mäki M, Keyriläinen O, et al. Selective IgA deficiency and coeliac disease. Scand J Gastroenterol. 1992;27(367) doi: 10.3109/00365529209000089. [DOI] [PubMed] [Google Scholar]
- 21.Heneghan MA, Stevens FM, Cryan EM, et al. Celiac sprue and immunodeficiency states: a 25-year review. J Clin Gastroenterol. 1997;25(421) doi: 10.1097/00004836-199709000-00004. [DOI] [PubMed] [Google Scholar]
- 22.Meini A, Pillan NM, Villanacci V, et al. Prevalence and diagnosis of celiac disease in IgA-deficient children. Ann Allergy Asthma Immunol. 1996;77(333) doi: 10.1016/S1081-1206(10)63329-7. [DOI] [PubMed] [Google Scholar]
- 23.Vorechovský I, Cullen M, Carrington M, et al. Fine mapping of IGAD1 in IgA deficiency and common variable immunodeficiency: identification and characterization of haplotypes shared by affected members of 101 multiple-case families. J Immunol. 2000;164(4408) doi: 10.4049/jimmunol.164.8.4408. [DOI] [PubMed] [Google Scholar]
- 24.Cataldo F, Marino V, Ventura A, et al. Prevalence and clinical features of selective immunoglobulin A deficiency in coeliac disease: an Italian multicentre study. Italian Society of Paediatric Gastroenterology and Hepatology (SIGEP) and “Club del Tenue” Working Groups on Coeliac Disease. Gut. 1998;42(362) doi: 10.1136/gut.42.3.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Savilahti E. IgA deficiency in children. Immunoglobulin-containing cells in the intestinal mucosa, immunoglobulins in secretions and serum IgA levels. Clin Exp Immunol. 1973;13(395) [PMC free article] [PubMed] [Google Scholar]
- 26.Villalta D, Alessio MG, Tampoia M, et al. Diagnostic accuracy of IgA anti-tissue transglutaminase antibody assays in celiac disease patients with selective IgA deficiency. Ann N Y Acad Sci. 2007;1109(212) doi: 10.1196/annals.1398.025. [DOI] [PubMed] [Google Scholar]
- 27.Korponay-Szabó IR, Dahlbom I, Laurila K, et al. Elevation of IgG antibodies against tissue transglutaminase as a diagnostic tool for coeliac disease in selective IgA deficiency. Gut. 2003;52(1567) doi: 10.1136/gut.52.11.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar V, Jarzabek-Chorzelska M, Sulej J, et al. Celiac disease and immunoglobulin A deficiency: how effective are the serological methods of diagnosis? Clin Diagn Lab Immunol. 2002;9(1295) doi: 10.1128/CDLI.9.6.1295-1300.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McGowan KE, Lyon ME, Butzner JD. Celiac disease and IgA deficiency: complications of serological testing approaches encountered in the clinic. Clin Chem. 2008;54(1203) doi: 10.1373/clinchem.2008.103606. [DOI] [PubMed] [Google Scholar]
- 30.Klemola T. Immunohistochemical findings in the intestine of IgA-deficient persons: number of intraepithelial T lymphocytes is increased. J Pediatr Gastroenterol Nutr. 1988;7(537) doi: 10.1097/00005176-198807000-00010. [DOI] [PubMed] [Google Scholar]
- 31.Mann JG, Brown WR, Kern F., Jr The subtle and variable clinical expressions of gluten-induced enteropathy (adult celiac disease, nontropical sprue). An analysis of twenty-one consecutive cases. Am J Med. 1970;48(357) doi: 10.1016/0002-9343(70)90066-5. [DOI] [PubMed] [Google Scholar]
- 32.Chow MA, Lebwohl B, Reilly NR, et al. Immunoglobulin A deficiency in celiac disease. J Clin Gastroenterol. 2012;46(850) doi: 10.1097/MCG.0b013e31824b2277. [DOI] [PubMed] [Google Scholar]
- 33.Postgate A, Despott E, Talbot I, et al. An unusual cause of diarrhea: diffuse intestinal nodular lymphoid hyperplasia in association with selective immunoglobulin A deficiency (with video) Gastrointest Endosc. 2009;70(168) doi: 10.1016/j.gie.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 34.Jacobson KW, deShazo RD. Selective immunoglobulin A deficiency associated with modular lymphoid hyperplasia. J Allergy Clin Immunol. 1979;64(516) doi: 10.1016/0091-6749(79)90061-7. [DOI] [PubMed] [Google Scholar]
- 35.Piaścik M, Rydzewska G, Pawlik M, et al. Diffuse nodular lymphoid hyperplasia of the gastrointestinal tract in patient with selective immunoglobulin A deficiency and sarcoid-like syndrome–case report. Adv Med Sci. 2007;52(296) [PubMed] [Google Scholar]
- 36.Magrath I, O'Conor GT, Ramot B, et al. Pathogenesis of leukemias and lymphomas: environmental influences. Raven; New York: 1984. Fogarty International Center for Advanced Study in the Health Sciences. National Cancer Institute (U.S.), Ortho Diagnostics Systems. [Google Scholar]
- 37.Mir-Madjlessi SH, Vafai M, Khademi J, et al. Coexisting primary malignant lymphoma and adenocarcinoma of the large intestine in an IgA-deficient boy. Dis Colon Rectum. 1984;27(822) doi: 10.1007/BF02553947. [DOI] [PubMed] [Google Scholar]
- 38.Quigley EM, Carmichael HA, Watkinson G. Adult celiac disease (celiac sprue), pernicious anemia and IgA deficiency. Case report and review of the relationships between vitamin B12 deficiency, small intestinal mucosal disease and immunoglobulin deficiency. J Clin Gastroenterol. 1986;8(277) doi: 10.1097/00004836-198606000-00016. [DOI] [PubMed] [Google Scholar]
- 39.Ginsberg A, Mullinax F. Pernicious anemia and monoclonal gammopathy in a patient with IgA deficiency. Am J Med. 1970;48(787) doi: 10.1016/s0002-9343(70)80015-8. [DOI] [PubMed] [Google Scholar]
- 40.Spector JI. Juvenile achlorhydric pernicious anemia with IgA deficiency. A family study. JAMA. 1974;228(334) [PubMed] [Google Scholar]
- 41.Falchuk KR, Falchuk ZM. Selective immunoglobulin A deficiency, ulcerative colitis, and gluten-sensitive enteropathy–a unique association. Gastroenterology. 1975;69(503) [PubMed] [Google Scholar]
- 42.Hodgson HJ, Jewell DP. Selective IgA deficiency and Crohn's disease: report of two cases. Gut. 1977;18(644) doi: 10.1136/gut.18.8.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iizuka M, Itou H, Sato M, et al. Crohn's disease associated with selective immunoglobulin A deficiency. J Gastroenterol Hepatol. 2001;16(951) [PubMed] [Google Scholar]
- 44.Asada Y, Isomoto H, Shikuwa S, et al. Development of ulcerative colitis during the course of rheumatoid arthritis: association with selective IgA deficiency. World J Gastroenterol. 2006;12(5240) doi: 10.3748/wjg.v12.i32.5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsukada S, Saffran DC, Rawlings DJ, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. 1993;72(279) doi: 10.1016/0092-8674(93)90667-f. [DOI] [PubMed] [Google Scholar]
- 46.Conley ME, Rohrer J, Minegishi Y. X-linked agammaglobulinemia. Clin Rev Allergy Immunol. 2000;19(183) doi: 10.1385/CRIAI:19:2:183. [DOI] [PubMed] [Google Scholar]
- 47.Hermaszewski RA, Webster AD. Primary hypogammaglobulinaemia: a survey of clinical manifestations and complications. QJM. 1993;86(31) [PubMed] [Google Scholar]
- 48.Lederman HM, Winkelstein JA. X-linked agammaglobulinemia: an analysis of 96 patients. Medicine (Baltimore) 1985;64(145) [PubMed] [Google Scholar]
- 49.Winkelstein JA, Marino MC, Lederman HM, et al. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine (Baltimore) 2006;85(193) doi: 10.1097/01.md.0000229482.27398.ad. [DOI] [PubMed] [Google Scholar]
- 50.Atarod L, Raissi A, Aghamohammadi A, et al. A review of gastrointestinal disorders in patients with primary antibody immunodeficiencies during a 10-year period (1990–2000), in children hospital medical center. Iran J Allergy Asthma Immunol. 2003;2(75) [PubMed] [Google Scholar]
- 51.Cellier C, Foray S, Hermine O. Regional enteritis associated with enterovirus in a patient with X-linked agammaglobulinemia. N Engl J Med. 2000;342(1611) doi: 10.1056/NEJM200005253422113. [DOI] [PubMed] [Google Scholar]
- 52.Misbah SA, Spickett GP, Ryba PC, et al. Chronic enteroviral meningoencephalitis in agammaglobulinemia: case report and literature review. J Clin Immunol. 1992;12(266) doi: 10.1007/BF00918150. [DOI] [PubMed] [Google Scholar]
- 53.Quartier P, Foray S, Casanova JL, et al. Enteroviral meningoencephalitis in X-linked agammaglobulinemia: intensive immunoglobulin therapy and sequential viral detection in cerebrospinal fluid by polymerase chain reaction. Pediatr Infect Dis J. 2000;19(1106) doi: 10.1097/00006454-200011000-00020. [DOI] [PubMed] [Google Scholar]
- 54.Bachmeyer C, Monge M, Cazier A, et al. Gastric adenocarcinoma in a patient with X-linked agammaglobulinaemia. Eur J Gastroenterol Hepatol. 2000;12(1033) doi: 10.1097/00042737-200012090-00013. [DOI] [PubMed] [Google Scholar]
- 55.Lavilla P, Gil A, Rodríguez MC, et al. X-linked agammaglobulinemia and gastric adenocarcinoma. Cancer. 1993;72(1528) doi: 10.1002/1097-0142(19930901)72:5<1528::aid-cncr2820720506>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 56.van der Meer JW, Weening RS, Schellekens PT, et al. Colorectal cancer in patients with X-linked agammaglobulinaemia. Lancet. 1993;341(1439) doi: 10.1016/0140-6736(93)90883-i. [DOI] [PubMed] [Google Scholar]
- 57.Washington K, Stenzel TT, Buckley RH, et al. Gastrointestinal pathology in patients with common variable immunodeficiency and X-linked agammaglobulinemia. Am J Surg Pathol. 1996;20(1240) doi: 10.1097/00000478-199610000-00010. [DOI] [PubMed] [Google Scholar]
- 58.Davies EG, Thrasher AJ. Update on the hyper immunoglobulin M syndromes. Br J Haematol. 2010;149(167) doi: 10.1111/j.1365-2141.2010.08077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levy J, Espanol-Boren T, Thomas C, et al. Clinical spectrum of X-linked hyper-IgM syndrome. J Pediatr. 1997;131(47) doi: 10.1016/s0022-3476(97)70123-9. [DOI] [PubMed] [Google Scholar]
- 60.Quartier P, Bustamante J, Sanal O, et al. Clinical, immunologic and genetic analysis of 29 patients with autosomal recessive hyper-IgM syndrome due to activation-induced cytidine deaminase deficiency. Clin Immunol. 2004;110(22) doi: 10.1016/j.clim.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 61.Bhatia V, Garg PK, Agarwal V, et al. Inflammatory papillary stenosis due to Giardia lamblia in a patient with hyper-immunoglobulin M immunodeficiency syndrome. Gastrointest Endosc. 2007;66(181) doi: 10.1016/j.gie.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 62.Winkelstein JA, Marino MC, Ochs H, et al. The X-linked hyper-IgM syndrome: clinical and immunologic features of 79 patients. Medicine (Baltimore) 2003;82(373) doi: 10.1097/01.md.0000100046.06009.b0. [DOI] [PubMed] [Google Scholar]
- 63.Kutukculer N, Moratto D, Aydinok Y, et al. Disseminated cryptosporidium infection in an infant with hyper-IgM syndrome caused by CD40 deficiency. J Pediatr. 2003;142(194) doi: 10.1067/mpd.2003.41. [DOI] [PubMed] [Google Scholar]
- 64.Dimicoli S, Bensoussan D, Latger-Cannard V, et al. Complete recovery from Cryptosporidium parvum infection with gastroenteritis and sclerosing cholangitis after successful bone marrow transplantation in two brothers with X-linked hyper-IgM syndrome. Bone Marrow Transplant. 2003;32:733–737. doi: 10.1038/sj.bmt.1704211. [DOI] [PubMed] [Google Scholar]
- 65.Kerkar N, Miloh T. Sclerosing cholangitis: pediatric perspective. Curr Gastroenterol Rep. 2010;12:195–202. doi: 10.1007/s11894-010-0104-5. [DOI] [PubMed] [Google Scholar]
- 66.Hayward AR, Levy J, Facchetti F, et al. Cholangiopathy and tumors of the pancreas, liver, and biliary tree in boys with X-linked immunodeficiency with hyper-IgM. J Immunol. 1997;158:977–983. [PubMed] [Google Scholar]
- 67.Quinti I, Giovannetti A, Paganelli R, et al. HCV infection in a patient with hyper IgM syndrome. J Clin Immunol. 1996;16:321–325. doi: 10.1007/BF01541667. [DOI] [PubMed] [Google Scholar]
- 68.Zirkin HJ, Levy J, Katchko L. Small cell undifferentiated carcinoma of the colon associated with hepatocellular carcinoma in an immunodeficient patient. Hum Pathol. 1996;27:992–996. doi: 10.1016/s0046-8177(96)90232-4. [DOI] [PubMed] [Google Scholar]
- 69.Grimbacher B, Hutloff A, Schlesier M, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. 2003;4:261–268. doi: 10.1038/ni902. [DOI] [PubMed] [Google Scholar]
- 70.van Zelm MC, Reisli I, van der Burg M, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006;354:1901–1912. doi: 10.1056/NEJMoa051568. [DOI] [PubMed] [Google Scholar]
- 71.van Zelm MC, Smet J, Adams B, et al. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest. 2010;120:1265–1274. doi: 10.1172/JCI39748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Warnatz K, Salzer U, Rizzi M, et al. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci U S A. 2009;106:13945–13950. doi: 10.1073/pnas.0903543106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cunningham-Rundles C. Autoimmunity in primary immune deficiency: taking lessons from our patients. Clin Exp Immunol. 2011;164(Suppl 2):6–11. doi: 10.1111/j.1365-2249.2011.04388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cunningham-Rundles C. Lung disease, antibodies and other unresolved issues in immune globulin therapy for antibody deficiency. Clin Exp Immunol. 2009;157(Suppl 1):12–16. doi: 10.1111/j.1365-2249.2009.03952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.da Silva SP, Resnick E, Lucas M, et al. Lymphoid proliferations of indeterminate malignant potential arising in adults with common variable immunodeficiency disorders: unusual case studies and immunohistological review in the light of possible causative events. J Clin Immunol. 2011;31:784–791. doi: 10.1007/s10875-011-9565-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Resnick ES, Moshier EL, Godbold JH, et al. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119:1650–1657. doi: 10.1182/blood-2011-09-377945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chapel H, Lucas M, Lee M, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112:277–286. doi: 10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 78.Cunningham-Rundles C. How I treat common variable immune deficiency. Blood. 2010;116:7–15. doi: 10.1182/blood-2010-01-254417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hermans PE, Diaz-Buxo JA, Stobo JD. Idiopathic late-onset immunoglobulin deficiency. Clinical observations in 50 patients. Am J Med. 1976;61:221–237. doi: 10.1016/0002-9343(76)90173-x. [DOI] [PubMed] [Google Scholar]
- 80.Agarwal S, Smereka P, Harpaz N, et al. Characterization of immunologic defects in patients with common variable immunodeficiency (CVID) with intestinal disease. Inflamm Bowel Dis. 2011;17:251–259. doi: 10.1002/ibd.21376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mannon PJ, Fuss IJ, Dill S, et al. Excess IL-12 but not IL-23 accompanies the inflammatory bowel disease associated with common variable immunodeficiency. Gastroenterology. 2006;131:748–756. doi: 10.1053/j.gastro.2006.06.022. [DOI] [PubMed] [Google Scholar]
- 82.Daniels JA, Lederman HM, Maitra A, et al. Gastrointestinal tract pathology in patients with common variable immunodeficiency (CVID): a clinicopathologic study and review. Am J Surg Pathol. 2007;31:1800–1812. doi: 10.1097/PAS.0b013e3180cab60c. [DOI] [PubMed] [Google Scholar]
- 83.Daniels JA, Torbenson M, Vivekanandan P, et al. Hepatitis in common variable immunodeficiency. Hum Pathol. 2009;40:484–488. doi: 10.1016/j.humpath.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 84.Kalha I, Sellin JH. Common variable immunodeficiency and the gastrointestinal tract. Curr Gastroenterol Rep. 2004;6:377–383. doi: 10.1007/s11894-004-0053-y. [DOI] [PubMed] [Google Scholar]
- 85.Oksenhendler E, Gérard L, Fieschi C, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis. 2008;46:1547–1554. doi: 10.1086/587669. [DOI] [PubMed] [Google Scholar]
- 86.Quinti I, Soresina A, Spadaro G, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. 2007;27:308–316. doi: 10.1007/s10875-007-9075-1. [DOI] [PubMed] [Google Scholar]
- 87.Leung DY, Kelly CP, Boguniewicz M, et al. Treatment with intravenously administered gamma globulin of chronic relapsing colitis induced by Clostridium difficile toxin. J Pediatr. 1991;118:633–637. doi: 10.1016/s0022-3476(05)83393-1. [DOI] [PubMed] [Google Scholar]
- 88.Salcedo J, Keates S, Pothoulakis C, et al. Intravenous immunoglobulin therapy for severe Clostridium difficile colitis. Gut. 1997;41:366–370. doi: 10.1136/gut.41.3.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dhalla F, da Silva SP, Lucas M, et al. Review of gastric cancer risk factors in patients with common variable immunodeficiency disorders, resulting in a proposal for a surveillance programme. Clin Exp Immunol. 2011;165:1–7. doi: 10.1111/j.1365-2249.2011.04384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zullo A, Romiti A, Rinaldi V, et al. Gastric pathology in patients with common variable immunodeficiency. Gut. 1999;45:77–81. doi: 10.1136/gut.45.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Twomey JJ, Jordan PH, Jr, Laughter AH, et al. The gastric disorder in immunoglobulin-deficient patients. Ann Intern Med. 1970;72:499–504. doi: 10.7326/0003-4819-72-4-499. [DOI] [PubMed] [Google Scholar]
- 92.Luzi G, Zullo A, Iebba F, et al. Duodenal pathology and clinical-immunological implications in common variable immunodeficiency patients. Am J Gastroenterol. 2003;98:118–121. doi: 10.1111/j.1572-0241.2003.07159.x. [DOI] [PubMed] [Google Scholar]
- 93.Teahon K, Webster AD, Price AB, et al. Studies on the enteropathy associated with primary hypogammaglobulinaemia. Gut. 1994;35:1244–1249. doi: 10.1136/gut.35.9.1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Malamut G, Verkarre V, Suarez F, et al. The enteropathy associated with common variable immunodeficiency: the delineated frontiers with celiac disease. Am J Gastroenterol. 2010;105:2262–2275. doi: 10.1038/ajg.2010.214. [DOI] [PubMed] [Google Scholar]
- 95.Aslam A, Misbah SA, Talbot K, et al. Vitamin E deficiency induced neurological disease in common variable immunodeficiency: two cases and a review of the literature of vitamin E deficiency. Clin Immunol. 2004;112:24–29. doi: 10.1016/j.clim.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 96.Ward C, Lucas M, Piris J, et al. Abnormal liver function in common variable immunodeficiency disorders due to nodular regenerative hyperplasia. Clin Exp Immunol. 2008;153:331–337. doi: 10.1111/j.1365-2249.2008.03711.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Malamut G, Ziol M, Suarez F, et al. Nodular regenerative hyperplasia: the main liver disease in patients with primary hypogammaglobulinemia and hepatic abnormalities. J Hepatol. 2008;48:74–82. doi: 10.1016/j.jhep.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 98.Gandhi K, Parikh P, Aronow WS, et al. A case of explosive progression of hepatocellular carcinoma in a patient with common variable immunodeficiency (CVID) J Gastrointest Cancer. 2010;41:281–284. doi: 10.1007/s12029-010-9158-8. [DOI] [PubMed] [Google Scholar]
- 99.Quiding-Järbrink M, Sundström P, Lundgren A, et al. Decreased IgA antibody production in the stomach of gastric adenocarcinoma patients. Clin Immunol. 2009;131:463–471. doi: 10.1016/j.clim.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 100.Cunningham-Rundles C, Siegal FP, Cunningham-Rundles S, et al. Incidence of cancer in 98 patients with common varied immunodeficiency. J Clin Immunol. 1987;7:294–299. doi: 10.1007/BF00915550. [DOI] [PubMed] [Google Scholar]
- 101.Borba de Arruda SM, do Rego Silva AM, Cezário de Barros KS, et al. Ulcerative colitis and common variable immunodeficiency: case report. Inflamm Bowel Dis. 2009;15:478–481. doi: 10.1002/ibd.20607. [DOI] [PubMed] [Google Scholar]
- 102.Chua I, Standish R, Lear S, et al. Anti-tumour necrosis factor-alpha therapy for severe enteropathy in patients with common variable immunodeficiency (CVID) Clin Exp Immunol. 2007;150:306–311. doi: 10.1111/j.1365-2249.2007.03481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nos P, Bastida G, Beltran B, et al. Crohn's disease in common variable immunodeficiency: treatment with antitumor necrosis factor alpha. Am J Gastroenterol. 2006;101:2165–2166. doi: 10.1111/j.1572-0241.2006.00763_5.x. [DOI] [PubMed] [Google Scholar]
- 104.Liston A, Enders A, Siggs OM. Unravelling the association of partial T-cell immunodeficiency and immune dysregulation. Nat Rev Immunol. 2008;8:545–558. doi: 10.1038/nri2336. [DOI] [PubMed] [Google Scholar]
- 105.Griffith LM, Cowan MJ, Notarangelo LD, et al. Improving cellular therapy for primary immune deficiency diseases: recognition, diagnosis, and management. J Allergy Clin Immunol. 2009;124:1152–1160e12. doi: 10.1016/j.jaci.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Verbsky J, Thakar M, Routes J. The Wisconsin approach to newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol. 2012;129:622–627. doi: 10.1016/j.jaci.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 107.Baker MW, Grossman WJ, Laessig RH, et al. Development of a routine newborn screening protocol for severe combined immunodeficiency. J Allergy Clin Immunol. 2009;124:522–527. doi: 10.1016/j.jaci.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 108.Antachopoulos C, Walsh TJ, Roilides E. Fungal infections in primary immunodeficiencies. Eur J Pediatr. 2007;166:1099–1117. doi: 10.1007/s00431-007-0527-7. [DOI] [PubMed] [Google Scholar]
- 109.Matute JD, Arias AA, Wright NA, et al. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40 phox and selective defects in neutrophil NADPH oxidase activity. Blood. 2009;114:3309–3315. doi: 10.1182/blood-2009-07-231498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Winkelstein JA, Marino MC, Johnston RB, Jr, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Med Baltimore. 2000;79:155–169. doi: 10.1097/00005792-200005000-00003. [DOI] [PubMed] [Google Scholar]
- 111.Huang A, Abbasakoor F, Vaizey CJ. Gastrointestinal manifestations of chronic granulomatous disease. Colorectal Dis. 2006;8:637–644. doi: 10.1111/j.1463-1318.2006.01030.x. [DOI] [PubMed] [Google Scholar]
- 112.Marciano BE, Rosenzweig SD, Kleiner DE, et al. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics. 2004;114:462–468. doi: 10.1542/peds.114.2.462. [DOI] [PubMed] [Google Scholar]
- 113.Foster CB, Lehrnbecher T, Mol F, et al. Host defense molecule polymorphisms influence the risk for immune-mediated complications in chronic granulomatous disease. J Clin Invest. 1998;102:2146–2155. doi: 10.1172/JCI5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rosh JR, Tang HB, Mayer L, et al. Treatment of intractable gastrointestinal manifestations of chronic granulomatous disease with cyclosporine. J Pediatr. 1995;126:143–145. doi: 10.1016/s0022-3476(95)70519-8. [DOI] [PubMed] [Google Scholar]
- 115.Yu JE, De Ravin SS, Uzel G, et al. High levels of Crohn's disease-associated anti-microbial antibodies are present and independent of colitis in chronic granulomatous disease. Clin Immunol. 2011;138:14–22. doi: 10.1016/j.clim.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Barton LL, Moussa SL, Villar RG, et al. Gastrointestinal complications of chronic granulomatous disease: case report and literature review. Clin Pediatr (Phila) 1998;37:231–236. doi: 10.1177/000992289803700403. [DOI] [PubMed] [Google Scholar]
- 117.Uzel G, Orange JS, Poliak N, et al. Complications of tumor necrosis factor-α blockade in chronic granulomatous disease-related colitis. Clin Infect Dis. 2010;51:1429–1434. doi: 10.1086/657308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang J, Mayer L, Cunningham-Rundles C. Use of GM-CSF in the treatment of colitis associated with chronic granulomatous disease. J Allergy Clin Immunol. 2005;115:1092–1094. doi: 10.1016/j.jaci.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 119.Leiding JW, Freeman AF, Marciano BE, et al. Corticosteroid therapy for liver abscess in chronic granulomatous disease. Clin Infect Dis. 2012;54:694–700. doi: 10.1093/cid/cir896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lublin M, Bartlett DL, Danforth DN, et al. Hepatic abscess in patients with chronic granulomatous disease. Ann Surg. 2002;235:383–391. doi: 10.1097/00000658-200203000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hussain N, Feld JJ, Kleiner DE, et al. Hepatic abnormalities in patients with chronic granulomatous disease. Hepatology. 2007;45:675–683. doi: 10.1002/hep.21524. [DOI] [PubMed] [Google Scholar]
- 122.Székely A, Péter M, Jr, Erdos M, et al. Hepatic abscess as the single manifestation of X-linked chronic granulomatous disease. Pediatr Blood Cancer. 2012;58:828–829. doi: 10.1002/pbc.23362. [DOI] [PubMed] [Google Scholar]
- 123.Feld JJ, Hussain N, Wright EC, et al. Hepatic involvement and portal hypertension predict mortality in chronic granulomatous disease. Gastroenterology. 2008;134:1917–1926. doi: 10.1053/j.gastro.2008.02.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Margolis DM, Melnick DA, Alling DW, et al. Trimethoprim-sulfamethoxazole prophylaxis in the management of chronic granulomatous disease. J Infect Dis. 1990;162:723–726. doi: 10.1093/infdis/162.3.723. [DOI] [PubMed] [Google Scholar]
- 125.Gallin JI, Alling DW, Malech HL, et al. Itraconazole to prevent fungal infections in chronic granulomatous disease. N Engl J Med. 2003;348:2416–2422. doi: 10.1056/NEJMoa021931. [DOI] [PubMed] [Google Scholar]
- 126.Kang EM, Marciano BE, DeRavin S, et al. Chronic granulomatous disease: overview and hematopoietic stem cell transplantation. J Allergy Clin Immunol. 2011;127:1319–1326. doi: 10.1016/j.jaci.2011.03.028. quiz 1327–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Segal BH, Veys P, Malech H, et al. Chronic granulomatous disease: lessons from a rare disorder. Biol Blood Marrow Transplant. 2011;17:S123–S131. doi: 10.1016/j.bbmt.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dinauer MC, Li LL, Björgvinsdóttir H, et al. Long-term correction of phagocyte NADPH oxidase activity by retroviral-mediated gene transfer in murine X-linked chronic granulomatous disease. Blood. 1999;94:914–922. [PubMed] [Google Scholar]
- 129.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 130.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. [PubMed] [Google Scholar]
- 131.Khattri R, Cox T, Yasayko SA, et al. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- 132.Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 133.Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked: forkhead box protein 3 mutations and lack of regulatory T cells. J Allergy Clin Immunol. 2007;120:744–750. doi: 10.1016/j.jaci.2007.08.044. quiz 751–752. [DOI] [PubMed] [Google Scholar]
- 134.d'Hennezel E, Ben-Shoshan M, Ochs HD, et al. FOXP3 forkhead domain mutation and regulatory T cells in the IPEX syndrome. N Engl J Med. 2009;361:1710–1713. doi: 10.1056/NEJMc0907093. [DOI] [PubMed] [Google Scholar]
- 135.Patey-Mariaud de Serre N, Canioni D, Ganousse S, et al. Digestive histopathological presentation of IPEX syndrome. Mod Pathol. 2009;22:95–102. doi: 10.1038/modpathol.2008.161. [DOI] [PubMed] [Google Scholar]
- 136.Powell BR, Buist NR, Stenzel P. An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J Pediatr. 1982;100:731–737. doi: 10.1016/s0022-3476(82)80573-8. [DOI] [PubMed] [Google Scholar]
- 137.Wildin RS, Smyk-Pearson S, Filipovich AH. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J Med Genet. 2002;39:537–545. doi: 10.1136/jmg.39.8.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Baud O, Goulet O, Canioni D, et al. Treatment of the immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) by allogeneic bone marrow transplantation. N Engl J Med. 2001;344:1758–1762. doi: 10.1056/NEJM200106073442304. [DOI] [PubMed] [Google Scholar]
- 139.Rao A, Kamani N, Filipovich A, et al. Successful bone marrow transplantation for IPEX syndrome after reduced-intensity conditioning. Blood. 2007;109:383–385. doi: 10.1182/blood-2006-05-025072. [DOI] [PubMed] [Google Scholar]
- 140.Levy-Lahad E, Wildin RS. Neonatal diabetes mellitus, enteropathy, thrombocytopenia, and endocrinopathy: further evidence for an X-linked lethal syndrome. J Pediatr. 2001;138:577–580. doi: 10.1067/mpd.2001.111502. [DOI] [PubMed] [Google Scholar]
- 141.Kobayashi I, Kawamura N, Okano M. A long-term survivor with the immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. N Engl J Med. 2001;345:999–1000. doi: 10.1056/NEJM200109273451314. [DOI] [PubMed] [Google Scholar]
- 142.Battaglia M, Stabilini A, Migliavacca B, et al. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol. 2006;177:8338–8347. doi: 10.4049/jimmunol.177.12.8338. [DOI] [PubMed] [Google Scholar]
- 143.Yong PL, Russo P, Sullivan KE. Use of sirolimus in IPEX and IPEX-like children. J Clin Immunol. 2008;28:581–587. doi: 10.1007/s10875-008-9196-1. [DOI] [PubMed] [Google Scholar]
- 144.Bindl L, Torgerson T, Perroni L, et al. Successful use of the new immune-suppressor sirolimus in IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome) J Pediatr. 2005;147:256–259. doi: 10.1016/j.jpeds.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 145.Lucas KG, Ungar D, Comito M, et al. Epstein Barr virus induced lymphoma in a child with IPEX syndrome. Pediatr Blood Cancer. 2008;50:1056–1057. doi: 10.1002/pbc.21341. [DOI] [PubMed] [Google Scholar]
- 146.Taddio A, Faleschini E, Valencic E, et al. Medium-term survival without haematopoietic stem cell transplantation in a case of IPEX: insights into nutritional and immunosuppressive therapy. Eur J Pediatr. 2007;166:1195–1197. doi: 10.1007/s00431-006-0395-6. [DOI] [PubMed] [Google Scholar]
- 147.Berg DJ, Kühn R, Rajewsky K, et al. Interleukin-10 is a central regulator of the response to LPS in murine models of endotoxic shock and the Shwartzman reaction but not endotoxin tolerance. J Clin Invest. 1995;96:2339–2347. doi: 10.1172/JCI118290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Spencer SD, Di Marco F, Hooley J, et al. The orphan receptor CRF2-4 is an essential subunit of the interleukin 10 receptor. J Exp Med. 1998;187:571–578. doi: 10.1084/jem.187.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kühn R, Löhler J, Rennick D, et al. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 150.Kotlarz D, Beier R, Murugan D, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology. 2012;143:347–355. doi: 10.1053/j.gastro.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 151.Glocker E, Grimbacher B. Inflammatory bowel disease: is it a primary immunodeficiency? Cell Mol Life Sci. 2012;69:41–48. doi: 10.1007/s00018-011-0837-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Glocker EO, Kotlarz D, Klein C, et al. IL-10 and IL-10 receptor defects in humans. Ann N Y Acad Sci. 2011;1246:102–107. doi: 10.1111/j.1749-6632.2011.06339.x. [DOI] [PubMed] [Google Scholar]
- 153.Ochs HD, Filipovich AH, Veys P, et al. Wiskott-Aldrich syndrome: diagnosis, clinical and laboratory manifestations, and treatment. Biol Blood Marrow Transplant. 2009;15:84–90. doi: 10.1016/j.bbmt.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 154.Blaese RM, Strober W, Brown RS, et al. The Wiskott-Aldrich syndrome. A disorder with a possible defect in antigen processing or recognition. Lancet. 1968;1:1056–1061. doi: 10.1016/s0140-6736(68)91411-6. [DOI] [PubMed] [Google Scholar]
- 155.Ozcan E, Notarangelo LD, Geha RS. Primary immune deficiencies with aberrant IgE production. J Allergy Clin Immunol. 2008;122:1054–1062. doi: 10.1016/j.jaci.2008.10.023. quiz 1063–1064. [DOI] [PubMed] [Google Scholar]
- 156.Blaese RM, Strober W, Levy AL, et al. Hypercatabolism of IgG, IgA, IgM, and albumin in the Wiskott-Aldrich syndrome. A unique disorder of serum protein metabolism. J Clin Invest. 1971;50:2331–2338. doi: 10.1172/JCI106731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ochs HD, Slichter SJ, Harker LA, et al. The Wiskott-Aldrich syndrome: studies of lymphocytes, granulocytes, and platelets. Blood. 1980;55:243–252. [PubMed] [Google Scholar]
- 158.Cannioto Z, Berti I, Martelossi S, et al. IBD and IBD mimicking enterocolitis in children younger than 2 years of age. Eur J Pediatr. 2009;168:149–155. doi: 10.1007/s00431-008-0721-2. [DOI] [PubMed] [Google Scholar]
- 159.Dupuis-Girod S, Medioni J, Haddad E, et al. Autoimmunity in Wiskott-Aldrich syndrome: risk factors, clinical features, and outcome in a single-center cohort of 55 patients. Pediatrics. 2003;111:e622–e627. doi: 10.1542/peds.111.5.e622. [DOI] [PubMed] [Google Scholar]
- 160.Folwaczny C, Ruelfs C, Walther J, et al. Ulcerative colitis in a patient with Wiskott-Aldrich syndrome. Endoscopy. 2002;34:840–841. doi: 10.1055/s-2002-34272. [DOI] [PubMed] [Google Scholar]
- 161.Filipovich AH, Stone JV, Tomany SC, et al. Impact of donor type on outcome of bone marrow transplantation for Wiskott-Aldrich syndrome: collaborative study of the international bone marrow transplant Registry and the National Marrow Donor program. Blood. 2001;97:1598–1603. doi: 10.1182/blood.v97.6.1598. [DOI] [PubMed] [Google Scholar]
- 162.Pai SY, DeMartiis D, Forino C, et al. Stem cell transplantation for the Wiskott-Aldrich syndrome: a single-center experience confirms efficacy of matched unrelated donor transplantation. Bone Marrow Transplant. 2006;38:671–679. doi: 10.1038/sj.bmt.1705512. [DOI] [PubMed] [Google Scholar]
- 163.Ozsahin H, Cavazzana-Calvo M, Notarangelo LD, et al. Long-term outcome following hematopoietic stem-cell transplantation in Wiskott-Aldrich syndrome: collaborative study of the European Society for Immunodeficiencies and European Group for Blood and Marrow Transplantation. Blood. 2008;111:439–445. doi: 10.1182/blood-2007-03-076679. [DOI] [PubMed] [Google Scholar]