

Abstract
For this Perspectives article, we explore a hypothesis involving the possible role of reduction/oxidation (redox) state in cancer. We hypothesize many modifications in cellular macromolecules observed in cancer progression may be caused by redox imbalance. Recent biochemical data suggest human prostate cancer cell lines demonstrate a redox imbalance (oxidizing) compared to benign primary prostate epithelial cells; the degree of oxidation varied with aggressive behavior of each cell line. Our recent data suggest human breast cancer tissues demonstrate a redox imbalance (reducing) compared to benign adjacent breast tissues. Accumulating data summarized in this article suggest redox imbalance may regulate gene expression and alter protein stability by posttranslational modifications, in turn modulating existing cellular programs. Despite significant improvements in cancer therapeutics, resistance occurs, and redox imbalance may play a role in this process. Studies demonstrate some cancer therapeutic agents increase generation of reactive oxygen/nitrogen species and antioxidant enzymes, which may alter total antioxidant capacity, cause cellular adaptation, and result in reduced effectiveness of treatment modalities. Approaches involving modulations of intra- and extracellular redox states in combination with other therapies may lead to new treatment options, especially for patients resistant to standard treatments.
Keywords: reduction/oxidation (redox), posttranslational modifications (PTM)
Introduction
Redox homeostasis is maintained by the net physiologic balance between reducing and oxidizing equivalents within subcellular compartments, with important components being reactive oxygen species and antioxidant enzymes (AEs) (1). The free radical theory of cancer traditionally encompassed the view that oxidative stress by reactive oxygen/nitrogen species (ROS/RNS) caused DNA damage and promoted genetic instability (1). However, ROS/RNS are now thought to be involved in both direct DNA damage and modulations of redox regulated signaling pathways, which may be beneficial or detrimental in cancers. For example, data show BCR/ABL oncogenes induce ROS, causing regulation of gene expression patterns of the transcription factor (TF) nuclear factor erythroid 2-related factor 2 (NRF2), a major regulator of cellular redox homeostasis (2). Adaptations resulting from NRF2 activation may have beneficial effects under stress conditions through modulation of antioxidant pathways, but may also contribute to cancer therapy resistance mechanisms (3). A recent study measured tissue redox activity in an animal model of neuroblastoma or glioma brain cancers using nitroxide-enhanced magnetic resonance imaging (MRI) in living mice or electron paramagnetic resonance spectroscopy (EPRS) in excised organs (4). Brain tissues in mice with late stage cancers differed in their reducing activity for the nitroxide radical measured by MRI or EPRS, tissue total antioxidant capacity, and plasma levels of matrix metalloproteinases (4), with combined results indicating late stage brain cancers were in an oxidized redox state. Further, abundant data have demonstrated cancer cells showed altered epigenetic patterns relative to benign cells; epigenetic changes have been demonstrated to be caused by redox imbalance in certain instances, resulting in alterations of gene expression patterns (5).
Several distinguishing hallmarks of cancer were originally proposed by Weinberg with underlying genetic instability as a major feature of cancer (6). Currently, the role of changes in energy metabolism and interactions between tumor intra- and extracellular redox environments are being investigated. For example, as proposed by Warburg, tumor cells often exhibit increased aerobic glycolysis compared to benign cells. Recent data indicate that the mitochondrial AE manganese superoxide dismutase (MnSOD) may contribute to the Warburg effect by regulating redox flux and glucose consumption in transition from quiescence to proliferation in human breast cancer cells (7). In contrast, recently proposed as the Reverse Warburg effect, tumor-associated stromal secrete energy rich metabolites, which epithelial cancer cells take up and use in mitochondrial oxidative phosphorylation, promoting proliferation and tumor growth (8). Increased levels of glutamine and loss of Caveolin-1 (CAV-1) expression in human breast cancer-associated fibroblasts correlated with recurrence and poor patient prognosis, and, therefore, absence of CAV-1 may be a new biomarker for the Reverse Warburg effect (8).
While studies of genetic instability and metabolism were major milestones in our understanding of cancer, oxidative stress and redox imbalance have been suggested to be present in cancer tissues. Oxidative stress is often putatively identified by analysis of fluorescent dyes that are sensitive to ROS, while redox imbalance is usually measured by biochemical analysis of stoichiometry of redox couples (NAD/NADH, NADP/NADPH, cysteine (Cys)/cystine (Cyss), glutatathione (GSH)/glutathione disulfide, thioredoxin (TRX)/thioredoxin disulfide). Unfortunately, analyses of ROS levels and/or redox imbalance do not provide mechanistic insights concerning cancer. What is missing is an understanding of how increased ROS and/or redox imbalance affect key macromolecules involved in cancer development and progression. Posttranslational modifications (PTMs) involving redox-sensing Cys residues or addition/deletion of low molecular weight metabolites to macromolecules induced by ROS and/or redox imbalance needs to be further investigated in cancer development and progression. While PTMs regulated by redox are the subject of active investigation in cancer, the possible relationship of redox to other biochemical changes in cancer is less frequently studied. This article briefly summarizes links between redox imbalance and biochemical changes described in cancer.
Posttranslational Modifications
For this Perspectives article, we focus on PTMs controlled by redox reactions, including the formation of intramolecular protein disulfide bonds between two closely spaced Cys residues containing two paired sulfhydryl (−SH) groups, a reaction which may be reversed by reduction (9). Oxygen is required for cellular metabolism and ROS are produced as bioproducts (10). Physiologic (balanced) levels of oxidants and antioxidants are required for proper cellular functions, which are regulated by redox biochemistry and show alterations in cancers. Increased levels of oxidants for a required period of time may oxidize intramolecular closely spaced −SH groups to disulfides, potentially affecting protein function. For example, TRX is an antioxidant protein, which provides reducing equivalents to proteins involved in cancer, including ribonucleotide reductase and TFs. TRX may undergo reversible redox reactions involving Cys32 and Cys35 located within the active site, or other modifications involving Cys62, Cys69 and Cys73 outside of the active site (10). TRX with two −SH groups at Cys32, Cys35 and Cys62, Cys69 is enzymatically active, while protein with disulfides at these two positions is irreversibly inactivated. Oxidation in the active site cysteines only is reversible by TRX reductase. Data in human prostate cell lines show increased TRX1 protein oxidation with decreased enzyme activity in more invasive cancer cell lines (11). Studies showed increased TRX1 oxidation in human prostate cancer tissues with high Gleason scores in comparison to tissues with low Gleason scores (unpublished data). In contrast, recent preliminary studies in human breast cancer tissues using western blot analysis of tissue lysates demonstrated 1.5-fold increased levels of TRX1 in infiltrating ductal carcinomas compared to adjacent benign tissue (data not shown). The reduced form of TRX1 was predominant over the oxidized, demonstrating a 4.7-fold increase (p ≤ 0.04, Figure 1). Furthermore, TRX1 enzymatic activities were 2.1-fold increased (Figure 2). Interestingly, these data suggest while human prostate cancer cell lines may be in a state of redox imbalance favoring an oxidizing environment and under oxidative stress, human breast cancer tissues may be in a state of imbalance favoring a reducing environment and reductive stress. Further investigations in human breast tissues and mechanistic studies in a cell culture model of human breast cancer progression are in progress. It will be important to determine redox state in metastatic breast cancer tissues.
Figure 1.

The reduced form of TRX1 was predominant in human breast cancer tissues. The redox state of TRX1 was quantified relative to total TRX1 and demonstrated a 4.7-fold significant increase (p ≦ 0.04) * expressed as the reduced form of TRX1 relative to total in infiltrating ductal carcinomas (cancer) versus benign/adjacent tissues (benign). Data from paired tissues obtained on biopsy or mastectomy were averaged from two patients from the University of Wisconsin School of Medicine and Public Health and two from Capital Biosciences. Redox western blotting involves derivatization of proteins with a thiol-reactive reagent such as iodoacetic acid (IAA), followed by native gel electrophoresis and western blotting.
Figure 2.
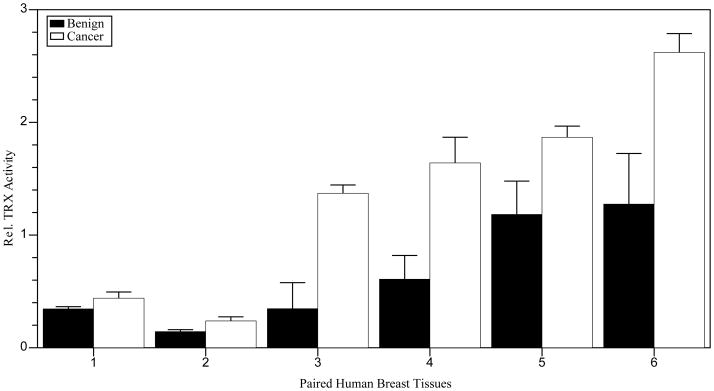
Human breast cancer tissues demonstrated increased TRX activities. Data demonstrated a 2.1-fold increased trend in TRX activities in infiltrating ductal carcinomas (cancer) versus benign/adjacent tissues (benign). Paired tissues obtained on biopsy/mastectomy from six patients were obtained from the University of Wisconsin School of Medicine and Public Health and Capital Biosciences. The insulin disulfide reduction assay was utilized to assess total TRX activity of six paired human tissues relative to GAPDH in triplicate. The assay measures the rate of which TRX (1 × 10 −8 M TRX) catalyzes reduction of insulin disulfides by dithiothreitol.
Redox state in extracellular spaces is largely regulated by the Cys/Cyss redox couple. Data in prostate cancer cell lines show that modulating the extracellular redox state by changing the concentration of Cys/Cyss in cell culture media altered prostate cancer invasiveness (12). It is likely that the Cys/Cyss redox couple regulates −SH to disulfide conversion in key proteins of the extracellular matrix and plasma membranes. Studies in head and neck squamous cell carcinoma cells suggest activities of Cyss transporters control redox status and may be modulated to improve therapeutic efficacy (13). Studies have demonstrated the importance of Cyss metabolism in extra- and intracellular interactions between stromal and chronic lymphocytic leukemia (CLL) cells, with modulations of redox state altering viability and drug sensitivity (14). Primary cells from CLL patients expressed low levels of the active subunit of the Cyss/glutamate antiporter (xCT), resulting in limited ability to transport Cyss into the cell for GSH synthesis. Stromal bone marrow cells were demonstrated to actively convert Cyss to Cys and release the molecule to the extracellular environment which may then be taken up by CLL cells and result in increased GSH (14). Increased GSH is thought to inhibit cell death and protect CLL cells from drug toxicity, a mechanism which could potentially be modulated as a therapeutic strategy to overcome resistance.
Several other important PTMs are being actively investigated in cancers. Examples of PTMs shown to be influenced by redox in cancer or under conditions causing oxidative/nitrosative stress are summarized in Table 1, and include phosphorylation (15), methylation (16), acetylation (17), sumolyation (18, 19), nitrosylation (20, 21), nitration (22), and glutathionylation (23, 24). Potential important targets for PTMs in cancers include TFs, oncogenes, tumor suppressors, proteins involved in metabolism, and proteins controlling antioxidant/prooxidant balance. Further studies are needed to determine whether alterations of PTMs in cancers are a cause or consequence of changes in ROS, redox state, or redox imbalance. However, it has been shown ataxia-telangiectasia mutated (ATM) protein kinase activation is regulated by ROS under oxidative stress (15). As mentioned above, BCR/ABL oncogenes have been shown to induce ROS, causing regulation of gene expression patterns of the TF nuclear factor erythroid 2-related factor 2 (NRF2), a major regulator of cellular redox homeostasis (2).
Table 1.
Posttranslational modifications documented in cancer or oxidative/nitrosative stress
| Modification | Addition | Cancer | Stress | Ref. |
|---|---|---|---|---|
| Phosphorylation | Phosphate to serine, threonine, tyrosine | ATM activation in response to double strand breaks; altered MnSOD (SOD2) activities | ATM activation under oxidative stress in absence of double strand breaks by oxidation of cysteine | (15, 22) |
| Methylation | Methyl to arginine, lysine | Altered MnSOD (SOD2) activities | Loss of MnSOD associated with chronic oxidative stress and aberrant epigenetic regulation | (16, 22) |
| Acetylation | Acetyl to lysine | Changes in HAT and HDAC activities modulate transcription; altered MnSOD (SOD2) activities | Under caloric restriction and resveratrol, SIRT1 and AMPK synergize to establish physiologic conditions; dysregulated MnSOD (SOD2) activities with oxidative stress | (17, 22) |
| Sumoylation | SUMO to lysine | eEF2 and SUMO-1 overexpression correlated with cisplatin resistance | Global effects involving inhibition of SUMO conjugating enzymes by oxidation of cysteine | (18, 19) |
| Nitrosylation | Nitric oxide to cysteine | Inflammatory mediators inhibit BRCA1 | NRF2 activation by oxidation of cysteines within KEAP1 | (20, 21) |
| Nitration | Nitric oxide to tyrosine | Altered levels of nitrated nucleotides, fatty acids, and proteins | Nitration of MnSOD results in loss of activity | (22) |
| Glutathionylation | Glutathione to cysteine | GST overexpression associated with drug resistance | Protection from irreversible oxidation | (23, 24) |
Epigenetic and Transcriptional Changes
Epigenetics may be defined as heritable environmental or life-style dependent alterations to DNA or chromatin acquired post birth (or in utero) without changes within the underlying DNA sequence; epigenetic changes may involve the reversible addition of methyl groups to DNA (methylation) or reversible PTMs involving addition of acetyl and/or methyl groups to histone proteins (acetylation/methylation). These reactions are catalyzed by DNA-histone acetyltransferases or methyltransferases (6). Additionally, micro RNAs bind to the 5′ or 3′ untranslated regions (UTRs) of target mRNAs, which in turn modulate gene expression. Several effects on specific genes or subsets of genes have been characterized as being regulated by micro RNA modifiers, including roles in cancer progression (6). For example, recent studies demonstrated that repression of miR-200a in breast cancer cells, which targets Keap1 mRNA, subsequently leads to its degradation and contributes to dysregulation of NRF2 (25). The Keap1 levels were restored upon epigenetic treatment with a histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA). Recent literature suggests epigenetic changes may be driven by redox imbalance through metabolic reprogramming of the existing cellular epigenetic program (5). Transcription may be modulated by redox imbalance through epigenetic mechanisms, which are linked to metabolism through variations of required intermediates, such as NAD+. For example, modulations of cellular processes may occur in any instance where there is limited availability of enzymes required for modifications within subcellular compartments, such as class III histone deacetylases (HDACs), which are dependent on NAD+ (commonly referred to as sirtuins [SIRT]) (17). Therefore, any alteration that depletes cellular NAD+ will affect the activity of SIRTs. In addition, xanthine oxidoreductase activity is increased in human serum obtained from patients with leukemia compared to healthy controls and is further increased in relapse (2). These data suggest a role for the xanthine oxidoreductase system in disease recurrence/resistance, of note since this system is known to use the cofactor NAD+ in drug metabolism with concomitant increased generation of ROS (2).
It has been suggested that an increased reducing environment provides conditions necessary for the optimal rate of electron transfer and enzymatic activity required for TF binding to DNA. Transcription occurs within the nucleus of a cell and is a process by which nucleic acid sequences of DNA are copied to ribonucleic acid (RNA) (6). Transcription is dependent upon structural components and regulatory sequences located internally within the complete nucleic acid sequence. Splicing occurs concurrently or afterwards and is a process which removes introns and joins exons to produce mRNA required for proper protein synthesis (6). Mutations in coding regions, gene promoters, and consensus binding sites are involved in regulation of gene expression, while modifications in splicing may affect protein function. Epigenetic modifications to DNA, histone modifications, and micro RNA modifiers may modulate gene expression patterns (6).
Transcriptional regulation often involves redox-sensing Cys residues located within the region required for DNA binding of redox-regulated TFs. The promoter regions of redox-regulated genes contain an ARE (antioxidant responsive element), a CRE (cyclic AMP responsive element), and SP-1 sites required for activation of redox-regulated TFs such as AP-1, NF-κB, NRF2, TP53, glucocorticoid and other hormone receptors, among others (3, 10). The presence of AREs in so many TFs provides strong evidence that redox imbalance is a major factor in regulating cellular adaptation to redox injury. Reduced forms of TRX are active enzymatically, while partially oxidized forms are inactive and must be reduced by TRX reductases for activation (10). Therefore, modulations of nuclear compartment redox state may alter activity of TFs that are reduced by TRX and ultimately gene expression patterns. Interestingly, recent data suggest the redox state of human TRX1 may be changed by treatment with the histone deacetylase inhibitor SAHA in cell culture, and this compound is currently in clinical trials for treatment of acute myeloid leukemia (AML) (26).
Several examples document complex genetic and epigenetic influences in cancer progression. For example, mutational status of VHL in renal cell carcinoma was not correlated with clinical outcome, although alterations in DNA methylation were associated with metastasis (27). Monozygotic (MZ) and dizygotic twin studies estimate relative importance of complex genetic and epigenetic factors influencing disease phenotypes (28). One study comparing MZ twins found increased promoter methylation at specific CpG sites with no DNA mutations of the disease-associated genes (28). Another study demonstrated MZ twins discordant for childhood leukemia (previously considered only a genetic disease) showed increased BRCA promoter methylation in somatic cells (fibroblasts of saliva) of the female diagnosed with childhood leukemia (12%) compared to her healthy sister (3%) (29). Interestingly, mutations in IDH1 and IDH2 result in complex genetic and epigenetic cellular and biochemical changes, such as increased hematopoietic stem cell progenitors, alterations in cell differentiation, changes in DNA methylation, and metabolism (30). Thus, single mutations affecting cell biochemistry can have complex effects on cell behavior, epigenetic changes, and metabolism.
Protein Stability
Changes in protein stability within subcellular compartments may influence protein levels and function. Protein stability is regulated by changes in protein folding/unfolding, conformational changes, hydrophobic interactions, hydrogen bonding, redox potential, pH, temperature, and metal binding (6). Oxidizing conditions favor unfolding of proteins and stabilization, with disulfide bond formation through oxidation of two Cys residues forming covalent sulfur-sulfur bonds (9). Reducing conditions favor highly reactive free −SH groups (9). Subcellular oxygenation levels and cellular responses to oxidative stress are important factors regulating protein stability, which are altered in cancers. For example, dysregulation of the TFs hypoxia inducible factor alpha (HIF1α) and NRF2 have been identified in several cancers and therapeutic approaches involving these TFs are being investigated (3, 6). Under normoxic conditions HIF1α is ubiquinated and subsequently degraded. However, under hypoxic conditions it may be stabilized and associates with the VHL protein, which in turn activates HIF1α regulated genes (6). Similar to HIF1α, under normoxic cellular conditions NRF2 is ubiquinated and subsequently degraded, potentially regulated by biochemical reactions involving Cys residues as redox sensors within the scaffold protein Kelch-like ECH-associated protein 1 (Keap1) (3). In response to oxidative/nitrosative stresses NRF2 is stabilized, with subsequent activation of genes responsible for stimulating antioxidant genes and cytoprotective enzymes (3). Modulations to NRF2 compartmentalization and trafficking also occur, which play an important role in mediating crosstalk with other molecular signaling pathways, including nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), aryl hydrocarbon receptor (AHR), tumor protein 53 (TP53), and notch homolog 1 (Notch1) (3). Importantly, high levels of NRF2 have been identified in several cancers, which result in constitutive activation of NRF2 and redox-regulated adaptations. The adaptations protect cells from cytotoxic effects of anticancer or chemoprevention compounds and cause resistance to chemo- and radiation therapy; therefore, controversy exists whether therapeutic approaches may be useful for activating or inhibiting NRF2 (3).
Conclusion and Discussion
It is undisputed that genetic and epigenetic factors influence cancer initiation and progression. It has been shown that oxidation may cause mutations and promote genetic instability. Despite an initial belief that rapid sequencing of DNA from cancer families and further extensive knowledge about the biochemistry of cancer would perhaps result in cancer cures, metastatic cancer remains a formidable challenge today. Two mechanistic reasons for failure are genomic instability and development of resistance to therapeutic agents. While the latter feature of the metastatic cancer phenotype may be related to genomic instability, it may also be caused by the other biochemical changes of cancer described in this article. Treatment failure may be due to a lack of understanding of an underlying feature that drives all of the biochemical changes described in cancer progression. This article provides beginning evidence that sequential changes in redox balance drive cancer development and progression. To prove this hypothesis, combined analyses of already described biochemical changes and biochemical and biomarker studies of redox imbalance need to be performed in human benign and cancer tissues of varying degrees of progression. Subsequently, animal models will need to be developed in which key proteins regulating redox balance are mutated at active sites or overexpressed to determine effects on cancer formation.
Studies demonstrate some cancer therapeutic agents increase generation of ROS/RNS and AEs, which may alter total antioxidant capacity and cause cellular adaptations. Adaptations resulting from HIF1α and NRF2 pathway activation may have beneficial effects through modulating antioxidant pathways under stress conditions, but may also contribute to cancer therapy resistance. Accumulating data show oxidative stress and redox imbalance cause epigenetic changes. Regulation of gene expression is influenced by several factors, including posttranslational modifications and micro RNA, which may in turn modulate existing cellular programs. Recent data suggest cancers may be in a state of redox imbalance, which may increase with progression. Preliminary studies in our laboratory suggest redox imbalance may be either oxidizing (prostate) or reducing (one pathologic type of breast cancer before metastasis). Many modifications in cellular macromolecules observed in cancer progression may be caused by redox imbalance. Proteomic studies are needed to identify protein modifications in cancer progression and will determine whether redox imbalance precedes or follows these modifications or both in a vicious cycle. Perhaps future cancer diagnostics may include an analysis of redox state with progression or provide a new method for monitoring response to therapy. New treatment options may involve modulating intra- or extracellular redox state of cancer cells at varying stages of progression, which may involve metabolic reprogramming. New clinical trials might involve attempts at modulating redox imbalance in cancers resistant to anti-hormonal treatments, radiation, or standard chemotherapy or sensitizing resistant cancers to drug treatment.
Acknowledgments
The contents do not represent the views of the U.S. Department of Veterans Affairs. This work was supported in part by funds from the University of Wisconsin, Department of Pathology and Laboratory Medicine, NCI UW Carbone Comprehensive Cancer Center (NIH grant P30 CA014520), NIH grants RO1CA139844, RO1CA139843, RO1CA073599 (TDO-CoI), and resources and facilities at the William S. Middleton Memorial Veterans Hospital (Madison, WI, USA). Tonia C. Jorgenson is supported in part by PF-13-286-01-TBE from the American Cancer Society. The authors would like to thank Weihua Shan for technical assistance.
References
- 1.Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3(4):276–85. doi: 10.1038/nrc1046. 04//print. [DOI] [PubMed] [Google Scholar]
- 2.Irwin ME, Rivera-Del Valle N, Chandra J. Redox control of leukemia: from molecular mechanisms to therapeutic opportunities. Antioxidants & redox signaling. 2013 Apr 10;18(11):1349–83. doi: 10.1089/ars.2011.4258. Epub 2012/08/21. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang DD. The Nrf2-Keap1-ARE signaling pathway: The regulation and dual function of Nrf2 in cancer. Antioxidants & redox signaling. 2010 Dec 1;13(11):1623–6. doi: 10.1089/ars.2010.3301. Epub 2010/05/22. eng. [DOI] [PubMed] [Google Scholar]
- 4.Bakalova R, Zhelev Z, Aoki I, Saga T. Tissue redox activity as a hallmark of carcinogenesis: from early to terminal stages of cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013 May 1;19(9):2503–17. doi: 10.1158/1078-0432.CCR-12-3726. Epub 2013/03/28. eng. [DOI] [PubMed] [Google Scholar]
- 5.Hitchler MJ, Domann FE. Redox regulation of the epigenetic landscape in Cancer: A role for metabolic reprogramming in remodeling the epigenome. Free Radical Biology and Medicine. 2012;53(11):2178–87. doi: 10.1016/j.freeradbiomed.2012.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weinberg RA. The biology of cancer Garland Science. Taylor & Francis Grouop, LLC; 2007. [Google Scholar]
- 7.Sarsour EH, Kalen AL, Xiao Z, Veenstra TD, Chaudhuri L, Venkataraman S, et al. Manganese superoxide dismutase regulates a metabolic switch during the mammalian cell cycle. Cancer Research. 2012 Aug 1;72(15):3807–16. doi: 10.1158/0008-5472.CAN-11-1063. Epub 2012/06/20. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell cycle (Georgetown, Tex) 2009 Dec;8(23):3984–4001. doi: 10.4161/cc.8.23.10238. Epub 2009/11/20. eng. [DOI] [PubMed] [Google Scholar]
- 9.Klomsiri C, Karplus PA, Poole LB. Cysteine-based redox switches in enzymes. Antioxidants & redox signaling. 2011 Mar 15;14(6):1065–77. doi: 10.1089/ars.2010.3376. Epub 2010/08/31. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee S, Kim SM, Lee RT. Thioredoxin and thioredoxin target proteins: from molecular mechanisms to functional significance. Antioxidants & redox signaling. 2013 Apr 1;18(10):1165–207. doi: 10.1089/ars.2011.4322. Epub 2012/05/23. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shan W, Zhong W, Zhao R, Oberley TD. Thioredoxin 1 as a subcellular biomarker of redox imbalance in human prostate cancer progression. Free radical biology & medicine. 2010 Dec 15;49(12):2078–87. doi: 10.1016/j.freeradbiomed.2010.10.691. Epub 2010/10/20. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaiswing L, Zhong W, Liang Y, Jones DP, Oberley TD. Regulation of prostate cancer cell invasion by modulation of extra- and intracellular redox balance. Free radical biology & medicine. 2012 Jan 15;52(2):452–61. doi: 10.1016/j.freeradbiomed.2011.10.489. Epub 2011/11/29. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshikawa M, Tsuchihashi K, Ishimoto T, Yae T, Motohara T, Sugihara E, et al. xCT Inhibition Depletes CD44v-Expressing Tumor Cells That Are Resistant to EGFR-Targeted Therapy in Head and Neck Squamous Cell Carcinoma. Cancer Research. 2013 Mar 15;73(6):1855–66. doi: 10.1158/0008-5472.CAN-12-3609-T. [DOI] [PubMed] [Google Scholar]
- 14.Zhang W, Trachootham D, Liu J, Chen G, Pelicano H, Garcia-Prieto C, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nature cell biology. 2012 Mar;14(3):276–86. doi: 10.1038/ncb2432. Epub 2012/02/22. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM Activation by Oxidative Stress. Science. 2010 Oct 22;330(6003):517–21. doi: 10.1126/science.1192912. [DOI] [PubMed] [Google Scholar]
- 16.Cyr AR, Hitchler MJ, Domann FE. Regulation of SOD2 in Cancer by Histone Modifications and CpG Methylation: Closing the Loop Between Redox Biology and Epigenetics. Antioxidants & redox signaling. 2012 Oct 18; doi: 10.1089/ars.2012.4850. Epub 2012/09/06. Eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morris BJ. Seven sirtuins for seven deadly diseases ofaging. Free Radical Biology and Medicine. 2013;56:133–71. doi: 10.1016/j.freeradbiomed.2012.10.525. [DOI] [PubMed] [Google Scholar]
- 18.Bossis G, Melchior F. Regulation of SUMOylation by Reversible Oxidation of SUMO Conjugating Enzymes. Molecular Cell. 2006;21(3):349–57. doi: 10.1016/j.molcel.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 19.Chen C-Y, Fang H-Y, Chiou S-H, Yi S-E, Huang C-Y, Chiang S-F, et al. Sumoylation of eukaryotic elongation factor 2 is vital for protein stability and anti-apoptotic activity in lung adenocarcinoma cells. Cancer Science. 2011;102(8):1582–9. doi: 10.1111/j.1349-7006.2011.01975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yakovlev VA. Nitric Oxide-Dependent Downregulation of BRCA1 Expression Promotes Genetic Instability. Cancer Res. 2013 Jan 15;73(2):706–15. doi: 10.1158/0008-5472.CAN-12-3270. Epub 2012/10/31. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fourquet S, Guerois R, Biard D, Toledano MB. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. The Journal of biological chemistry. 2010 Mar 12;285(11):8463–71. doi: 10.1074/jbc.M109.051714. Epub 2010/01/12. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dhar SK, St Clair DK. Manganese superoxide dismutase regulation and cancer. Free radical biology & medicine. 2012 Jun 1;52(11–12):2209–22. doi: 10.1016/j.freeradbiomed.2012.03.009. Epub 2012/05/09. eng. [DOI] [PubMed] [Google Scholar]
- 23.Townsend DM, Manevich Y, He L, Hutchens S, Pazoles CJ, Tew KD. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. The Journal of biological chemistry. 2009 Jan 2;284(1):436–45. doi: 10.1074/jbc.M805586200. Epub 2008/11/08. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mieyal JJ, Chock PB. Posttranslational modification of cysteine in redox signaling and oxidative stress: Focus on s-glutathionylation. Antioxidants & redox signaling. 2012 Mar 15;16(6):471–5. doi: 10.1089/ars.2011.4454. Epub 2011/12/06. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eades G, Yang M, Yao Y, Zhang Y, Zhou Q. miR-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. The Journal of biological chemistry. 2011 Nov 25;286(47):40725–33. doi: 10.1074/jbc.M111.275495. Epub 2011/09/20. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ungerstedt J, Du Y, Zhang H, Nair D, Holmgren A. In vivo redox state of Human thioredoxin and redox shift by the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) Free Radical Biology and Medicine. 2012;53(11):2002–7. doi: 10.1016/j.freeradbiomed.2012.09.019. [DOI] [PubMed] [Google Scholar]
- 27.Vanharanta S, Shu W, Brenet F, Hakimi AA, Heguy A, Viale A, et al. Epigenetic expansion of VHL-HIF signal output drives multiorgan metastasis in renal cancer. Nat Med. 2013;19(1):50–6. doi: 10.1038/nm.3029. 01//print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Dongen J, Slagboom PE, Draisma HH, Martin NG, Boomsma DI. The continuing value of twin studies in the omics era. Nature reviews Genetics. 2012 Sep;13(9):640–53. doi: 10.1038/nrg3243. Epub 2012/08/01. eng. [DOI] [PubMed] [Google Scholar]
- 29.Galetzka D, Hansmann T, El Hajj N, Weis E, Irmscher B, Ludwig M, et al. Monozygotic twins discordant for constitutive BRCA1 promoter methylation, childhood cancer and secondary cancer. Epigenetics : official journal of the DNA Methylation Society. 2012 Jan 1;7(1):47–54. doi: 10.4161/epi.7.1.18814. Epub 2011/12/31. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010 Dec 14;18(6):553–67. doi: 10.1016/j.ccr.2010.11.015. Epub 2010/12/07. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]