

Abstract
Semi-synthesis of natural product derivatives combines the power of fermentation with orthogonal chemical reactions. Yet, chemical modification of complex structures represents an unmet challenge, as poor selectivity often undermines efficiency. The complex antibiotic teicoplanin eradicates bacterial infections. However, as resistance emerges, the demand for improved analogs grows. We have discovered chemical reactions that achieve site-selective alteration of teicoplanin. Utilizing peptide-based additives that alter reaction selectivities, certain bromo-teicoplanins are accessible. These new compounds are also scaffolds for selective cross-coupling reactions, enabling further molecular diversification. These studies enable two-step access to glycopeptide analogs not available through either biosynthesis or rapid total chemical synthesis alone. The new compounds exhibit a spectrum of activities, revealing that selective chemical alteration of teicoplanin may lead to analogs with attenuated or enhanced antibacterial properties, in particular against vancomycin and teicoplanin resistance strains.
Keywords: teicoplanin, site-selective, bromination, antibiotic, peptide-catalyzed
Introduction
Complex glycopeptides are important compounds in the ongoing campaign against antibiotic resistance.1-4 Vancomycin (1, Figure 1) in particular has been studied intensively due to its efficacy against resistant strains of bacteria.1-4 Even so, as vancomycin-resistance has emerged (e.g., vancomycin resistant enterococcus (VRE) and vancomycin resistant S. aureus (VRSA)), the pursuit of more effective antibiotics assumes even greater significance.5 Teicoplanin (2), a structurally more intricate glycopeptide, exhibits enhanced potency and a different spectrum of activities relative to vancomycin.6-8 In fact, 2 offers some promise in the context of resistant strains with analogs advancing in clinical trials.9
Figure 1.

Structure of glycopeptide antibiotics vancomycin (1), teicoplanin (2) and teicoplanin A2-2 (3).
Synthesis of analogs of 1 or 2 for biological evaluation continues to fuel medicinal discoveries.10,11 Total synthesis,11-19 and semi-synthesis3,20-24 (employing enzymatic catalysts, synthetic catalysts, or chemical reagents) all provide important avenues for research. In the case of 2, a number of particular challenges exist. The structure is substantially more complex than 1, rendering analog access by total synthesis more difficult. To date, while a total synthesis of the teicoplanin aglycone has been reported,14,16,17 the complete total chemical synthesis has not yet been reported. Moreover, the enhanced complexity renders direct and selective functionalization of 2 more challenging than the related goal of direct functionalization of 1. Also, whereas 1 is readily available in homogeneous form through a fermentation process,25 2 is obtained as a complex mixture.26 Further purification is therefore necessary to obtain a homogeneous starting material for study of selective chemical reactions. Our laboratory has been pursuing a program that tests the capacity of small-molecule catalysts to functionalize complex molecules in a selective manner.27-29 This chemistry-based approach offers the promise of enhancing access to analogs of complex natural products, and also presents challenges in the study of catalysis in environments of extreme molecular complexity. One tenet of these studies involves documentation of the intrinsic reactivity of the substrate towards a reagent of interest, in the absence of a catalyst or promoter. Then, the goal becomes the identification of catalysts, conditions or reagents that alter the intrinsic reactivity to produce alternative compounds.
Our recent study of the site-selective bromination of vancomycin provides a case in point (Figure 2).29 We evaluated selective halogenation as a way of mimicking the chemistry of the tailoring halogenase enzymes with small molecule catalysts.30,31 The selective introduction of halide into the glycopeptides offers a unique way to tune the properties of the antibiotics. In addition, controlled introduction of halide offers an opportunity to examine further chemical functionalization of these scaffolds, for example with metal-catalyzed cross-coupling reactions.32 As shown in Figure 2A, when native vancomycin (with no protecting groups) is exposed to N-bromophthalimide (NBP), a very sluggish reaction takes place in which one observes, after 2 h, mostly unreacted 1, but also small, nearly equimolar amounts of several products. However, in the presence of a designed peptide-based catalyst (4, Figure 2B), substantial rate-acceleration is observed, the starting material is completely consumed, and selective formation of mono-bromo-vancomycin derivative 6 is observed. Alternatively, as shown in Figure 2C, when peptide 4 is replaced with guanidine as an additive, the selectivity is reversed, and alternative mono-bromo-vancomycin derivative 5 is obtained.
Figure 2.

Summary of studies for the bromimation of 1. HPLC traces are shown for (a) the control reaction of 1 with no catalyst, (b) the reaction conducted in the presence of peptide 4, and (c) the reaction conducted in the presence of guanidine.
Given the heightened biological activity and higher level of molecular complexity of teicoplanin compared to vancomycin, we wished to establish whether site-selective halogenation of teicoplanin might be possible chemically. The structure of teicoplanin presents the additional challenge of tuning the site of bromination to either the 5,7-biaryl region of the structure (2, Figure 1, red) or the 1,3-biaryl-ether region (2, Figure 1, blue). The intrinsic reactivity of these moieties, relative to one another, was not completely clear at the outset. Thus, one of the key goals of this study was to evaluate catalysts/conditions that could provide site-selective bromination of either biaryl ring system. To initiate these studies, it was necessary to undertake the purification of teicoplanin.8,22 We targeted teicoplanin A2-2 (3, Figure 1) from the readily available mixture of teicoplanins, which is a composite of approximately six to nine molecular forms of teicoplanin (Figure S1). The assignment of the aromatic region of the NMR spectrum of 3 with modern NMR techniques was also essential at the outset of these studies,22,33-35 as a critical step for analysis and determination of the site of bromination within new products (Figures S1-S3).
Results and Discussion
Studies of the site-selective bromination of 3 began with an evaluation of its reaction in the presence of various quantities of N-bromophthalimide (NBP). As shown in Figure 2A, when 3 was dissolved in MeOH/H2O (1:1), and 1.1 equiv of NBP was employed, a major product was observed, with unreacted 3 also prominent in the HPLC trace (Figure 3A(a)). When multiple equivalents were employed, a highly complex mixture of products was observed (Figure S4). The reaction with 1.1 equiv allowed for isolation and purification of the major brominated species (7; Figure 3B). On preparative scale, 50.0 mg of 3 could be converted to 10.0 mg of analytically pure 7 with 6.0 mg of recovered 3 in a single operation. LC-MS analysis revealed that the new compound was a mono-brominated form of teicoplanin A2-2. Figure 2C shows one of the most revealing pieces of NMR data, an overlay of a diagnostic region of the HSQC spectrum for both 3 and 7. Most notably, the cross peak that we had assigned to the ring 7f (C–H) correlation is absent in 7. At the same time, there is excellent overlay of the overwhelming majority of the other peaks, suggesting a minimum structural alteration. The cross peak for the 7d and 5b (C–H) correlation has shifted slightly, as highlighted in Figure 2C, suggesting substitution within the 5,7-biaryl substructure. Additional data also supports the assignment of 7 as the ring 7f-Br variant of teicoplanin A2-2 (Figures S6-S8).
Figure 3.

Effect of peptides on the site-selectivity of teicoplanin bromination compared to the control reaction (data collected on HPLC). B) Structure of brominated analogs 7 and 9. C) Overlay of HSQC spectrum of 7 (gray) with 3 (blue).
We then turned our attention to perturbation of the inherent site-selectivity exhibited by 3 under our initial conditions. We initially targeted catalysts like 4, designed in our earlier studies29 to mimic the binding of 1 and 2 to their biological target,36,37 but outfitted with functional groups that might accelerate brominaton.38 When 3 was exposed to bromination conditions in the presence of peptide 4, we observed that indeed bromination occurs, but the analytical HPLC trace exhibits broad peaks, as shown in Figure 3A(b). The peak shape renders analysis of reaction mixture and isolation of pure materials difficult. We interpreted these features as the formation of robust (i.e., too robust) 3-peptide complexes. This assertion is consistent with the known, very high affinity of 3 for dAla-dAla-based peptides (Ka=1.6 × 106 for Ac-Lys-dAla-dAla at pH = 5.0).39 To remedy this issue in the context of peptide-mediated brominations, we elected to replace one of the dAla units with a dLeu. This strategy was projected to attenuate the binding affinity of substrate and peptide, in accord with previous binding studies of dXaa-dXaa peptides with glycopeptides like 1.37,40 Indeed, this strategy proved effective. The corresponding bromination of 3 with Boc-Asn(Me)2-dLeu-dAla-OH (8) as a promoter of the reaction produces a reaction mixture that was readily analyzed by HPLC/LC-MS (Figure 3A(c)). Strikingly, peptide 8 diverts the reaction to give a new mono-brominated teicoplanin, which was not observed in significant quantities in the absence of 8. As detailed below, the structure of the new, 8-dependent mono-brominated teicoplanin may be assigned as the ring 3b-Br analog 9. The capacity of peptide 8 to re-direct the site of bromination away from the intrinsically more reactive 5,7-ring system to the less reactive 1,3-ring system is a manifestation of nonenzymatic control of site-selectivity in a highly complex molecular environment. The reaction specificity is also highly dependent on peptide structure and stereochemistry. Alteration of the configuration of the dLeu residue of the peptide to the L-configuration, as in 11 (Boc-Asn(Me)2-Leu-dAla-OH), produced a very different result (Figure 3A(d)). In this case, the major product was reverted to 7f-Br compound 7. Presumably, these results are due to the reduced binding affinity of the stereochemically mismatched peptide 11 to 3 during the bromine transfer reaction.40 On preparative scale, in the presence of peptide 12 (Figure 5), 160.0 mg of 3 provided 47.0 mg of analytically pure 9 (with 35.0 mg of recovered 3 and 16.0 mg of 10) in a single step. We note parenthetically that while we employ a full equivalent of the peptide-based promoter in these experiments, and therefore that the “catalytic” species does not rigorously demonstrate turnover under these conditions, the capacity of the peptide to alter product selectivity is unambiguous. Moreover, when the reaction is conducted with 50 mol% peptide 8, similar selectivity is observed but the reaction proceeds to lower conversion (~90% conv. with 100 mol%, ~75% conv. with 50 mol%; Figure 3A(e)).
Figure 5.

Effect of varying position of catalytic residue on product distribution.
The assignment of the structure for 9 is based on the following NMR experiments. Shown in Figure 4A is the overlay of the HSQC spectra for 3 (blue) and 9 (gray). Notably, the correlation for the 3b (C–H) positions was not apparent in compound 9. At the same time, we observed minor changes in the chemical shift for the correlations corresponding to the 3f and 3d (C–H) positions. A more dramatic chemical shift perturbation was observed for the X3 (α-C–H) correlation (the α-position the residue 3 aryl glycine; Figure S11). These features could be induced by a change in the ring current of aryl ring 3 upon bromination. To further illustrate the basis of the assignment for structure 9, we show the key HMBC (blue) and NOESY (green) correlations in Figure 4B. These observations taken together culminate in our assignment of 9 as the 3b-Br teicoplanin. It is interesting to note that a more detailed analysis of the HSQC spectrum of 9 reveals a doubling of certain correlations. Figure 4C illustrates the presence of a putative minor conformation, with the chemical shift perturbations for the minor species most prominent for the 4b and X3 (α-C–H) correlations. These observations suggest possible conformational heterogeneity in the peptide backbone region between residue 5 and residue 3. This observation has also been made previously for teicoplanin A2-2 itself, albeit with quite low intensity for the putative minor conformer.33,41 The amplification of conformational heterogeneities as a function of site-selective modification of complex natural products is a phenomenon that appears to be quite common, based on these and other recent studies.28
Figure 4.
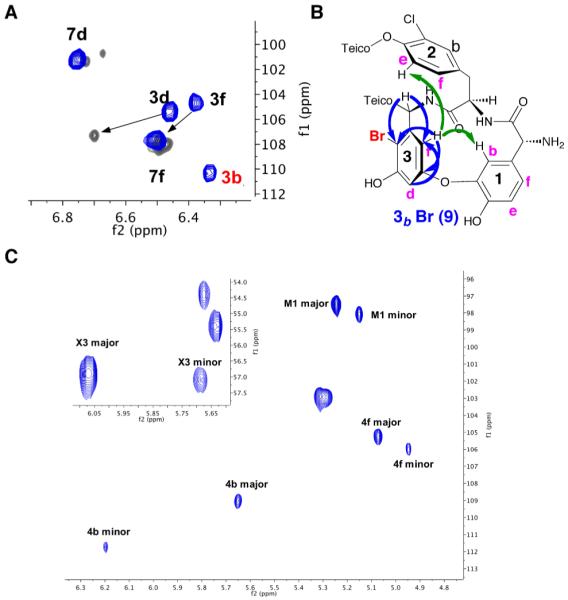
A) Overlay of HSQC spectrum of 9 (gray) with 3 (blue). B) Key NOESY (green) and HMBC (blue) correlation supportive of the structural assignment of 9. C) HSQC spectrum of 9 showcasing presence of minor conformer.
To further assess the capacity of peptide-based catalysts/promoters to influence the site of bromination, we examined different sequences wherein the position of the Asn(Me)2 moiety was varied within the tripeptide. Our initial hypothesis was that localization of a putative bromine-directing side chain might alter the site of bromination, affording access to alternative teicoplanin A2-2-derived bromides. However, as shown in Figure 4, our observations did not support this initial assertion. Instead, both peptides 12 and 13, with the Asn(Me)2 as the central residue (12, Figure 5B) or as the residue in the C-terminal position (13, Figure 5C), deliver highly selective brominations of teicoplanin to deliver the 3b-Br teicoplanin 9, as is observed in the presence of peptide 8 (Figure 5A). Notably, all three peptides in the series offer the same striking reversal of selectivity relative to the control reaction in the absence of peptide.
Our interpretation of these observations is that the binding of dXaa-dYaa peptide fragments to teicoplanin may occur in accord with well-established models in the literature (Figure 6, Adapted from PDB# 3VFJ). If so, this event may result in a conformational change of the glycopeptide that renders the 3b-position of the substrate more reactive towards uncatalyzed bromination, relative to the 7f-position, which reacts first in the absence of peptide. In this situation, the possible bromination-accelerating effects of the Asn(Me)2 side chain may be muted, relative to the intrinsically high reactivity of the resorcinol moieties in the glycopeptide. It is also possible that other amide bond carbonyl groups of the ligand, or the urethane carbonyl group, may also assist in directing the delivery of Br. It is also difficult to exclude the possible direction of bromination from carbonyl groups in teicoplanin A2-2 itself. In each scenario, the unique selectivity exhibited by the putative teicoplanin A2-2/peptide adduct does support a delivery event that derives from supramolecular complex formation.
Figure 6.

X-ray crystal structure of 3 bound to Lys-dAla-dAla-OH. Adapted from PDB# 3VFJ.
Pronounced peptide-dependent effects were also observed as one examines further bromination of teicoplanin A2-2. As shown in Figure 7A, when 3 was exposed to an excess of NBP (3.3 equiv), either in the absence or in the presence of different peptides (8, 12 and 13, with variable loci of the Asn(Me)2 side chain) different product distributions were obtained. In the absence of a peptide-based promoter, a highly complex mixture of products was obtained (Figure 7A(a)). One implication is that there appears to be a comparable level of reactivity for many sites within teicoplanin A2-2 as further functionalization occurs. Even so, the reaction mixtures are substantially less complex when peptide-based promoters were evaluated (Figure 7A(b)-(d)), exhibiting their capacity to perturb site-selectivity among the less reactive sites. When a peptide bearing the N-terminal Asn(Me)2 side chain was employed (8), the reaction mixture contains a number of products, including a prominent HPLC peak that contains an inseparable mixture of di- and tribromides (Figure 7A(b)). When the Asn(Me)2 is central to the tripeptide (12), two peaks were observed in the HPLC trace that once again contain mixtures (Figure 7A(c)). Yet, when the Asn(Me)2 is in the C-terminal position (13), a striking, peptide-dependent outcome was observed, with a new, homogeneous peaks apparent in the HPLC trace (Figure 7A(d)). LC-MS analysis reveals the compound to be a new tribrominated species.
Figure 7.

A) Effect of peptides on the site-selectivity of teicoplanin tribromination compared to the control reaction (data collected on HPLC). B) Assigned structure of tribrominated teicoplanin (14). C) Overlay of HSQC spectrum of 14 (gray) with 3 (blue).
Our analysis of the NMR spectra revealed the new compound to be the illustrated 7d,3b,3d-Br3 teicoplanin analog (14, Figure 7B). A first order analysis of the overlayed HSQC spectra of 3 (blue) and 14 (gray) reveals a larger number of chemical shift perturbations for many of the C–H correlations than we had observed for compounds 7 and 9, possibly reflecting greater structural reorganization upon tribromination. Among the most diagnostic observations were the absence of the C–H correlations for the 7d, 3b and 3d positions. In addition, the perturbation of chemical shifts for nearest neighbors was also apparent for all three of these positions. For example, X3 (α-C–H), 7f (C–H) and 3f (C–H) correlations perturbations may all be observed. Notably, significant changes in the chemical shifts of the unique mannose ring (M) were apparent, consistent with bromination of 7d-position (Figure S20). These features further suggested a significant conformational reorganization and prompted additional NMR studies to unambiguously assign tribromide analog. NOESY and HMBC experiments were performed, and the data support the assignment of 14 (Figure S21). On a preparative scale, 15 mg of 14 could be obtained from 80 mg of 3 in a single operation.
At this stage, we wished to establish whether the new brominated variants of 3 were substrates for metal-catalyzed Suzuki cross-coupling reactions.32,42 At the beginning of the study, we encountered poor results due to the apparent binding of Pd reagents to teicoplanin that resulted in inhibition of catalysis. This hypothesis derived from the observation of molecular ions corresponding to [substrate+Pd] in the LC-MS of the reaction mixture. However, we found that use of higher loadings of Pd (50 mol%) along with the water-soluble phosphine ligand 15 (100 mol%) allowed cross-coupling to occur with useful efficiencies. For example, when bromo-teicoplanin 9 was subjected to these conditions, furan-containing teicoplanin analog 16 was obtained in 28% yield (73% conversion, Figure S39), in a single step, with high purity after reverse phase HPLC purification (Figure 8A). Intriguingly, analysis of the reaction mixture prior to purification by LC-MS revealed the presence of a minor, doubly functionalized product with two furyl groups, and possessing only a chlorine atom, suggesting functionalization of one of the indigenous chlorines of 3. This compound was assigned as 17, and notably it can be isolated in 35% yield under conditions optimized for its formation. In this vein, we note that bromo-teicoplanin 7, under analogous conditions, may be converted to compound 18 or 19, where substitution of the typically less reactive (towards metal-catalyzed cross-coupling) C–Cl bond has occurred, rather than at the generally more reactive C–Br bond (Figure 8B). The unexpected high reactivity of the ring 2c position of 3 stimulated examination of the Pd-catalyzed cross coupling of native 3 under related conditions. Indeed, we found that compounds 20, 21 and 22 could be obtained in 43% (55% conv., Figure S24), 30% (79% conv., Figure S28) and 31% (58% conv., Figure S31) isolated yield, respectively (Figure 8C). We conclude at this stage that metalcatalyzed cross-coupling reactions of bromo-teicoplanins is possible in a site-selective manner, but we also recognize that normative hierarchies of haloarene reactivity are subject to case-specific study as molecular complexity alters reactivity.
Figure 8.

A) Cross-coupling of compounds 9; Conditions: a50 mol% Pd(OAc)2, 100 mol% Water-soluble-SPHOS, K2CO3 (10 equiv), Boronic acid (10 equiv), H2O:MeCN (2:1), 35 °C for 16 and 50 °C for 17. B) Cross-coupling of compounds 7. b50 mol% Pd(OAc)2, 100 mol% Water-soluble-SPHOS, K2CO3 (10 equiv), Boronic acid (10 equiv), H2O:MeCN (2:1), 35 °C 18 and 50 °C for 19. C) Cross-coupling of compounds 3. c100 mol% Pd(OAc)2, 100 mol% Water-soluble-SPHOS, K2CO3 (10 equiv), Boronic acid (10 equiv), H2O:MeCN (2:1), 50 °C. Conversions are not corrected for response factor.
The chemistry described above enabled direct synthesis of eleven previously unknown analogs of 3. Given that several exhibit conformational perturbations, and all possess altered functional group display on the teicoplanin scaffold, we elected to determine minimum inhibitory concentrations (MICs) for the new analogs as antibacterial agents. We tested the compounds against five bacterial strains, including methicillin-resistant S. aureus (MRSA) and vancomycin-resistant enterococcus (VRE; VanB exhibits vancomycin resistance, but it is teicoplanin susceptible; VanA is both vancomycin and teicoplanin resistant). As shown in Table 1, in comparison to control compounds (entry 1-3), the newly synthesized analogs 7, 9 and 10 (entries 4-6) are quite similar in potency to 3 against all five bacterial strains. In contrast, tribrominated teicoplanin A2-2 (14, entry 7) exhibits a decrease in activity with four of the five strains. It is perhaps related that we observed the most dramatic conformational perturbations with 14 relative to native teicoplanin A2-2, as exhibited by NMR spectroscopy.
| Entry | Compound | MSSAa,b | MRSAC | VSEd | VRE (VanB)e | VRE (VanA)f |
|---|---|---|---|---|---|---|
| 1 | Vancomycin | 0.5 | 1 | 2 | 16 | >64 |
| 2 | Teicoplanin | 0.5 | 0.5 | 0.25 | 0.25 | >64 |
| 3 | Teicoplanin A2-2 | 0.5 | 0.5 | 0.25 | 0.25 | >64 |
| 4 | 7 | 0.5 | 1 | 0.5 | 1 | >64 |
| 5 | 9 | 0.5 | 1 | 0.25 | 0.5 | >64 |
| 6 | 10 | 1 | 1 | 0.5 | 1 | >64 |
| 7 | 14 | 2 | 2 | 4 | 8 | >64 |
| 8 | 16 | 0.25 | 0.25 | 0.25 | 0.5 | >64 |
| 9 | 20 | 0.25 | 0.25 | 0.12 | 0.12 | 32 |
| 10 | 17 | 0.25 | 0.25 | 0.12 | 0.25 | >32 |
| 11 | 18 | 0.5 | 0.5 | 0.25 | 0.5 | >64 |
| 12 | 19 | 4 | 2 | 1 | 0.5 | 32 |
| 13 | 21 | 8 | 4 | 0.5 | 0.25 | 8 |
| 14 | 22 | 8 | 4 | 0.5 | 0.25 | 1 |
| 15 | Linezolid | 4 | 4 | 2 | 2 | 2 |
MIC values reported in μg/mL.
MSSA = methicillin-susceptible S. aureus, ATOC 29213.
MRSA = methicillin-resistant S. aureus, ATCC 43300.
VSE = vancomycin-susceptible enterococci, ATCC 29212.
VRE = vancomycin-resistant enterococci, ATCC 51299.
MMX 486.
See Supporting Information for additional details.
On the other hand, the analogs obtained through cross-coupling (entries 8-14) demonstrated comparable or increased potency against several of the bacterial strains, in comparison to vancomycin and teicoplanin. Compound 16, for example, with furyl substitution at the 3b-position, exhibited higher potency against the MRSA strain (entries 1-3 vs. 8). Relocation of furyl substituent from the 3b-position to the 2c-position (compound 20, entry 9) resulted in enhancement of activity against VRE strains (entries 1-3 vs. 9). Compound 17 (entry 10), with both 2c- and 3b-positions substituted with a furyl group, exhibits a similar activity profile in comparison to 16 and 20 (entry 8 and 9). Compound 18 (entry 11) possessing the 7f-bromine substituent and the 2c-furyl group also maintains an analogous profile. A striking and different profile was observed with compounds 19, 21 and 22 (entries 12-14). Substitution of the 2c-position of 3 with biphenyl functionality (compound 21) results in substantial activity against VRE (VanA) strain. Simultaneously, however, compound 19 exhibits a loss of potency when evaluated against the MSSA and MRSA strains. Compound 19 (entry 12), with a 7f-Br and a 2c-biphenyl functionality, also exhibits this trend. Compound 22, with 2c-octenyl substitution, exhibits the trend as well, while showing significant potency against the vancomcin- and teicoplanin-resistant strain (Van A, entries 1-3 vs. 14). These data are compared to antibacterial behaviors of the antibiotic Linezolid in entry 15. The unique behaviors of biphenyl-containing compounds 19, 21 and 22 may suggest change in the mechanism of action.1,43-45 The data presented in Table 1 demonstrate that altering the structure of teicoplanin with either bromination, or cross-coupling reactions of either brominated teicoplanins (7 and 9) or teicoplanin A2-2, itself (3), can lead to compounds with significant anti-bacterial activity against strains that exhibit vancomycin and teicoplanin resistance.
Conclusions
In summary, we have identified small molecule promoters that enable access to unique brominated forms of teicoplanin A2-2. The approach allows control over whether the 5,7-biaryl ring system, or the 1,3-biaryl ether sector of the natural product undergoes bromination. NMR studies facilitated assignments of the site of bromination, and analysis of conformational consequences. Metal-catalyzed cross-coupling reactions of brominated teicoplanin analogs and teicoplanin A2-2 itself have enabled access to further diversified compounds. These studies also unveil unexpected hierarchies of site-selectivity for cross-coupling reactions in complex molecular environments (C–Cl site over C–Br site). Evaluation of the antibacterial properties of the new compounds reveals both positive and negative modulation of activity against various strains of resistant bacteria. The activity of compound 22 may be particularly notable for its activity against the VanA-type strain of VRE. Chemistry-dependent, site-selective alteration of teicoplanin has thus led to unique compounds of altered and notable biological activity. Generally, this approach may allow for diversification and analysis of SAR for quite complex molecules.
Supplementary Material
ACKNOWLEDGMENT
Funding Sources We are grateful to the National Institutes of General Medical Sciences of the National Institute of Health (GM-068649) for support.
Footnotes
Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes The authors declare no competing financial interest.
Author Contributions The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
REFERENCES
- (1).Kahne D, Leimkuhler C, Lu W, Walsh C. Chem. Rev. 2005;105:425. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- (2).Jeya M, Moon H-J, Lee K-M, Kim I-W, Lee J-K. Curr. Pharm. Biotechnol. 2011;12:1194. doi: 10.2174/138920111796117382. [DOI] [PubMed] [Google Scholar]
- (3).Ashford P-A, Bew SP. Chem. Soc. Rev. 2012;41:957. doi: 10.1039/c1cs15125h. [DOI] [PubMed] [Google Scholar]
- (4).Li T-L, Liu Y-C, Lyu S-Y. Curr. Opin. Chem. Biol. 2012;16:170. doi: 10.1016/j.cbpa.2012.01.017. [DOI] [PubMed] [Google Scholar]
- (5).Payne DJ, Gwynn MN, Holmes DJ. Nat. Rev. Drug Discov. 2007;6:29. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- (6).Bardone MR, Paternoster M, Coronelli C. J. Antibiot. 1978;31:170. doi: 10.7164/antibiotics.31.170. [DOI] [PubMed] [Google Scholar]
- (7).Parenti F, Beretta G, Berti M, Arioli V. J. Antibiot. 1978;31:276. doi: 10.7164/antibiotics.31.276. [DOI] [PubMed] [Google Scholar]
- (8).Borghi A, Coronelli C, Faniuolo L, Allievi G, Pallanza R, Gallo GG. J. Antibiot. 1984;37:615. doi: 10.7164/antibiotics.37.615. [DOI] [PubMed] [Google Scholar]
- (9).Malabarba A, Goldstein BP. J. Antimicrob. Chemother. 2005;55:ii15. doi: 10.1093/jac/dki005. [DOI] [PubMed] [Google Scholar]
- (10).Guskey MT, Tsuji BT. Pharmacotherapy. 2010;30:80. doi: 10.1592/phco.30.1.80. [DOI] [PubMed] [Google Scholar]
- (11).James RC, Pierce JG, Okano A, Xie J, Boger DL. ACS Chem. Bio. 2012;7:797. doi: 10.1021/cb300007j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Evans DA, Wood MR, Trotter BW, Richardson TI, Barrow JC, Katz JL. Angew. Chem, Int. Ed. 1998;37:2700. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2700::AID-ANIE2700>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- (13).Nicolaou KC, Mitchell HJ, Jain NF, Bando T, Hughes R, Winssinger N, Natarajan S, Koumbis AE. Chem– Eur. J. 1999;5:2648. [Google Scholar]
- (14).Boger DL, Kim SH, Miyazaki S, Strittmatter H, Weng J-H, Mori Y, Rogel O, Castle SL, McAtee JJ. J. Am. Chem. Soc. 2000;122:7416. [Google Scholar]
- (15).Boger DL. Med. Res. Rev. 2001;21:356. doi: 10.1002/med.1014. [DOI] [PubMed] [Google Scholar]
- (16).Boger DL, Kim SH, Mori Y, Weng J-H, Rogel O, Castle SL, McAtee JJ. J. Am. Chem. Soc. 2001;123:1862. doi: 10.1021/ja003835i. [DOI] [PubMed] [Google Scholar]
- (17).Evans DA, Katz JL, Peterson GS, Hintermann T. J. Am. Chem. Soc. 2001;123:12411. doi: 10.1021/ja011943e. [DOI] [PubMed] [Google Scholar]
- (18).Xie J, Okano A, Pierce JG, James RC, Stamm S, Crane CM, Boger DL. J. Am. Chem. Soc. 2011;134:1284. doi: 10.1021/ja209937s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Okano A, James RC, Pierce JG, Xie J, Boger DL. J. Am. Chem. Soc. 2012;134:8790. doi: 10.1021/ja302808p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Losey HC, Jiang J, Biggins JB, Oberthür M, Ye X-Y, Dong SD, Kahne D, Thorson JS, Walsh CT. Chem. Biol. 2002;9:1305. doi: 10.1016/s1074-5521(02)00270-3. [DOI] [PubMed] [Google Scholar]
- (21).Pinter G, Batta G, Keki S, Mandi A, Komaromi I, Takacs-Novak K, Sztaricskai F, Roth E, Ostorhazi E, Rozgonyi F, Naesens L, Herczegh P. J. Med. Chem. 2009;52:6053. doi: 10.1021/jm900950d. [DOI] [PubMed] [Google Scholar]
- (22).Liu Y-C, Li Y-S, Lyu S-Y, Hsu L-J, Chen Y-H, Huang Y-T, Chan H-C, Huang C-J, Chen G-H, Chou C-C, Tsai M-D, Li T-L. Nat. Chem. Biol. 2011;7:304. doi: 10.1038/nchembio.556. [DOI] [PubMed] [Google Scholar]
- (23).Jeya M, Moon H-J, Lee K-M, Kim I-W, Lee J-K. Curr. Pharm. Biotechnol. 2011;12:1194. doi: 10.2174/138920111796117382. [DOI] [PubMed] [Google Scholar]
- (24).Losey HC, Peczuh MW, Chen Z, Eggert US, Dong SD, Pelczer I, Kahne D, Walsh CT. Biochemistry. 2001;40:4745. doi: 10.1021/bi010050w. [DOI] [PubMed] [Google Scholar]
- (25).Jung H-M, Kim S-Y, Moon H-J, Oh D-K, Lee J-K. Appl. Microbiol. Biotechnol. 2007;77:789. doi: 10.1007/s00253-007-1221-4. [DOI] [PubMed] [Google Scholar]
- (26).Jung H-M, Jeya M, Kim S-Y, Moon H-J, Kumar SR, Zhang Y-W, Lee J-K. Appl. Microbiol. Biotechnol. 2009;84:417. doi: 10.1007/s00253-009-2107-4. [DOI] [PubMed] [Google Scholar]
- (27).Lewis CA, Miller SJ. Angew. Chem, Int. Ed. 2006;118:5744. [Google Scholar]
- (28).Fowler BS, Laemmerhold KM, Miller SJ. J. Am. Chem. Soc. 2012;134:9755. doi: 10.1021/ja302692j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Pathak TP, Miller SJ. J. Am. Chem. Soc. 2012;134:6120. doi: 10.1021/ja301566t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Neumann CS, Fujimori DG, Walsh CT. Chem. Biol. 2008;15:99. doi: 10.1016/j.chembiol.2008.01.006. [DOI] [PubMed] [Google Scholar]
- (31).Dong C, Flecks S, Unversucht S, Haupt C, van Pée K-H, Naismith JH. Science. 2005;309:2216. doi: 10.1126/science.1116510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Nakama Y, Yoshida O, Yoda M, Araki K, Sawada Y, Nakamura J, Xu S, Miura K, Maki H, Arimoto H. J. Med. Chem. 2010;53:2528. doi: 10.1021/jm9017543. [DOI] [PubMed] [Google Scholar]
- (33).Heald SL, Mueller L, Jeffs PW. J. Magn. Reson. 1987;72:120. [Google Scholar]
- (34).Hunt AH, Molloy RM, Occolowitz JL, Marconi GG, Debono M. J. Am. Chem. Soc. 1984;106:4891. [Google Scholar]
- (35).Barna JCJ, Williams DH, Stone DJM, Leung TWC, Doddrell DM. J. Am. Chem. Soc. 1984;106:4895. [Google Scholar]
- (36).Nitanai Y, Kikuchi T, Kakoi K, Hanamaki S, Fujisawa I, Aoki K. J. Mol. Bio. 2009;385:1422. doi: 10.1016/j.jmb.2008.10.026. [DOI] [PubMed] [Google Scholar]
- (37).Economou NJ, Nahoum V, Weeks SD, Grasty KC, Zentner IJ, Townsend TM, Bhuiya MW, Cocklin S, Loll PJ. J. Am. Chem. Soc. 2012;134:4637. doi: 10.1021/ja208755j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Denmark SE, Burk MT. Proc. Nat. Acad. Sci. 2010;107:20655. doi: 10.1073/pnas.1005296107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Malabarba A, Trani A, Tarzia G, Ferrari P, Pallanza R, Berti M. J. Med. Chem. 1989;32:783. doi: 10.1021/jm00124a010. [DOI] [PubMed] [Google Scholar]
- (40).Nieto M, Perkins HR. Biochem. J. 1971;124:845. doi: 10.1042/bj1240845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Westwell MS, Gerhard U, Williams DH. J. Antibiot. 1995;48:1292. doi: 10.7164/antibiotics.48.1292. [DOI] [PubMed] [Google Scholar]
- (42).Roy AD, Grüschow S, Cairns N, Goss RJM. J. Am. Chem. Soc. 2010;132:12243. doi: 10.1021/ja1060406. [DOI] [PubMed] [Google Scholar]
- (43).Ge M, Chen Z, Russell H, Onishi, Kohler J, Silver LL, Kerns R, Fukuzawa S, Thompson C, Kahne D. Science. 1999;284:507. doi: 10.1126/science.284.5413.507. [DOI] [PubMed] [Google Scholar]
- (44).Goldman RC, Baizman ER, Longley CB, Branstrom AA. FEMS Microbio. Lett. 2000;183:209. doi: 10.1111/j.1574-6968.2000.tb08959.x. [DOI] [PubMed] [Google Scholar]
- (45).Chen L, Walker D, Sun B, Hu Y, Walker S, Kahne D. Proc. Nat. Acad. Sci. 2003;100:5658. doi: 10.1073/pnas.0931492100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.