

Abstract
In many types of tumours, especially pancreatic adenocarcinoma, miR-301a is over-expressed. This over-expression results in negative regulation of the target gene of miR-301a, the nuclear factor-κB (NF-κB) repressing factor (NKRF), increasing the activation of NF-κB and production of NF-κB-responsive pro-inflammatory cytokines such as interleukin-8, interferon-β, nitric oxide synthase 2A and cytochrome oxidase subunit 2 (COX-2). However, in immune cells, mechanisms that regulate miR-301a have not been reported. Similar to tumour cells, Toll-like receptor (TLR) -activated macrophages produce NF-κB-responsive pro-inflammatory cytokines. Therefore, it is of considerable interest to determine whether miR-301a regulates the secretion of cytokines by immune cells. In the present study, we demonstrate that the expression of miR-301a was decreased in TLR-triggered macrophages. Through targeting NKRF, miR-301a affected the activity of NF-κB and the expression of pro-inflammatory genes downstream of NF-κB such as COX-2, prostaglandin E2 and interleukin-6. In addition, when lipopolysaccharide-treated macrophages were simultaneously stimulated with trichostatin A, an inhibitor of histone deacetylases, the expression of miR-301a increased, whereas NKRF and pro-inflammatory cytokine expression decreased. However, further investigation revealed that there was no correlation between the induction of miR-301a and the inhibitory effect of trichostatin A on lipopolysaccharide-induced gene expression in macrophages. In summary, our study indicates a new mechanism by which miR-301a regulates inflammatory cytokine expression in macrophages, which may clarify the regulatory role of microRNAs in immune-mediated inflammatory responses.
Keywords: histone deacetylase inhibitor, inflammatory response, macrophage, miR-301a, nuclear factor-κB-repressing factor, Toll-like receptor
Introduction
Toll-like receptors (TLRs), are important pattern-recognition receptors and are widely expressed in antigen-presenting cells, especially on the surface of macrophages and dendritic cells. The TLRs recognise conserved structural motifs of pathogenic microorganisms (also called pathogen-associated molecular patterns, PAMPs), such as lipopolysaccharide (LPS), flagellin, double-stranded RNA, CpG motifs and mannose in the cell wall of yeast. The innate immune response is rapidly triggered by the TLR-mediated recognition of PAMPs, and this pathway is an integral part of host defence against microorganisms. The innate immune response requires the rapid and co-ordinated control of multiple inflammatory genes in immune cells such as macrophages and dendritic cells.1
The early immune inflammatory response to PAMPs is achieved mainly through activation of the nuclear factor-κB (NF-κB) signalling pathway.2 Once NF-κB signalling has been activated, the NF-κB-p65/p50 heterodimer translocates to the nucleus and induces the expression of various inflammatory genes,2 including those encoding pro-inflammatory cytokines, cell surface receptors, transcription factors, neuropeptides, chemokines and adhesion molecules. The localisation and activity of NF-κB in cells is subject to strict regulation at multiple levels by a series of transcriptional and post-translational events.3,4 The ongoing activation of NF-κB in patients may lead to microbial infection, autoimmune diseases, developmental abnormalities, tumours and other pathological phenomena, which suggests that inhibition of NF-κB activity may be an anti-inflammatory therapy.
MicroRNAs (miRNAs) are a class of highly conserved non-coding small RNAs. The commonly accepted mechanism of miRNA regulation is that the ‘seed region’ (two to eight nucleotides at the 5′ end) of an miRNA is complementary to the 3′ untranslated region (3′-UTR) of an mRNA, resulting in either translation inhibition or mRNA degradation.5 It has been demonstrated that miRNAs affect a wide variety of physiological and pathological processes, including cell differentiation and development, metabolism, immunity and inflammation.6 As miRNAs are small molecules without antigenic properties, they have been considered effective targets with potential clinical applications. Recent research has revealed that miRNAs may play both a negative and a positive modulatory role in the innate immune response, especially in the regulation of NF-κB signalling.6–9 It has been confirmed that miR-146, let-7b, miR-124a, miR-155, miR-125b, miR-220, miR-199b and miR-301a down-regulate the expression of different proteins in the NF-κB pathway in immune cells, thereby affecting the production of inflammatory mediators such as interleukin-1β (IL-1β) and tumour necrosis factor-α (TNF-α).
MiR-301a negatively regulates its target gene, NF-κB repressing factor (NKRF), resulting in the induction of NF-κB. When NKRF was first discovered, it was thought to interact with the specific negative regulatory elements (NREs) of some genes [IL-8, interferon-β (IFN-β) and nitric oxide synthase 2A (NOS2A)] to mediate the transcriptional activity of NF-κB.10–12 However, more recent evidence suggests that NKRF also broadly inhibits the expression of other NF-κB-dependent genes, such as those for matrix metalloproteinase 2 (MMP2) and cytochrome oxidase subunit 2 (COX-2), in a non-NRE-dependent manner.13 Because miR-301a is specifically over-expressed in a number of cancers, including pancreatic adenocarcinoma13,14 and hepatocellular carcinoma,15 it is considered to be a potent activator of NF-κB.13 Moreover, in a mouse model of pancreatic cancer, the inhibition of miR-301a or the up-regulation of NKRF results in attenuated NF-κB signalling and tumour growth.13 Similar to tumour cells, immune cells also produce NF-κB-responsive cytokines. Therefore, we are interested to determine whether miR-301a affects the expression of such cytokines by regulating NKRF in the innate immune response. Here, we observed that miR-301a expression was significantly decreased in murine macrophages upon TLR ligand stimulation. MiR-301a over-expression directly targeted NKRF and increased NF-κB activity, resulting in the increased expression of cytokines such as COX-2 and IL-6 in macrophages. Conversely, decreased miR-301a expression suppressed pro-inflammatory cytokine expression in TLR-triggered macrophages. Unexpectedly, when LPS-treated macrophages were stimulated with trichostatin A (TSA), an inhibitor of histone deacetylases, the expression of miR-301a increased, but the production of pro-inflammatory cytokines decreased, indicating that, in macrophages, TSA represses the expression of LPS-induced cytokines independently of miR-301a.
Materials and methods
Mice and reagents
C57BL/6J mice were obtained from the Institute of Zoological Sciences at the Chinese Academy of Medical Sciences (Beijing, China). All animal procedures were approved by the committee on the use and care of animals at the Chinese Academy of Medical Sciences. Animals were housed in specific pathogen-free conditions. Lipopolysaccharide (from Escherichia coli serotype 0111:B4), CpG ODN and poly (I:C) were as previously described.16 The TSA was purchased from Sigma-Aldrich (St Louis, MO). The antibody against IκBα phosphorylated at Ser32/36 (5A5) was purchased from Cell Signaling Technology (Beverly, MA). The polyclonal anti-NKRF antibody was obtained from Proteintech (Chicago, IL); anti-COX-2 and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The si-NKRF RNA (sc-72276) was obtained from Santa Cruz Biotechnology.
Cell culture, treatment and transfection
RAW264.7 cells, a mouse macrophage cell line, were obtained from the American Type Culture Collection (Manassas, VA) and were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum. Thioglycollate-elicited mouse peritoneal macrophages were obtained and cultured in endotoxin-free RPMI-1640 medium supplemented with 10% fetal calf serum (Invitrogen, Carlsbad, CA).17 The miR-301a mimics (dsRNA oligonucleotides) and inhibitors (single-stranded chemically modified oligonucleotides) (Life Technologies Corporation, Shanghai, China) were used for the over-expression and inhibition, respectively, of miR-301a in mouse peritoneal macrophages. Negative control mimics or inhibitors were transfected as matched controls. Cells were transfected with RNAs using INTERFERin (Polyplus-Transfection SA, Illkirch, France) according to the manufacturer's instructions.
Treatment with TSA in macrophages
The miR-301a inhibitors or control inhibitors were used to knock down the expression of miR-301a in macrophages. Macrophages were first transfected with miR-301a inhibitors or control inhibitors to knock down miR-301a expression. After overnight incubation, cells were cultured for 48 hr with or without 3 nmol/l of TSA (Sigma-Aldrich). Six hours before cell harvest, 100 ng/ml LPS was added to the cell cultures. The transcription level of miR-301a was detected by quantitative PCR (qPCR), and the expression of p-IκBα, COX-2 and NKRF protein was detected by Western blot.
Measurement of miRNA and mRNA expression
Total RNA was extracted from each cell sample using an mirVana miRNA Isolation Kit (Ambion Inc, Austin, TX) according to the manufacturer's instructions. The Taqman miRNA assay system (Applied Biosystems, Foster City, CA) was used according to the manufacturer's instructions to quantitatively detect the expression of miR-301a. The relative expression level of miRNA was normalised to that of the internal control U6. The SYBR Green PCR Master Mix (Toyobo, Osaka, Japan) was used to analyse the expression of mRNA. The quantification of miRNA and mRNA by qPCR was performed using an ABI 7300HT thermocycler at 95° for 10 min, followed by 40 cycles of 95° for 15 seconds and 60° for 1 min. Reactions were performed in triplicate, with mouse GAPDH as an internal control. Cycle threshold values were converted to relative gene expression levels using the 2−ΔΔCT method.18
Luciferase reporter assay
293T cells were co-transfected with the miR-301a mimics or inhibitors, pMIR-REPORT-NKRF 3′UTR plasmid, or pMIR-REPORT-NKRF-mutant 3′UTR plasmid13, pMIR-REPORT and pTK-RL plasmid at a final plasmid concentration of 1 μg/well. Forty-eight hours after transfection, the cells were lysed with lysis buffer. A luciferase activity assay was performed using a Dual-luciferase™ Reporter System (Promega, Madison, WI) according to the manufacturer's instructions. Light units from luciferase activity were measured using a microplate luminometer (Berthold Centro LB 960). Data representing firefly luciferase activity were normalised against Renilla luciferase activity.
Western blot analysis
Total cell lysates were prepared and subjected to SDS–PAGE, transferred onto a PVDF membrane, and blotted as previously described.17
Cytokine detection
The secretion of prostaglandin E2 (PGE2) and IL-6 into the cell medium was measured using ELISA kits (R&D Systems, Minneapolis, MN).
Statistical analysis
All of the data were obtained from at least three independent experiments. The data are expressed as the mean values ± SD and were compared between two groups using Student's t-test. Statistical significance was defined as a P-value of < 0·05.
Results
Stimulation with LPS, CpG or poly (I:C) significantly reduces the expression of miR-301a in macrophages
To determine whether miR-301a is involved in the regulation of TLR-triggered responses in macrophages, we measured the expression of miR-301a in LPS-, CpG- or poly (I:C)-stimulated macrophages. As shown in Fig. 1(a), miR-301a was expressed in a concentration-dependent manner in mouse peritoneal macrophages stimulated by LPS for 24 hr. At 10 ng/ml LPS, miR-301a was slightly decreased compared with the control group; with 10–100 ng/ml LPS, miR-301a sharply decreased to a point at which it reached a plateau. Furthermore, miR-301a expression decreased with time, reaching its lowest level of expression (approximately 25% of the level observed in the control group) at 6–12 hr and then increasing slightly over the next 12 hr (Fig. 1b). Similar results were obtained when macrophages were treated with CpG or poly (I:C): miR-301a levels had decreased significantly at 6 hr, reaching levels of 20% and 25% of the control group, respectively (Fig. 1c). Moreover, a similar pattern of miR-301a expression was observed using RAW264.7 cells (Fig. 1d–f). The data above suggest that miR-301a may be a regulator of TLR signalling pathways in macrophages.
Figure 1.

Down-regulation of miR-301a expression in primary macrophages and RAW264.7 cells stimulated with Toll-like receptor (TLR) agonists. Mouse peritoneal macrophages (a–c) and RAW264.7 cells (d–f) were stimulated with different concentrations of lipopolysaccharide (LPS) for 24 hr (a and d) or at the indicated time-points with 100 ng/ml LPS (b and e), and 10 μg/ml CpG or poly (I:C) for 24 hr (c and f). The expression of miR-301a was measured using quantitative PCR and normalised to the expression of U6. Data are presented as the mean ± SD (n = 3) of three independent experiments. *P < 0·05; **P < 0·01.
NKRF is a target of miR-301a in macrophages
As previously described, miR-301a regulates NF-κB activity by targeting NKRF in tumour cells13. To test whether miR-301a acts similarly in macrophages, we constructed a reporter plasmid by cloning the mouse NKRF 3′-UTR or mutant NKRF 3′-UTR into the pMIR-REPORT™ luciferase vector. In co-transfection experiments with the reporter plasmids and miR-301a mimics in 293T cells, luciferase activity was markedly decreased in cells transfected with the NKRF 3′-UTR vector compared with the cells transfected with the mutant NKRF 3′-UTR and pMIR-REPORT empty plasmid, indicating that NKRF could be targeted by miR-301a (Fig. 2a). In addition, in RAW264.7 cells, transfection with miR-301a mimics down-regulated NKRF protein expression, whereas transfection with miR-301a inhibitors up-regulated NKRF protein expression (Fig. 2b), suggesting that miR-301a negatively regulates endogenous mouse NKRF expression in macrophages.
Figure 2.
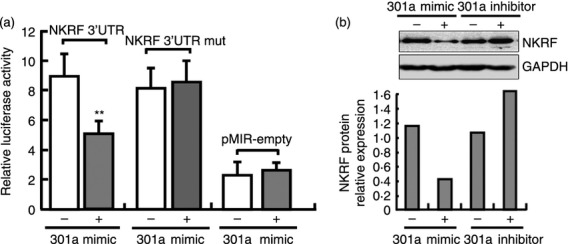
MiR-301a targets mouse nuclear factor-κB (NF-κB) repressing factor (NKRF). (a) 293T cells were co-transfected with the NKRF-3′ untranslated region (3′UTR) reporter plasmid, the mutant NKRF-3′UTR reporter plasmid (NFRF-3′UTR mut) or the empty pMIR-REPORT plasmid and the pTK-RL plasmid, together with miR-301a mimics or inhibitors. After 24 hr, firefly luciferase activity was measured and normalised to Renilla luciferase activity. (b) RAW264.7 cells were transfected with mimics or inhibitors of miR-301a and their controls. After 48 hr, NKRF protein expression at 6 hr after lipopolysaccharide (LPS) stimulation was detected by Western blot. The glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as a loading control, and the densitometry analysis is shown. Data are presented as the mean ± SD (n = 3) of three independent experiments. *P < 0·05; **P < 0·01.
NKRF inhibits NF-κB activation and the production of IL-6 in LPS-stimulated macrophages
Previous work has demonstrated that NKRF plays an essential role in the NF-κB signalling pathway, which leads to the expression of pro-inflammatory cytokines in various cell lines and tissues.10–12 To test whether NKRF performs the same function in macrophages, RAW264.7 cells were transfected with NKRF-specific small interfering RNA (si-NKRF). As shown in Fig. 3(a,b), si-NKRF down-regulated NKRF expression at both the mRNA and the protein level. We then examined the effects of the knockdown of NKRF in RAW264.7/NF-κB-RE cells. RAW264.7/NF-κB-RE cells were stably transfected with the plasmid pGL4.32(luc2P/NF-κB-RE/Hygro), which contained five copies of an NF-κB response element (NF-κB-RE) that drives the transcription of the luciferase reporter gene luc2P. As shown in Fig. 3(c), luciferase activity was increased in a dose-dependent manner in the LPS-stimulated macrophages transfected with si-NKRF from 10 to 100 nmol/l, suggesting that NKRF significantly inhibits the activation of NF-κB in an NRE-independent manner in TLR-triggered macrophages. Then, we tested whether NKRF regulates the production of NF-κB-dependent cytokines. As shown in Fig. 3(d–g), si-NKRF markedly enhanced the expression of COX-2 and IL-6 in LPS-stimulated macrophages, indicating that the NKRF was involved in the negative regulation of COX-2 and IL-6 in macrophages.
Figure 3.
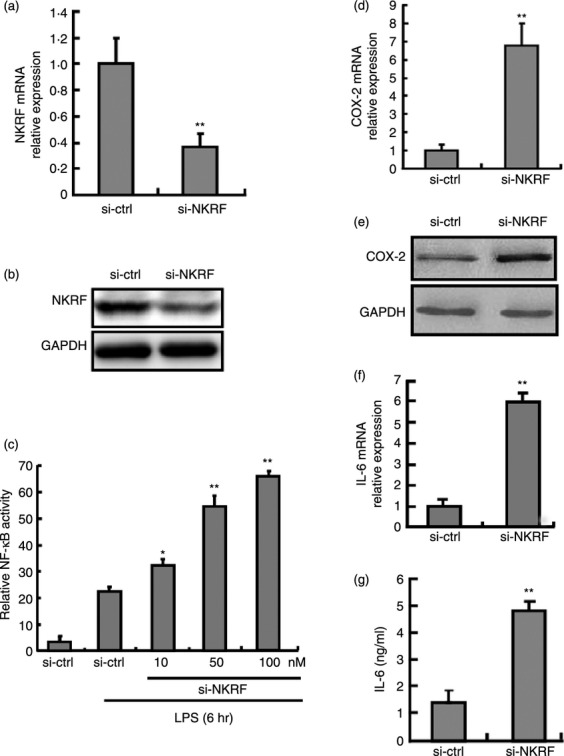
Knockdown of nuclear factor-κB (NF-κB) repressing factor (NKRF) inhibits lipopolysaccharide (LPS) -induced NKRF expression, NF-κB activity, the expression of cytochrome oxidase subunit 2 (COX-2) and interleukin-6 (IL-6). (a, b) RAW264.7 cells were transfected with si-NKRF or control small interfering (si) RNA at a final concentration of 100 nmol/l (M). After 48 hr, the cells were stimulated with LPS for 6 hr, and then NKRF expression was detected by quantitative PCR (qPCR) (a) and Western blot assay (b). (c) RAW264.7 cells stably transfected with pGL4.32[luc2P/NF-κB-RE/Hygro] were transfected with si-NKRF for 48 hr. After the cells were stimulated with LPS (100 ng/ml) for 6 hr, firefly luciferase activity was measured and was normalised to Renilla luciferase activity. (d, e) The mRNA and protein levels of COX-2 were determined by qPCR and Western blot. (f, g) The mRNA and protein levels of IL-6 were analysed by qPCR (f) and ELISA (g) in LPS-treated macrophages. Data are presented as the mean ± SD (n = 3) of three independent experiments. *P < 0·05; **P < 0·01.
MiR-301a enhances NF-κB activation and inflammatory genes in TLR-triggered macrophages
To further determine the consequences of miR-301a down-regulation in macrophages, RAW264.7 cells were transfected with miR-301a mimics or miR-301a inhibitors. The results showed that miR-301a mimics increased NF-κB activation twofold in RAW264.7/NF-κB-RE cells after LPS stimulation for 6 hr (Fig. 4a). Furthermore, miR-301a mimics markedly increased the expression of COX-2 and IL-6 (Fig. 4b–e) and enhanced the production of PGE2, which is a protein downstream of COX-2 in the arachidonic acid pathway (Fig. 4d). The above results were consistent with the effects of si-NKRF on inflammatory gene expression. Conversely, COX-2 and IL-6 expression was decreased in RAW264.7 cells treated with miR-301a inhibitors (Fig. 4a–f). These results indicate that the down-regulation of miR-301a negatively regulates COX-2 and IL-6 expression by targeting NKRF in TLR-triggered macrophages.
Figure 4.
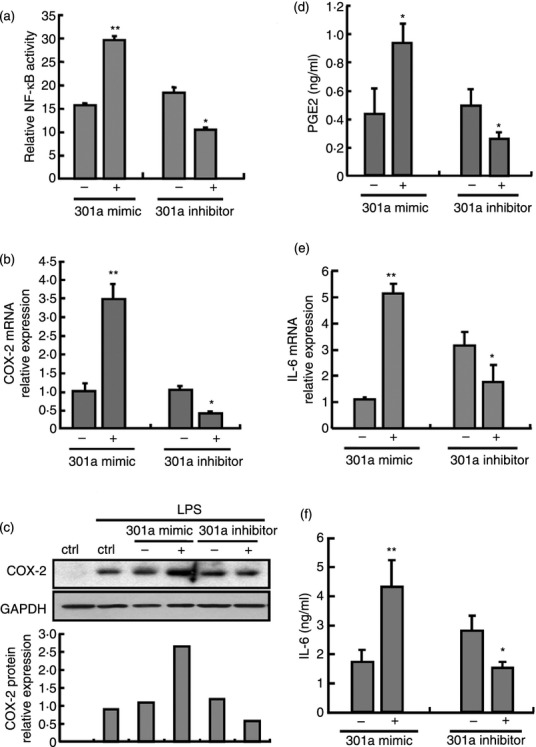
MiR-301a regulates nuclear factor-κB (NF-κB) activity, the expression of cytochrome oxidase 2 (COX-2) and interleukin-6 (IL-6) in lipopolysaccharide (LPS) -triggered macrophages. RAW264.7 cells were transfected with mimics or inhibitors of miR-301a and their controls. After 24 hr, cells were stimulated with or without LPS (100 ng/ml) for 6 hr. NF-κB activity was measured by Renilla luciferase activity (a). Messenger RNA levels of COX-2 (b) and IL-6 (e) were detected by quantitative PCR. The protein expression of COX-2 was determined by Western blot (c). Supernatants were collected to measure PGE2 (d) and IL-6 (f) by ELISA. Data are presented the mean ± SD (n = 3) of three independent experiments. *P < 0·05; **P < 0·01.
MiR-301a does not regulate the expression of TSA-suppressed inflammatory cytokines in macrophages
It has previously been demonstrated that TSA suppresses the production of various pro-inflammatory cytokines, such as TNF-α, IL-1 and IL-6.19 Because miR-301a is involved in the regulation of inflammatory cytokine expression, we hypothesised that miR-301a would also play a role in TSA-regulated inflammatory cytokine production. Peritoneal macrophages were transfected with miR-301a inhibitors following TSA stimulation for 48 hr and LPS stimulation for 6 hr. As shown in Fig. 5(a), TSA induced the expression of miR-301a by 20-fold compared with LPS-treatment alone, whereas the expression of NKRF mRNA and protein was significantly reduced (Fig. 5b,c). To determine the direct causal relationship between miR-301a and NKRF in TSA-treated cells, the peritoneal macrophages were transfected with miR-301a inhibitors or with controls and were then treated with TSA. As shown in Fig. 5(b,c), miR-301a inhibitors significantly increased the expression of NKRF mRNA and protein, indicating that the ability of TSA to inhibit NKRF expression was dependent on the up-regulation of miR-301a expression in macrophages. Then, we tested whether miR-301a could modulate the expression of inflammatory cytokines in TSA-treated macrophages. As shown in Fig. 5(d,e), TSA significantly inhibited the LPS-induced expression of COX-2 and IL-6 (TSA-LPS-treated cells) compared with LPS-treated cells. However, miR-301a knockdown had no effect on the expression of COX-2 and IL-6 in TSA-LPS-miR-301a inhibitor-treated cells compared with TSA-LPS-treated cells. To further analyse the transcriptional regulation of these two genes, we examined the activation of NF-κB in four different groups of cells (control, LPS-, TSA-LPS- and TSA-LPS-miR-301a inhibitor-treated macrophages). The phosphorylation of IκBα in the last three groups of cells was significantly higher than that of the control group, but there was no significant difference among these three exposures. In contrast to the above results for the expression of COX-2 and IL-6, the activity of NF-κB was not consistent with that of NF-κB-dependent genes among the four types of cells (Fig. 5f). Therefore, we concluded that miR-301a was not involved in the regulation of the NF-κB responsive inflammatory genes that were blocked by TSA in TLR-triggered macrophages.
Figure 5.
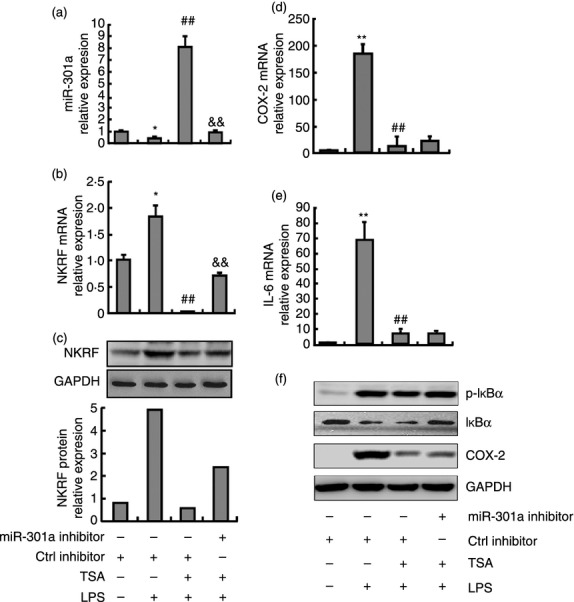
The effect of trichostatin A (TSA) on gene expression in lipopolysaccharide (LPS) -treated macrophages. Mouse peritoneal macrophages were transfected with the miR-301a inhibitor and its control. After overnight incubation, macrophages were treated with TSA for 48 hr, and then treated with media or LPS for 6 hr. The mRNA levels of miR-301a (a), nuclear factor-κB (NF-κB) repressing factor (NKRF) (b), cytochrome oxidase 2 (COX-2) (d) and interleukin-6 (IL-6) (e) were detected by quantitative PCR. The protein levels of NKRF (c), COX-2 and phosphorylated (p-) IκBα (f) were determined by Western blot. Data are presented as the mean ± SD (n = 3) of three independent experiments. *P < 0·05 and **P < 0·01 compared with neither LPS nor TSA stimulation; ##, P < 0·01 compared with LPS but without TSA stimulation; &&, P < 0·01 compared with both LPS and TSA stimulation.
Discussion
Our study first confirmed that decreased miR-301a expression resulted in a suppressed TLR-dependent innate immune response by macrophages. The expression of miR-301a decreased in macrophages stimulated with TLR activators [LPS, CpG and poly (I: C)]. Decreased miR-301a resulted in increased NKRF expression, thereby inhibiting the activation of NF-κB and resulting in the suppressed expression of pro-inflammatory genes.
Previous work has shown that miR-301a is over-expressed in pancreatic,13,14 hepatocellular,15 small cell lung20 and breast cancers.21 The miR-301a has also been shown to down-regulate NKRF, activate NF-κB target genes (MMP2, COX-2, MYC, VEGFC and FAM33A) and increase xenograft tumour growth. 13 NKRF is a nuclear repressor that is detectable in most human cell lines, indicating that NKRF is abundant and available to participate in transcriptional regulation of target genes. In the regulation of NF-κB, NKRF works through two primary molecular mechanisms: one is NRE dependent, and the other is NRE independent. Some NF-κB-responsive genes (e.g. IL-8, IFN-β and inducible nitric oxide synthase)10,11,12 contain NRE in their promoter regions. NKRF represses the basal transcription of these genes by targeting their NREs; other promoters of NF-κB-responsive genes (MMP2, MYC and COX-2) lack NRE sequences.13 NKRF inhibits these genes by interacting with RelA protein, which can bind to DNA.13 In non-cancerous cell lines such as 293T cells, NKRF also modulates the activation of NF-κB and the expression of NF-κB-responsive genes (NOS2A, IL8, IFNB and FAM33A).13 It has been proposed that NKRF functions in a similar way to IκBα in regulating NF-κB, i.e. by binding p65 in the cytoplasm, which then inhibits the nuclear translocation of NF-κB. In immune cells, we demonstrate that NKRF affects the binding of NF-κB and DNA in an NRE-independent manner in LPS-treated macrophages, thereby regulating the expression levels of the pro-inflammatory mediators COX-2 and IL-6.
Because miR-301a regulates NF-κB-related gene expression by targeting NKRF in both tumour and 293 cells, we deduced that miR-301a may serve a similar regulatory function in immune cells. As expected, we found that miR-301a is involved in the regulation of COX-2, PGE2 and IL-6 expression in macrophages by targeting the NKRF gene, and so blocking the ability of NF-κB to bind DNA.
Histone deacetylases (HDACs) are widely expressed in immune tissues and cells and participate in the differentiation and maturation of a variety of immune cells, including T lymphocytes,22 dendritic cells23 and monocytes-macrophages24. HDAC inhibitors (HDACi, such as TSA) have anti-inflammatory effects in a number of animal models of several inflammatory diseases, including systemic lupus erythematosus, septic shock, rheumatoid arthritis and ulcerative colitis.25–28 The HDACi generally reduce the production of pro-inflammatory cytokines such as IL-6, IL-12, TNF-α and IFN-β,25,29–31 which implies that they act by inhibiting NF-κB activation.
Because miR-301a modulates NF-κB transcriptional activity and regulates the expression of NF-κB-dependent genes, we investigated whether miR-301a also plays an important role in TSA-suppressed inflammatory responses. In LPS-TSA-treated macrophages, the expression of miR-301a was significantly induced, NKRF was down-regulated, and pro-inflammatory genes (COX-2 and IL-6) were suppressed by TSA. However, further experiments indicated that the inhibitory effect of TSA on the products of pro-inflammatory genes is unrelated to miR-301a expression or activity in TLR ligand-stimulated macrophages.
Trichostatin A inhibits multiple HDAC enzymes. Owing to the pleiotropic effects of different HDAC enzymes in innate immune functions, broad-spectrum HDACi (TSA) most probably suppresses the expression of cytokine genes in macrophages through multiple mechanisms. For example, HDAC1 and HDAC3 specifically mediated the repressive effects of the NF-κB homodimer on promoters of inflammatory genes.32–35 However, HDACi reduced the TLR-mediated recruitment of NF-κB p65 to inflammatory gene promoters.25,30 Hence, the biological function of HDACi in the TLR-mediated activation of NF-κB remains unclear. In addition, HDACi are required for nucleosome remodelling and for the recruitment of Rel family transcription factors, a subset of TLR-target genes. Because HDACi affect chromatin remodelling, the binding of RelA to the promoter and its transcriptional activation were abrogated.30 Therefore, it is perhaps not surprising that although miR-301a is an NF-κB activator, it has no effect on the expression of NF-κB-dependent cytokines in macrophages treated with both LPS and TSA in combination.
Taken together, our findings identify a new miRNA that is responsible for the negative regulation of NF-κB activation and the expression of downstream pro-inflammatory genes in TLR-triggered macrophages. This study may indicate a new molecular strategy for the treatment of inflammatory diseases by knockdown of miR-301a. Given that miR-301a inhibition or NKRF up-regulation in cancer cells led to reduced NF-κB target gene expression and attenuated xenograft tumour growth, the modulation of miR-301a may provide a therapeutic option for controlling the inflammatory response with a low risk of cancer.
Acknowledgments
This work was supported by a grant from the National Natural Science Foundation of China (Grant No. 81001315).
Disclosure
The authors have declared that no competing interests exist.
References
- 1.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 2.Vallabhapurapu S, Karin M. Regulation and function of NF-κB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 3.Hoffmann A, Baltimore D. Circuitry of nuclear factor κB signaling. Immunol Rev. 2006;210:171–86. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 4.Li Q, Verma IM. NF-κB regulation in the immune system. Nat Rev Immunol. 2002;2:725–34. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 5.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 6.O'Neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol. 2011;11:163–75. doi: 10.1038/nri2957. [DOI] [PubMed] [Google Scholar]
- 7.Boldin MP, Baltimore D. MicroRNAs, new effectors and regulators of NF-κB. Immunol Rev. 2012;246:205–20. doi: 10.1111/j.1600-065X.2011.01089.x. [DOI] [PubMed] [Google Scholar]
- 8.Wendlandt EB, Graff JW, Gioannini TL, McCaffrey AP, Wilson ME. The role of microRNAs miR-200b and miR-200c in TLR4 signaling and NF-κB activation. Innate Immun. 2012;18:846–55. doi: 10.1177/1753425912443903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma X, Becker Buscaglia LE, Barker JR, Li Y. MicroRNAs in NF-κB signaling. J Mol Cell Biol. 2011;3:159–66. doi: 10.1093/jmcb/mjr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nourbakhsh M, Hauser H. Constitutive silencing of IFN-β promoter is mediated by NRF (NF-κB-repressing factor), a nuclear inhibitor of NF-κB. EMBO J. 1999;18:6415–25. doi: 10.1093/emboj/18.22.6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nourbakhsh M, Kalble S, Dorrie A, Hauser H, Resch K, Kracht M. The NF-κb repressing factor is involved in basal repression and interleukin (IL)-1-induced activation of IL-8 transcription by binding to a conserved NF-κb-flanking sequence element. J Biol Chem. 2001;276:4501–8. doi: 10.1074/jbc.M007532200. [DOI] [PubMed] [Google Scholar]
- 12.Feng X, Guo Z, Nourbakhsh M, Hauser H, Ganster R, Shao L, Geller DA. Identification of a negative response element in the human inducible nitric-oxide synthase (hiNOS) promoter: the role of NF-κB-repressing factor (NRF) in basal repression of the hiNOS gene. Proc Natl Acad Sci USA. 2002;99:14212–7. doi: 10.1073/pnas.212306199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu Z, Li Y, Takwi A, Li B, Zhang J, Conklin DJ, Young KH, Martin R. miR-301a as an NF-κB activator in pancreatic cancer cells. EMBO J. 2011;30:57–67. doi: 10.1038/emboj.2010.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee EJ, Gusev Y, Jiang J, et al. Expression profiling identifies microRNA signature in pancreatic cancer. Int J Cancer. 2007;120:1046–54. doi: 10.1002/ijc.22394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang J, Gusev Y, Aderca I, Mettler TA, Nagorney DM, Brackett DJ, Roberts LR, Schmittgen TD. Association of MicroRNA expression in hepatocellular carcinomas with hepatitis infection, cirrhosis, and patient survival. Clin Cancer Res. 2008;14:419–27. doi: 10.1158/1078-0432.CCR-07-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu X, Zhan Z, Li D, Xu L, Ma F, Zhang P, Yao H, Cao X. Intracellular MHC class II molecules promote TLR-triggered innate immune responses by maintaining activation of the kinase Btk. Nat Immunol. 2011;12:416–24. doi: 10.1038/ni.2015. [DOI] [PubMed] [Google Scholar]
- 17.Jiang M, Xiang Y, Wang D, Gao J, Liu D, Liu Y, Liu S, Zheng D. Dysregulated expression of miR-146a contributes to age-related dysfunction of macrophages. Aging Cell. 2012;11:29–40. doi: 10.1111/j.1474-9726.2011.00757.x. [DOI] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(–ΔΔCT) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Cao W, Bao C, Padalko E, Lowenstein CJ. Acetylation of mitogen-activated protein kinase phosphatase-1 inhibits Toll-like receptor signaling. J Exp Med. 2008;205:1491–503. doi: 10.1084/jem.20071728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shi W, Gerster K, Alajez NM, et al. MicroRNA-301 mediates proliferation and invasion in human breast cancer. Cancer Res. 2011;71:2926–37. doi: 10.1158/0008-5472.CAN-10-3369. [DOI] [PubMed] [Google Scholar]
- 21.Miko E, Czimmerer Z, Csanky E, Boros G, Buslig J, Dezso B, Clynes M, Barron N. Differentially expressed microRNAs in small cell lung cancer. Exp Lung Res. 2009;35:646–64. doi: 10.3109/01902140902822312. [DOI] [PubMed] [Google Scholar]
- 22.Beier UH, Akimova T, Liu Y, Wang L, Hancock WW. Histone/protein deacetylases control Foxp3 expression and the heat shock response of T-regulatory cells. Curr Opin Immunol. 2011;23:670–8. doi: 10.1016/j.coi.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ge Z, Da Y, Xue Z, et al. Vorinostat, a histone deacetylase inhibitor, suppresses dendritic cell function and ameliorates experimental autoimmune encephalomyelitis. Exp Neurol. 2013;241:56–66. doi: 10.1016/j.expneurol.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 24.Wu C, Li A, Leng Y, Li Y, Kang J. Histone deacetylase inhibition by sodium valproate regulates polarization of macrophage subsets. DNA Cell Biol. 2012;31:592–9. doi: 10.1089/dna.2011.1401. [DOI] [PubMed] [Google Scholar]
- 25.Roger T, Lugrin J, Le Roy D, et al. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood. 2011;117:1205–17. doi: 10.1182/blood-2010-05-284711. [DOI] [PubMed] [Google Scholar]
- 26.Hancock WW, Akimova T, Beier UH, Liu Y, Wang L. HDAC inhibitor therapy in autoimmunity and transplantation. Ann Rheum Dis. 2012;71(Suppl 2):i46–54. doi: 10.1136/annrheumdis-2011-200593. [DOI] [PubMed] [Google Scholar]
- 27.Hancock WW. Rationale for HDAC inhibitor therapy in autoimmunity and transplantation. Handb Exp Pharmacol. 2011;206:103–23. doi: 10.1007/978-3-642-21631-2_6. [DOI] [PubMed] [Google Scholar]
- 28.Halili MA, Andrews MR, Sweet MJ, Fairlie DP. Histone deacetylase inhibitors in inflammatory disease. Curr Top Med Chem. 2009;9:309–19. doi: 10.2174/156802609788085250. [DOI] [PubMed] [Google Scholar]
- 29.Aung HT, Schroder K, Himes SR, et al. LPS regulates proinflammatory gene expression in macrophages by altering histone deacetylase expression. FASEB J. 2006;20:1315–27. doi: 10.1096/fj.05-5360com. [DOI] [PubMed] [Google Scholar]
- 30.Bode KA, Schroder K, Hume DA, Ravasi T, Heeg K, Sweet MJ, Dalpke AH. Histone deacetylase inhibitors decrease Toll-like receptor-mediated activation of proinflammatory gene expression by impairing transcription factor recruitment. Immunology. 2007;122:596–606. doi: 10.1111/j.1365-2567.2007.02678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brogdon JL, Xu Y, Szabo SJ, An S, Buxton F, Cohen D, Huang Q. Histone deacetylase activities are required for innate immune cell control of Th1 but not Th2 effector cell function. Blood. 2007;109:1123–30. doi: 10.1182/blood-2006-04-019711. [DOI] [PubMed] [Google Scholar]
- 32.Elsharkawy AM, Oakley F, Lin F, Packham G, Mann DA, Mann J. The NF-κB p50:p50:HDAC-1 repressor complex orchestrates transcriptional inhibition of multiple pro-inflammatory genes. J Hepatol. 2010;53:519–27. doi: 10.1016/j.jhep.2010.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen L, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-κB action regulated by reversible acetylation. Science. 2001;293:1653–7. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 34.Ashburner BP, Westerheide SD, Baldwin AS., Jr The p65 (RelA) subunit of NF-κB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol. 2001;21:7065–77. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi YS, Jeong S. PI3-kinase and PDK-1 regulate HDAC1-mediated transcriptional repression of transcription factor NF-κB. Mol Cells. 2005;20:241–6. [PubMed] [Google Scholar]