

Abstract
The effect of Pam3CSK4, a Toll-like receptor 2 (TLR2) ligand, on interferon-γ (IFN-γ) -induced nitric oxide (NO) production in mouse vascular endothelial END-D cells was studied. Pre-treatment or post-treatment with Pam3CSK4 augmented IFN-γ-induced NO production via enhanced expression of an inducible NO synthase (iNOS) protein and mRNA. Pam3CSK4 augmented phosphorylation of Janus kinase 1 and 2, followed by enhanced phosphorylation of signal transducer and activator of transcription 1 (STAT1) at tyrosine 701. Subsequently, the enhanced STAT1 activation augmented IFN-γ-induced IFN-regulatory factor 1 expression leading to the iNOS expression. Pam3CSK4 also induced the activation of p38 and subsequent phosphorylation of STAT1 at serine 727. A pharmacological p38 inhibitor abolished the augmentation of IFN-γ-induced NO production by Pam3CSK4. Surprisingly, Pam3CSK4 enhanced a physical association of MyD88 and IFN-γ receptor. Together, these findings suggest that Pam3CSK4 up-regulates IFN-γ signalling in vascular endothelial cells via the physical association between MyD88 and IFN-γ receptor α, and p38-dependent serine 727 STAT1 phosphorylation.
Keywords: interferon-γ receptor, MyD88, Pam3CSK4, Toll-like receptor 2, vascular endothelial cells
Introduction
Vascular endothelial cells are critical targets for microbial products and are directly exposed to them.1 A series of Toll-like receptors (TLRs), which are innate immune pattern recognition receptors, recognize a variety of microbial products and activate vascular endothelial cells in response to microbial products.2,3 Upon TLR stimulation, several TLR ligands trigger nuclear factor-κB (NF-κB) and mitogen-activated protein kinases (MAPKs) via MyD88 adaptor molecule, and subsequently leads to production of pro-inflammatory mediators.2 The TLR signalling is reported to cooperate with the interferon-γ (IFN-γ) signalling pathway by influencing the activity of transcription factors, such as signal transducer and activator of transcription 1 (STAT1).4 In fact, stimulation of macrophages with a series of TLR ligands [e.g. lipoteichoic acid, lipopolysaccharide (LPS), imiquimod, CpG DNA] followed by IFN-γ is well known to cause a significant increase of STAT1 activation, and it leads to higher STAT1-dependent transcription activities compared with ligands alone.5–8 Similarly, we have demonstrated that in vascular endothelial cells LPS as a TLR4 ligand augments IFN-γ-induced nitric oxide (NO) production via enhanced IFN-regulatory factor 1 (IRF1) expression in the IFN-γ signalling.9,10 However, the precise action of other TLR ligands on the IFN-γ signalling in vascular endothelial cells is unknown. In this study, we examined the effect of a series of TLR ligands on IFN-γ-induced NO production by using murine vascular endothelial END-D cells and found their enhancing effect on the NO production. Among the TLR ligands tested, we focused on the enhancing mechanism of Pam3CSK4, a TLR2 ligand, because TLR2 is expressed on the cell surface and uses the signalling pathway similar to LPS. Interestingly, we found that Pam3CSK4 caused a physical association between MyD88 and IFN-γ receptor, and further p38-mediated STAT1 at serine 727 (S727) phosphorylation in response to IFN-γ.
Materials and methods
Reagents
Mouse recombinant IFN-γ was purchased from PeproTech (Rocky Hill, NJ). Pam3CSK4, poly I:C, imiquimod and CpG DNA were obtained from Invivogen (San Diego, CA). Lipopolysaccharide from Escherichia coli O55: B5 and parthenolide were purchased from Sigma Chemicals (St Louis, MO). Bay 11-7082, SB 203580, SB 202474 and PD 98059 were obtained from Calbiochem (Gibbs-town, NJ). An anti-inducible NO synthase (anti-iNOS) antibody was obtained from BD Transduction Laboratories (Franklin Lakes, NJ). A series of antibodies to STAT1, Janus kinase 1 (JAK1), JAK2, p65, p38, extracellular signal-regulated kinase 1/2 (ERK1/2), c-Jun N-terminal kinase (JNK) and their phosphorylated forms were purchased from Cell Signaling Technology (Beverly, MA). Antibodies to IRF1, IFN-γ receptor α (IFN-γRα), MyD88, IκB-α and actin (C-11) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell culture
The murine aortic endothelial cell line END-D, which is positive for vascular cell adhesion molecule-1 and intracellular cell adhesion molecule-1, and negative for E-selectin,9,10 were maintained in Dulbecco's modified Eagle's medium (Sigma, St Louis, MO) containing 10% heat inactivated fetal calf serum (FCS; Gibco-BRL, Gaithersburg, MD) and antibiotic cocktail from Sigma (penicillin G, streptomycin and amphotericin B) at 37° under 5% CO2. The cells were treated with the 0·05% trypsin/EDTA solution (Gibco-BRL) for preparing the cell suspension. The murine macrophage cell line RAW 264.7 was obtained from the Riken Cell Bank (Tsukuba, Japan) and maintained with RPMI-1640 medium containing 5% FCS.
Determination of nitrite concentration
Nitrite, the end product of NO metabolism, was measured using the Griess reagent as described elsewhere.11 Briefly, END-D cells were seeded in a 96-well plastic plate, pre-treated with Pam3CSK4 and then stimulated with IFN-γ. To measure the nitrite concentration, the culture supernatant (50 μl) was mixed with an equal volume of Griess reagent for 2 min. The nitrite concentration in each culture supernatant was measured as absorbance at 570 nm (A570) with reference to the standard curve using sodium nitrite.
Luciferase reporter gene assay for iNOS activation
END-D cells (4 × 105 cells/well) were incubated in a 35-mm plastic dish for 14 hr. The cells were transfected with 0·2 μg/well of iNOS-luc luciferase reporter gene and 0·05 μg/well of pRL-TK plasmid (Promega, Madison, WI) as an internal control by lipofectamine transfection reagent (Invitrogen, Carlsbad, CA) for 24 hr. The transfection medium was then replaced with growth medium containing 5% FCS and no antibiotics, and the cells were maintained for 6 hr. The transfected cells were replaced into a 96-well plate, pre-treated with or without Pam3CSK4 for 1 hr and then stimulated with IFN-γ for 6 hr. The cells were treated with the lysis reagent and the luciferase activity was determined with the dual luciferase assay kit (Promega). The luciferase activity in the cell lysates was determined with a luminometer and the fold increase is expressed by the ratio in comparison to control cells.
Immunoblotting
Immunoblotting was carried out as described elsewhere.9,12 Briefly, the whole cell lysates were extracted by the lysis buffer (50 mm Tris–HCl, pH 8·0, 150 mm NaCl, 10% glycerol, 1 mm EGTA, 0·2 mm EDTA, 1% Nonidet P-40, 1 mm dithiothreitol) with protease inhibitors (1 mm PMSF, 5 μg/ml pepstatin, 10 μg/ml chymostatin and leupeptin) at 4°. The protein concentration of each sample was determined by the bicinchoninic acid protein assay reagent (Pierce, Rockford, IL). Equal amounts of protein were subjected to analysis with SDS–PAGE under reducing conditions. The proteins were electrically transferred to a membrane and the membranes were treated with a series of appropriately diluted antibodies. An anti-actin antibody was used for a negative control. The immune complexes were detected with horseradish peroxidase-conjugated second antibody (Cell Signaling Technology) at 1 : 2000 for 1 hr. The protein bands were visualized using a chemiluminescence reagent (Pierce) and the chemiluminescence was detected by a light capture system analyser AE6955 (Atto, Tokyo, Japan). For re-probing, the membranes were stripped with the restore Western blot stripping buffer (Thermo Scientific, Rockford, IL) for 20 min and treated with corresponding antibodies. The molecular sizes of the antigens were determined by comparison with a pre-stained protein size marker kit (Invitrogen). To quantify the expression of each molecule, an equal area of each band image on an immunoblotted membrane was gated and the intensity of the band was calculated using ImageJ software (National Institutes of Health, Bethesda, MA).
Immunoprecipitation
END-D cells were suspended in 0·5 ml of the lysis buffer at 4°. Lysates were separated by centrifugation at 4°. Aliquots of lysate (500 μg of protein) were mixed with 20 μl of protein G–Sepharose (Amersham Biosciences, Piscataway, NJ) for 1 hr with intermittent agitation at 4°. After centrifugation, the supernatant was incubated with 2 μg/ml of anti-IFN-γRα antibody overnight at 4° with shaking. Immune complexes were adsorbed on protein G-Sepharose and washed three times with the lysis buffer. Finally, immunoblotting was performed as described above.
Semi-quantitative RT-PCR
The semi-quantitative RT-PCR was performed as described previously.13 Briefly, RNA was extracted from the cells with an RNeasy mini kit (Qiagen, Valencia, CA). RT-PCR was carried out using the Access Quick RT-PCR system (Promega). Primers with iNOS sequence: forward 5'-GTCTTGCAAGCTGATGGTCA-3' and reverse 5'-TGTCTTGGGATCTGGCTCTT-3', and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) sequence: forward 5'-ATGGGGAAGGTGAAGGTCGGAGTC-3' and reverse 5'-GCTGATGATCTTGAGGCTGTTGTC-3' were obtained from Invitrogen. GAPDH was used as an equal loading control. Optimized RT-PCR conditions were 45° for 45 min followed by 95° for 2 min and 32 cycles at 95° for 45 seconds, 62° for 30 seconds, 70° for 30 seconds. The PCR products were analysed by electrophoresis on 2% agarose gels. The gels were stained with CYBR Gold nucleic acid gel stain (Molecular Probe, Invitrogen) and visualized under an ultraviolet transilluminator. The 100 base pair DNA size marker (Invitrogen) was also run to determine the approximate size of the product.
Transfection of small interfering RNA
MyD88-specific pool and a non-targeting small interfering RNA (siRNA) were obtained from Santa Cruz Biotechnology. END-D cells were seeded at a concentration of 1·2 × 105 cells/well in a 12-well culture plate in antibiotic-free complete growth medium with reduced FCS. After overnight incubation, the cells were transfected with TransIT-TKO transfection reagent (Mirus Bio, Madison, WI) according to the manufacturer's protocol. Briefly, MyD88 or control siRNA (100 nm) was diluted in 100 μl/well OPTI–MEM medium (Gibco-BRL) and transfection complexes were prepared by incubating for 30 min with 5 μl/well TransIT-TKO transfection reagent. After 24 hr of incubation, an additional half of culture medium containing FCS and antibiotics was added to each well. Finally, transfected and untreated cells were stimulated with Pam3CSK4 (10 μg/ml) and IFN-γ (100 ng/ml) and the efficiency of MyD88 silencing was evaluated by immunoblotting and nitrite determination, respectively.
Statistical analysis
Experimental values are represented as the mean ± standard deviation from at least three independent experiments. The significance of differences between experimental and control groups was determined by the Student's t-test. A value of P < 0·01 was considered statistically significant.
Results
Pam3CSK4 as a TLR2 ligand up-regulates IFN-γ-induced NO production in vascular endothelial END-D cells
First, the effect of a series of TLR ligands on IFN-γ-induced NO production was examined by using vascular endothelial END-D cells. Cells were pre-treated with each TLR ligand at the serial 10-fold dilution for 1 hr and then stimulated with IFN-γ at 100 ng/ml for 24 hr. All TLR ligands including Pam3CSK4 (a TLR2 ligand), poly I:C (a TLR3 ligand), LPS (a TLR4 ligand), imiquimod (a TLR7 ligand), and CpG DNA (a TLR9 ligand) up-regulated IFN-γ-induced NO production in a roughly concentration-dependent manner (Fig. 1a). Any TLR ligand alone and untreated control did not induce NO production in END-D cells. In this study we focused on the enhancing action of Pam3CSK4 because we previously reported the augmentation of IFN-γ-induced NO production by LPS,9,10 and both TLR2 and TLR4 are expressed on the cell surface and use the similar signal pathway using MyD88.14
Figure 1.
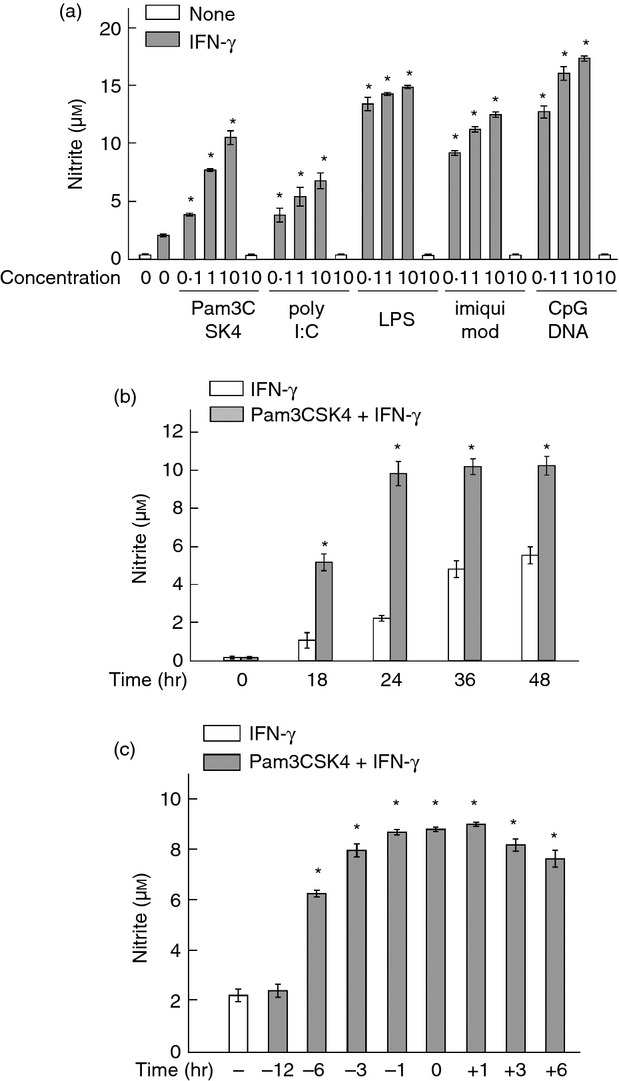
Effect of a series of Toll-like receptor (TLR) ligands on interferon-γ (IFN-γ) -induced nitric oxide (NO) production. (a) END-D cells were pre-treated with Pam3CSK4 (TLR2), poly I:C (TLR3), lipopolysaccharide (LPS; TLR4), imiquimod (TLR7) or CpG DNA (TLR9) at 0·1, 1 and 10 μg/ml for 1 hr and then stimulated with IFN-γ at 100 ng/ml for 24 hr. (b) Cells were pre-treated with Pam3CSK4 at 10 μg/ml for 1 hr and then stimulated with IFN-γ at 100 ng/ml for various numbers of hours. (c) Cells were treated with Pam3CSK4 at 10 μg/ml for various numbers of hours before and after stimulation with IFN-γ at 100 ng/ml for 24 hr. *P < 0·01 versus IFN-γ alone.
Second, the time course of IFN-γ-induced NO production in cells pre-treated with or without Pam3CSK4 was examined (Fig. 1b). Cells were pre-treated with or without Pam3CSK4 at 10 μg/ml for 1 hr and then stimulated with IFN-γ at 100 ng/ml for various lengths of time. Interferon-γ gradually increased the NO production up to 48 hr in untreated control cells. In contrast, Pam3CSK4 pre-treatment augmented and accelerated IFN-γ-induced NO production. The NO production in cells pre-treated with Pam3CSK4 reached a peak at 24 hr. In addition, Pam3CSK4 at 10 μg/ml exhibited no cytotoxic action against the cells by determining with an 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay (data not shown).
Third, the effect of pre-treatment or post-treatment with Pam3CSK4 on IFN-γ-induced NO production was examined (Fig. 1c). Cells were pre-treated or post-treated with Pam3CSK4 at 10 μg/ml for various hours before and after the stimulation with IFN-γ at 100 ng/ml for 24 hr. Treatment with Pam3CSK4 from − 6 hr to + 6 hr significantly augmented IFN-γ-induced NO production. However, pre-treatment with Pam3CSK4 for 12 hr did not augment it. For further characterization of the enhancing effect of Pam3CSK4, END-D cells were pre-treated with Pam3CSK4 at 10 μg/ml for 1 hr and then stimulated with IFN-γ at 100 ng/ml for 24 hr unless otherwise stated.
Pam3CSK4 augments the expression of iNOS protein and mRNA in response to IFN-γ
The effect of Pam3CSK4 on IFN-γ-induced iNOS protein expression was examined with immunoblotting. Cells were pre-treated with or without Pam3CSK4 and then stimulated with IFN-γ for 24 hr. Immunoblotting analysis showed the enhanced iNOS protein expression in cells pre-treated with Pam3CSK4 (approximately threefold increase, Fig. 2a). The effect of Pam3CSK4 on IFN-γ-induced iNOS mRNA expression was also examined with semi-quantitative RT-PCR. Cells pre-treated with or without Pam3CSK4 were stimulated with IFN-γ for 6 hr. RT-PCR analysis showed the enhanced expression of iNOS mRNA in cells pre-treated with Pam3CSK4 (approximately twofold increase, Fig. 2b).
Figure 2.
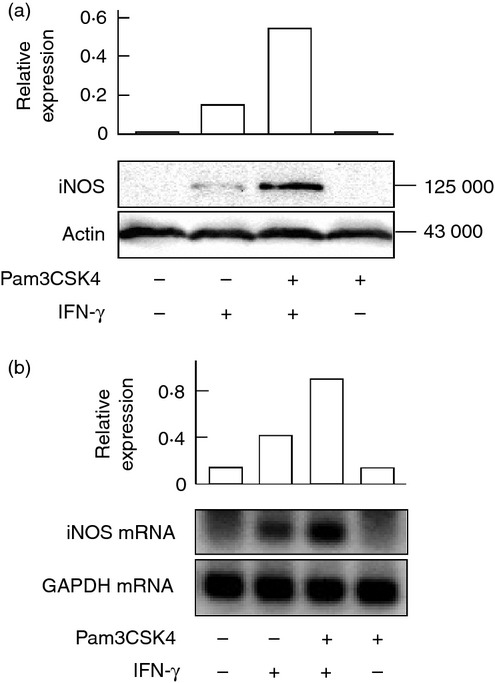
Effect of Pam3CSK4 on interferon-γ (IFN-γ) -induced inducible nitric oxide synthase (iNOS) protein and mRNA expression. END-D cells were pre-treated with or without Pam3CSK4 at 10 μg/ml for 1 hr and then stimulated with IFN-γ at 100 ng/ml for 24 hr (a) or 6 hr (b). The expression of iNOS protein (a) and mRNA (b) was determined by immunoblotting and RT-PCR, respectively. A typical experiment of three independent experiments is shown.
Pam3CSK4 augments IFN-γ-induced IRF1 expression
Interferon regulatory factor 1 is up-regulated by IFN-γ in a STAT1-dependent manner and is mainly involved in IFN-γ-induced iNOS expression.15 The effect of Pam3CSK4 on IFN-γ-induced IRF1 expression was examined with immunoblotting. Cells were pre-treated with or without Pam3CSK4 and then stimulated with IFN-γ for 2 hr. Although IFN-γ induced the expression of IRF1 protein, Pam3CSK4 further augmented it (approximately twofold increase, Fig. 3a), although it alone did not induce the IRF1 expression. The effect of Pam3CSK4 on the transcription of the iNOS gene was examined with a luciferase assay using iNOS-reporter gene (Fig. 3b). Pam3CSK4 enhanced the luciferase activity in response to IFN-γ, suggesting that Pam3CSK4-induced IRF1 enhancement might be efficient for the iNOS transcription.
Figure 3.
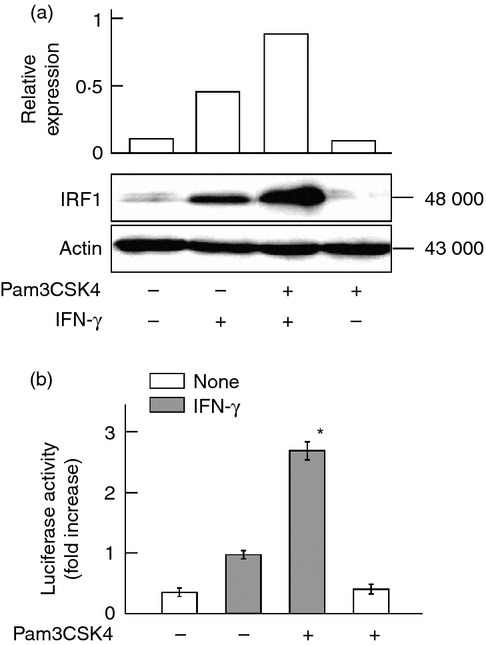
Effect of Pam3CSK4 on interferon-γ (IFN-γ) -induced interferon regulatory factor 1 (IRF1) expression. (a) END-D cells were pre-treated with or without Pam3CSK4 at 10 μg/ml for 1 hr and then stimulated with IFN-γ at 100 ng/ml for 2 hr. The expression of IRF1 protein was determined by immunoblotting. (b) Cells transfected with inducible nitric oxide synthase (iNOS) -luciferase reporter gene were pre-treated with or without Pam3CSK4 at 10 μg/ml for 1 hr and then stimulated with IFN-γ at 100 ng/ml for 6 hr. The iNOS-dependent luciferase activity in the cell lysates was determined with a luminometer. *P < 0·01 versus IFN-γ alone.
Pam3CSK4 augments the phosphorylation of STAT1 at tyrosine 701 (Y701) in response to IFN-γ
The activation of STAT1 is required for IFN-γ-induced IRF1 expression and is mediated by phosphorylation of STAT1 at Y701, leading to the dimerization.16 Therefore, the effect of Pam3CSK4 on IFN-γ-induced phosphorylation of STAT1 at Y701 was examined with immunoblotting. Cells were pre-treated with or without Pam3CSK4 and then stimulated with IFN-γ for 2 hr. Interferon-γ alone induced phosphorylation of STAT1 at Y701 and Pam3CSK4 pre-treatment significantly augmented it (approximately threefold increase, Fig. 4a).
Figure 4.
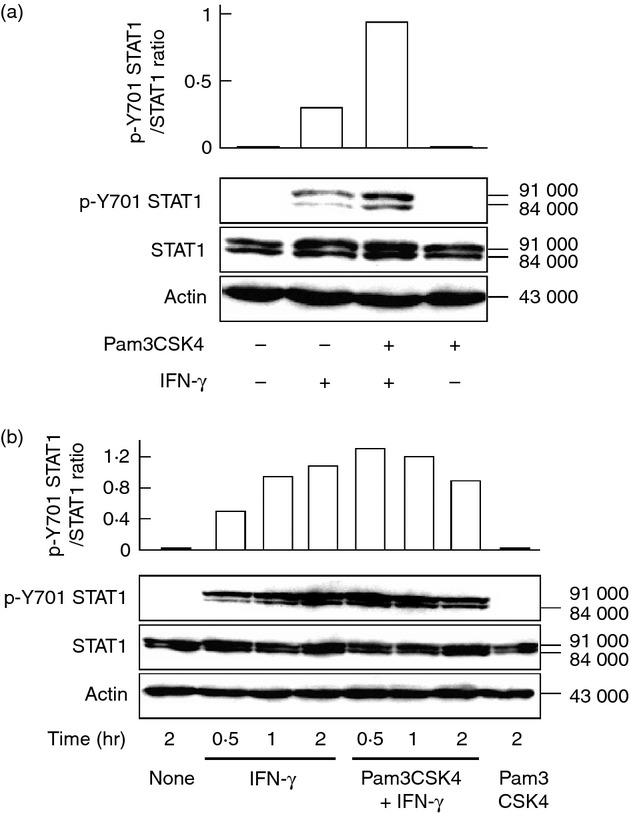
Effect of Pam3CSK4 on interferon-γ (IFN-γ) -induced phosphorylation of signal transducer and activator of transcription 1 (STAT1) at Y701. (a) END-D cells were pre-treated with or without Pam3CSK4 at 10 μg/ml for 1 hr and then stimulated with IFN-γ at 100 ng/ml for 2 hr. (b) Cells were pre-treated with or without Pam3CSK4 at 10 μg/ml for 1 hr and then stimulated with IFN-γ at 100 ng/ml for various hours. The expression of STAT1 and phosphorylated Y701 (p-Y701) STAT1 was determined by immunoblotting. A typical experiment of three independent experiments is shown.
The time course of the enhancing effect of Pam3CSK4 on IFN-γ-induced phosphorylation of STAT1 at Y701 was examined. Immunoblotting analysis showed that IFN-γ gradually induced phosphorylation of STAT1 at Y701 in a time-dependent manner. On the other hand, Pam3CSK4 rapidly augmented at 0·5 hr and accelerated IFN-γ-induced Y701 STAT1 phosphorylation (Fig. 4b).
Pam3CSK4 up-regulates IFN-γ-induced JAK1 and JAK2 activation
Interferon-γ-induced STAT1 phosphorylation is mediated by activation of JAK1 and JAK2 as upstream molecules of STAT1.4 Therefore, the effect of Pam3CSK4 on IFN-γ-induced JAK1 and JAK2 activation was examined with immunoblotting. Cells were pre-treated with or without Pam3CSK4 and then stimulated with IFN-γ for 0·5 hr. Immunoblotting analysis showed the enhanced phosphorylation of JAK1 and JAK2 in Pam3CSK4-pre-treated cells (Fig. 5), suggesting that Pam3CSK4 augments IFN-γ-induced phosphorylation of STAT1 at Y701 via enhanced JAK1 and JAK2 activation.
Figure 5.
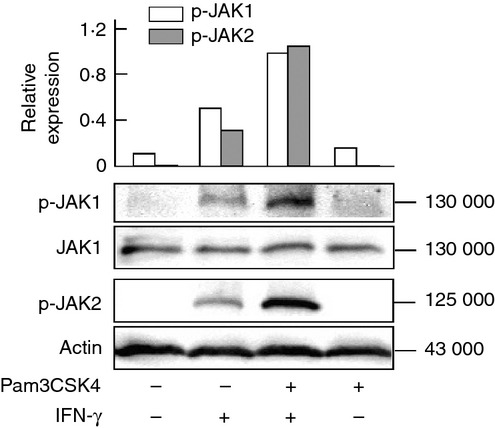
Effect of Pam3CSK4 on interferon-γ (IFN-γ) -induced Janus kinase 1/2 (JAK1/2) activation. END-D cells were pre-treated with or without Pam3CSK4 at 10 μg/ml for 1 hr and then stimulated with IFN-γ at 100 ng/ml for 0·5 hr. The expression of phosphorylated JAK1 (p-JAK1) and JAK2 (p-JAK2) was determined by immunoblotting. A typical experiment of three independent experiments is shown.
No augmentation of IFN-γRα expression in Pam3CSK4-treated cells
Because the activation of JAK1/2 is triggered by binding of IFN-γ to the receptor,17 a possibility was raised that Pam3CSK4-treated cells might express a higher level of IFN-γRα on the cell surface. Cells were treated with or without Pam3CSK4 and the cell surface expression of IFN-γRα was analysed with laser flow cytometry. There was no significant difference in the IFN-γRα expression on the cell surface between cells treated with or without Pam3CSK4 (see Supplementary material, Fig. S1).
Role of Pam3CSK4-mediated p38 activation in augmentation of IFN-γ-induced NO production
Pam3CSK4 binds to TLR2 and it leads to the induction of several transcription factors including NF-κB and MAPKs in a MyD88-dependent manner.2 The TLR2-mediated signal pathway might be involved in augmentation of IFN-γ-induced NO production in cells pre-treated with Pam3CSK4. First, the effect of Pam3CSK4 on activation of NF-κB, such as IκB degradation and p65 NF-κB phosphorylation, in END-D cells was examined in the presence or absence of IFN-γ. Cells were treated with Pam3CSK4 and/or IFN-γ for 0·5 hr. Pam3CSK4 enhanced neither degradation of IκB-α protein nor phosphorylation of p65 protein. There was no significant difference in Pam3CSK4-induced NF-κB activation between the presence and absence of IFN-γ (Fig. 6a). In addition, phosphorylated p65 was detected in untreated control cells.
Figure 6.
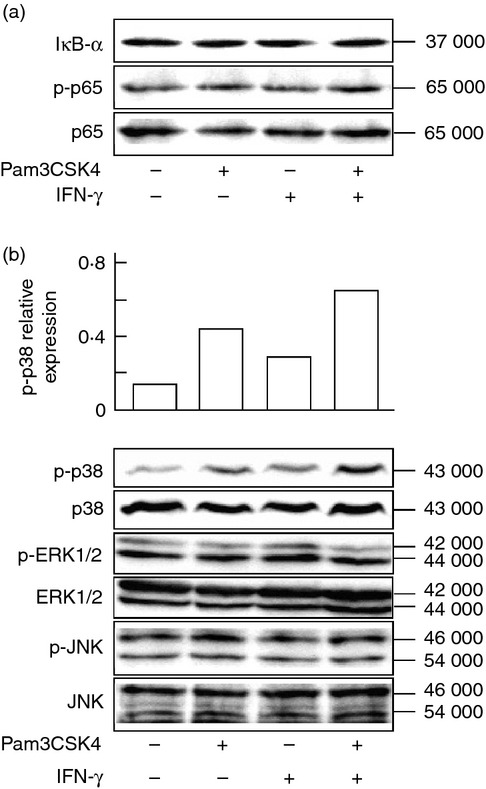
Effect of interferon-γ (IFN-γ) on Pam3CSK4-mediated nuclear factor-κB (NF-κB) and mitogen-activated protein kinases (MAPKs) activation. END-D cells were stimulated with IFN-γ at 100 ng/ml and/or Pam3CSK4 at 10 μg/ml for 0·5 hr. (a) The expression of IκB-α and phosphorylated p65 (p-p65). (b) The expression of phosphorylated p38 (p-p38), extracellular signal-regulated kinase 1/2 (p-ERK1/2) and Jun N-terminal kinase (p-JNK). Their expression was determined by immunoblotting. A typical experiment of three independent experiments is shown.
The effect of Pam3CSK4 on phosphorylation of p38, ERK1/2 and JNK was examined in the presence or absence of IFN-γ. Pam3CSK4 induced the phosphorylation of p38 but not ERK1/2 and JNK, and IFN-γ further augmented Pam3CSK4-induced p38 activation (Fig. 6b). On the other hand, IFN-γ did not affect Pam3CSK4-induced activation of ERK1/2 and JNK.
p38 activation is involved in the phosphorylation of STAT1 at S727 and augments IFN-γ-induced NO production
Toll-like receptor signalling induces the phosphorylation of STAT1 at S727 via p38 activation.6,7,18,19 As Pam3CSK4 induced p38 activation above, the effect of Pam3CSK4 on the p38-dependent phosphorylation of STAT1 at S727 was examined. Cells were treated with Pam3CSK4 at 10 μg/ml for various periods of time. Pam3CSK4 induced the phosphorylation of p38 and STAT1 at S727 30 min after treatment and their phosphorylation continued up to 2 hr (Fig. 7a), suggesting a close association between p38 activation and phosphorylation of STAT1 at S727.
Figure 7.
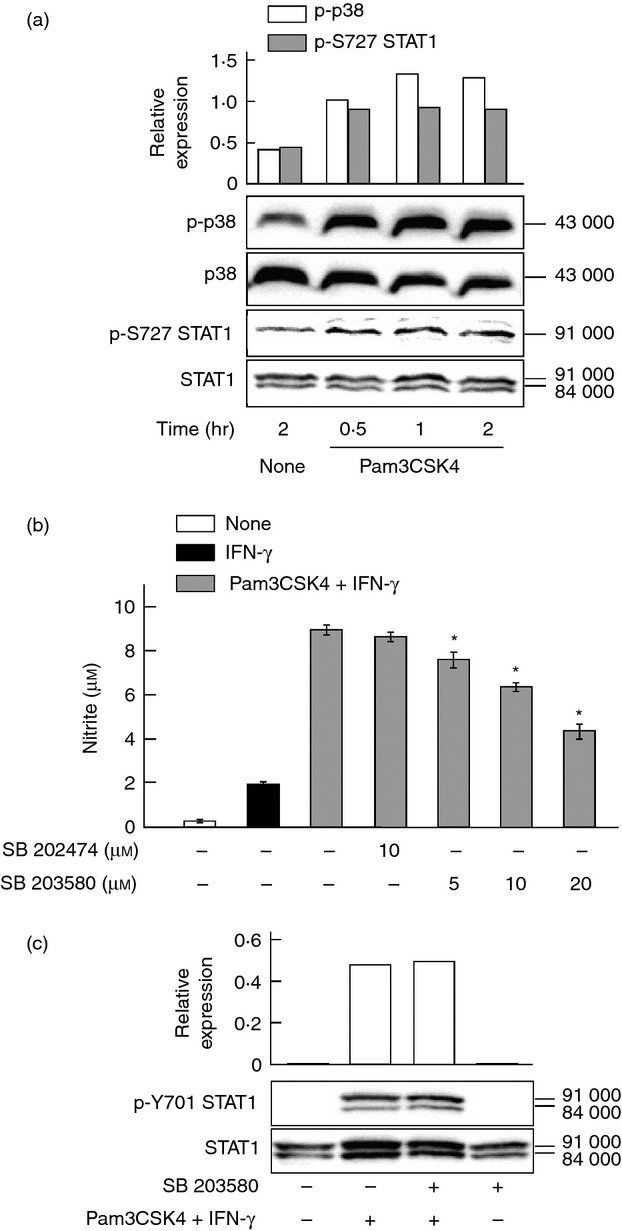
Effect of Pam3CSK4 on phosphorylation of p38 and signal transducer and activator of transcription (STAT1) at S727. (a) END-D cells were treated with Pam3CSK4 at 10 μg/ml for various hours. The expression of phosphorylated p38 (p-p38) and STAT1 at S727 (p-S727 STAT1) was determined by immunoblotting. (b) Cells were pre-treated with SB 203580 at various concentrations or SB 202474 at 10 μm for 1 hr, treated with Pam3CSK4 at 10 μg/ml for 1 hr and then stimulated with interferon-γ (IFN-γ) at 100 ng/ml for 24 hr. *P < 0·01 versus Pam3CSK4+ IFN-γ. (c) Cells were pre-treated with or without SB 203580 at 20 μm for 1 hr, treated with Pam3CSK4 at 10 μg/ml for 1 hr and then stimulated with IFN-γ at 100 ng/ml for 2 hr. The phosphorylation of STAT1 at Y701 (p-Y701 STAT1) was determined by immunoblotting. A typical experiment of three independent experiments is shown.
The effect of a pharmacological p38 inhibitor on the Pam3CSK4 plus IFN-γ-induced NO production was examined. Cells were pre-treated with SB 203580 at various concentrations or SB 202474 at 10 μm for 1 hr and then stimulated with Pam3CSK4 plus IFN-γ for 24 hr. Pre-treatment of SB 203580 inhibited Pam3CSK4 plus IFN-γ-induced NO production in a concentration-dependent manner (45% inhibition at 20 μm), whereas the negative control SB 202474 did not alter it (Fig. 7b).
The effect of SB 203580 on JAK1/2-dependent phosphorylation of STAT1 at Y701 in response to IFN-γ plus Pam3CSK4 was examined. Cells were pre-treated with SB 203580 at 20 μm for 1 hr, incubated with Pam3CSK4 at 10 μg/ml for 1 hr and then stimulated with IFN-γ at 100 ng/ml for 2 hr. SB 203580 did not affect the phosphorylation of STAT1 at Y701 (Fig. 7c), suggesting that Pam3CSK4-mediated p38 activation was not involved in the phosphorylation of STAT1 at Y701.
Pam3CSK4 induces a physical association between MyD88 and IFN-γRα
Interferon-γ stimulation triggers a physical association between IFN-γRα and MyD88 in macrophages.20 The effect of Pam3CSK4 on the association between MyD88 and IFN-γRα was examined. Cells were pre-treated with or without Pam3CSK4 and then stimulated with IFN-γ for 4 hr. Immunoblotting analysis showed a faint band of MyD88 in the IFN-γRα precipitates from untreated control cells. A higher amount of MyD88 was detected in both Pam3CSK4-pre-treated cells and IFN-γ-treated cells. Furthermore, the combined treatment with Pam3CSK4 and IFN-γ augmented the physical association between MyD88 and IFN-γRα (Fig. 8a). The physical association was also detected in RAW 264.7 macrophage cells in response to Pam3CSK4 (Fig. 8b). On the other hand, neither LPS nor poly I:C induced the physical association in those cells. Similarly, LPS and poly I:C did not induce the physical association in END-D cells (data not shown).
Figure 8.
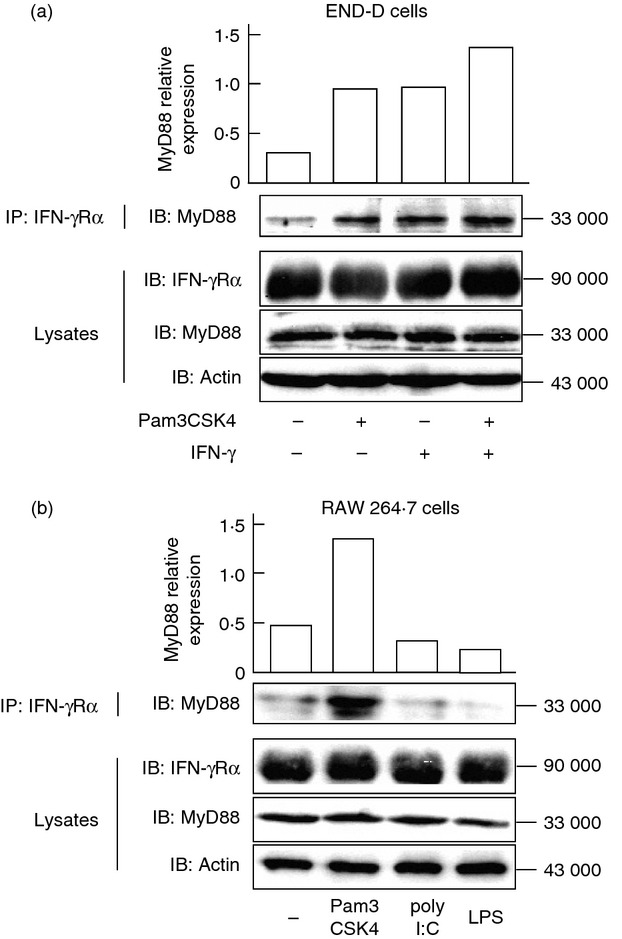
Effect of Pam3CSK4 and interferon-γ (IFN-γ) on the physical association between MyD88 and interferon-γ receptor α (IFN-γRα). (a) END-D cells were stimulated with Pam3CSK4 at 10 μg/ml and/or IFN-γ at 100 ng/ml for 4 hr. (b) RAW 264.7 cells were stimulated with Pam3CSK4 (10 μg/ml), poly I:C (10 μg/ml) or lipopolysaccharide (LPS; 10 μg/ml) for 2 hr. Cell lysates were immunoprecipitated (IP) with anti-IFN-γRα antibody and the immunoprecipitates were immunoblotted (IB) with anti-MyD88 antibody. Cell lysates were also analysed with anti-IFN-γRα, anti-MyD88 and anti-actin antibodies.
MyD88 siRNA abolishes Pam3CSK4-mediated enhancement of IFN-γ-induced NO production
The effect of MyD88 siRNA on IFN-γ-induced NO production was examined in END-D cells pre-treated with or without Pam3CSK4. Cells were transfected with MyD88 or control siRNA, pre-treated with Pam3CSK4 for 1 hr and then stimulated with IFN-γ for 24 hr. MyD88 siRNA but not control siRNA markedly reduced IFN-γ-induced NO production in cells pre-treated with Pam3CSK4 (Fig. 9). MyD88 siRNA also reduced to a lesser extent the NO production in untreated control cells.
Figure 9.
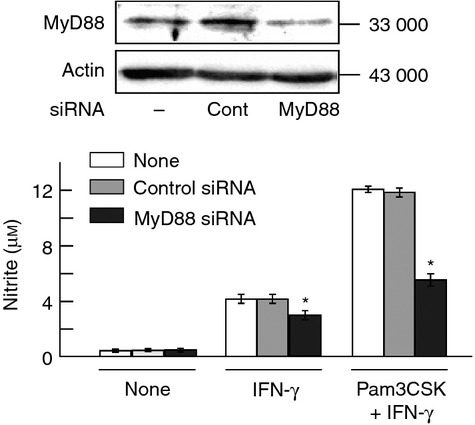
Effect of MyD88 small interfering RNA (siRNA) on interferon-γ (IFN-γ) -induced nitric oxide (NO) production in pre-treatment with or without Pam3CSK4. END-D cells were transfected with MyD88 siRNA or control siRNA for 24 hr. The expression of MyD88 protein was determined by immunoblotting (top). MyD88 siRNA and control siRNA-transfected cells were pre-treated with Pam3CSK4 at 10 μg/ml for 1 hr and then stimulated with IFN-γ at 100 ng/ml for 24 hr. The nitrite concentration was determined for NO production (bottom). *P < 0·01 versus control siRNA.
Discussion
We have demonstrated that Pam3CSK4, a TLR2 ligand, up-regulates IFN-γ-induced NO production via enhanced IFN-γ signalling in vascular endothelial cells. It is supported by the augmentation of IFN-γ-induced JAK1/2 and STAT1 phosphorylation by Pam3CSK4. Interestingly, Pam3CSK4 pre-treatment accelerates IFN-γ-induced STAT1 phosphorylation, suggesting that IFN-γ more rapidly activates the JAK/STAT signalling in cells pre-treated with Pam3CSK4. Therefore, Pam3CSK4 may up-regulate the IFN-γ signalling more rapidly and more strongly, followed by higher expression of IFN-γ-induced genes. Once again, Pam3CSK4 up-regulates IFN-γ-induced NO production in vascular endothelial cells via enhanced IFN-γ signalling.
The augmentation of IFN-γ signalling by Pam3CSK4 is mediated by two different mechanisms; one is a physical association between IFN-γRα and MyD88; the other is p38-dependent phosphorylation of STAT1 at S727. Surprisingly, Pam3CSK4 causes a physical association between IFN-γRα and MyD88 before IFN-γ stimulation and this association may lead to an augmentation of the phosphorylation of JAK1/2 in response to IFN-γ. Interferon-γ is reported to trigger a physical association between IFN-γR1 and MyD88, and stabilize IFN-γ-induced mRNA transcripts.20 However, our experimental result shows that Pam3CSK4 pre-treatment causes the physical association between IFN-γRα and MyD88 before IFN-γ stimulation and subsequently augments IFN-γ-induced JAK1/2 phosphorylation. It indicates that their association leads to enhanced IFN-γ signalling rather than transcript stabilization. On the other hand, the physical association occurs after IFN-γ stimulation in the absence of Pam3CSK4 and therefore IFN-γ signalling is not augmented. Although the involvement of transcript stabilization20 is not excluded, this is the first report that TLR2 stimulation up-regulates IFN-γ signalling via the physical association between IFN-γRα and MyD88. The phenomenon might occur in various cell types including vascular endothelial cells and macrophages.
The activation of STAT1 by phosphorylation at Y701 is mediated by the IFN-γRα/JAK complex.16 Pre-treatment with Pam3CSK4 enhances JAK1/2 activation via the association between MyD88 and IFN-γRα and augments the phosphorylation of STAT1 at Y701. The phosphorylation of STAT1 at Y701 is essential for STAT1 dimerization and IRF1 induction in IFN-γ-induced NO production.4 Therefore, the augmentation of IFN-γ-induced NO production with Pam3CSK4 is primarily mediated by the enhanced phosphorylation of STAT1 at Y701.
Besides the JAK1/2-dependent phosphorylation of STAT1 at Y701, Pam3CSK4 induces STAT1 phosphorylation at S727 via p38 activation. The STAT1 phosphorylation at S727 seems to be important for signal integration between TLR and IFN-γ signalling pathways. Signalling initiated by a number of TLR agonists (e.g. lipoteichoic acid, LPS, imiquimod, CpG DNA) is reported to induce S727 STAT1 phosphorylation via activation of the p38 MAPK pathway.6,7,18,19 Stimulation of macrophages with a series of TLR ligands followed by IFN-γ results in a significant increase of STAT1 phosphorylation at both Y701 and S727, and it leads to higher induction of STAT1-dependent transcription compared with agonists alone.5,6,21,22 It corresponds to our finding using vascular endothelial END-D cells, that STAT1 at S727 is more strongly phosphorylated in response to Pam3CSK4. Phosphorylation of STAT1 at both Y701 and S727 causes full activation of STAT1, leading to high expression of IFN-γ-induced genes.
Vascular endothelial cells are critical targets for microbial products.1 The present study demonstrates that a TLR2 ligand, Pam3CSK4, enhances NO production in vascular endothelial END-D cells. Besides Pam3CSK4 as a TLR2 ligand, a series of other TLR ligands such as poly I:C for TLR3, LPS for TLR4, imiquimod for TLR7, and CpG DNA for TLR9 augment IFN-γ-induced NO production, suggesting that TLR stimulation may regulate IFN-γ signalling in vascular endothelial cells. Previously we reported that LPS promoted IFN-γ-induced NO production in vascular endothelial END-D cells.9,10 Based on our finding, a possibility is raised that a series of TLR signalling may augment IFN-γ-induced NO production via a physical association between MyD88 and IFN-γRα. However, neither LPS nor poly I:C augments the physical association, suggesting that the physical association might be specific for TLR2 signalling with Pam3CSK4. It is of interest to clarify how each TLR ligand augments IFN-γ-induced NO production.
In conclusion, Pam3CSK4 up-regulates IFN-γ-induced NO production via enhanced IFN-γ signalling in vascular endothelial cells. Pam3CSK4 augments the IFN-γ signalling via a physical association between MyD88 and IFN-γRα before IFN-γ stimulation. Further, it augments the phosphorylation of STAT1 at S727 in the IFN-γ signalling via p38 activation. TLR signalling may regulate IFN-γ signalling in the expression of IFN-γ-induced genes in vascular endothelial cells.
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan and a grant of MEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2011–2015 (S1101027). We are grateful to K. Takahashi for the technical assistance.
Glossary
- ERK
extracellular signal-regulated kinase
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- IFN-γ
interferon-γ
- IFN-γRα
IFN-γ receptor α
- iNOS
inducible NO synthase
- IRF1
interferon regulatory factor 1
- JAK
Janus kinase
- JNK
c-Jun N-terminal kinase
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- MyD88
myeloid differentiation primary response gene 88
- NF-κB
nuclear factor-κB
- NO
nitric oxide
- RT-PCR
reverse transcription-polymerase chain reaction
- siRNA
small interfering RNA
- STAT1
signal transducer and activator of transcription 1
- TLR
Toll-like receptor
Disclosures
The authors report no conflicts of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Effect of Pam3CSK4 on the cell surface expression of interferon-γ receptor α (IFN-γRα) in END-D cells.
References
- 1.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–15. doi: 10.1038/nri2171. [DOI] [PubMed] [Google Scholar]
- 2.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 3.Sikorski K, Czerwoniec A, Bujnicki JM, Wesoly J, Bluyssen HA. STAT1 as a novel therapeutical target in pro-atherogenic signal integration of IFNγ, TLR4 and IL-6 in vascular disease. Cytokine Growth Factor Rev. 2011;22:211–9. doi: 10.1016/j.cytogfr.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 4.Schroder K, Sweet MJ, Hume DA. Signal integration between IFNγ and TLR signaling pathways in macrophages. Immunobiology. 2006;211:511–24. doi: 10.1016/j.imbio.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 5.Kovarik P, Stoiber D, Novy M, Decker T. Stat1 combines signals derived from IFN-γ and LPS receptors during macrophage activation. EMBO J. 1998;17:3660–8. doi: 10.1093/emboj/17.13.3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dalpke AH, Eckerle S, Frey M, Heeg K. Triggering of Toll-like receptors modulates IFN-γ signaling: involvement of serine 727 STAT1 phosphorylation and suppressors of cytokine signaling. Eur J Immunol. 2003;33:1776–87. doi: 10.1002/eji.200323621. [DOI] [PubMed] [Google Scholar]
- 7.Schroder K, Spille M, Pilz A, et al. Differential effects of CpG DNA on IFN-β induction and STAT1 activation in murine macrophages versus dendritic cells: alternatively activated STAT1 negatively regulates TLR signaling in macrophages. J Immunol. 2007;179:3495–503. doi: 10.4049/jimmunol.179.6.3495. [DOI] [PubMed] [Google Scholar]
- 8.Liljeroos M, Vuolteenaho R, Rounioja S, Henriques-Normark B, Hallman M, Ojaniemi M. Bacterial ligand of TLR2 signals Stat activation via induction of IRF1/2 and interferon-α production. Cell Signal. 2008;20:1873–81. doi: 10.1016/j.cellsig.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 9.Morikawa A, Koide N, Kato Y, Sugiyama T, Chakravortty D, Yoshida T, Yokochi T. Augmentation of nitric oxide production by γ interferon in a mouse vascular endothelial cell line and its modulation by tumor necrosis factor α and lipopolysaccharide. Infect Immun. 2000;68:6209–14. doi: 10.1128/iai.68.11.6209-6214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koide N, Mu MM, Hassan F, et al. Lipopolysaccharide enhances interferon-γ-induced nitric oxide (NO) production in murine vascular endothelial cells via augmentation of interferon regulatory factor-1 activation. J Endotoxin Res. 2007;13:167–75. doi: 10.1177/0968051907080894. [DOI] [PubMed] [Google Scholar]
- 11.Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR. Analysis of nitrate, nitrite, and [15N] nitrate in biological fluids. Anal Biochem. 1982;126:131–8. doi: 10.1016/0003-2697(82)90118-x. [DOI] [PubMed] [Google Scholar]
- 12.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dagvadorj J, Naiki Y, Tumurkhuu G, Noman AS, Iftakhar-E-Khuda I, Komatsu T, Yoshida T, Yokochi T. Tumor necrosis factor-α augments lipopolysaccharide-induced suppressor of cytokine signaling 3 (SOCS-3) protein expression by preventing the degradation. Immunology. 2010;129:97–104. doi: 10.1111/j.1365-2567.2009.03154.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signaling. Nat Rev Immunol. 2007;7:353–64. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 15.Ksienzyk A, Neumann B, Kröger A. IRF-1 is critical for IFNγ mediated immune surveillance. Oncoimmunology. 2012;1:533–4. doi: 10.4161/onci.19405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-γ: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–89. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 17.Hu X, Ivashkiv LB. Cross-regulation of signaling pathways by interferon-γ: implications for immune responses and autoimmune diseases. Immunity. 2009;31:539–50. doi: 10.1016/j.immuni.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takauji R, Iho S, Takatsuka H, et al. CpG-DNA-induced IFN-α production involves p38 MAPK-dependent STAT1 phosphorylation in human plasmacytoid dendritic cell precursors. J Leukoc Biol. 2002;72:1011–9. [PubMed] [Google Scholar]
- 19.Rhee SH, Jones BW, Toshchakov V, Vogel SN, Fenton MJ. Toll-like receptors 2 and 4 activate STAT1 serine phosphorylation by distinct mechanisms in macrophages. J Biol Chem. 2003;278:22506–12. doi: 10.1074/jbc.M208633200. [DOI] [PubMed] [Google Scholar]
- 20.Sun D, Ding A. MyD88-mediated stabilization of interferon-γ-induced cytokine and chemokine mRNA. Nat Immunol. 2006;7:375–81. doi: 10.1038/ni1308. [DOI] [PubMed] [Google Scholar]
- 21.Wen Z, Zhong Z, Darnell JE., Jr Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–50. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 22.Varinou L, Ramsauer K, Karaghiosoff M, Kolbe T, Pfeffer K, Müller M, Decker T. Phosphorylation of the Stat1 transactivation domain is required for full-fledged IFN-γ-dependent innate immunity. Immunity. 2003;19:793–802. doi: 10.1016/s1074-7613(03)00322-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.