

Abstract
B-lymphocyte activities are associated with allograft rejection. Interleukin-10 (IL-10) -expressing B cells, however, exhibit regulatory attributes. Human α1-antitrypsin (hAAT), a clinically available anti-inflammatory circulating glycoprotein that rises during acute-phase responses, promotes semi-mature dendritic cells and regulatory T (Treg) cells during alloimmune responses. Whether B lymphocytes are also targets of hAAT activity has yet to be determined. Here, we examine whether hAAT modulates B-cell responses. In culture, hAAT reduced the lipopolysaccharide-stimulated Ki-67+ B-cell population, IgM release and surface CD40 levels, but elevated IL-10-producing cells 1.5-fold. In CD40 ligand-stimulated cultures, hAAT promoted a similar trend; reduction in the Ki-67+ B-cell population and in surface expression of CD86, CD80 and MHCII. hAAT increased interferon-γ-stimulated macrophage B-cell activating factor (BAFF) secretion, and reduced BAFF-receptor levels. Draining lymph nodes of transgenic mice that express circulating hAAT (C57BL/6 background) and that received skin allografts exhibited reduced B-lymphocyte activation compared with wild-type recipients. BSA-vaccinated hAAT transgenic mice exhibited 2.9-fold lower BSA-specific IgG levels, but 2.3-fold greater IgM levels, compared with wild-type mice. Circulating Treg cells were 1.3-fold greater in transgenic hAAT mice, but lower in B-cell knockout (BKO) and chimeric hAAT–BKO mice, compared with wild-type mice. In conclusion, B cells are cellular targets of hAAT. hAAT-induced Treg cell expansion appears to be B-cell-dependent. These changes support the tolerogenic properties of hAAT during immune responses, and suggest that hAAT may be beneficial in pathologies that involve excessive B-cell responses.
Keywords: B-cell activating factor (BAFF), interleukin-10, regulatory B cells, regulatory T cells, tolerance
Introduction
Clinical pancreatic islet cell transplantation can restore normoglycaemia in diabetic patients and is increasingly considered to be a viable therapeutic option for patients with type 1 diabetes.1 However, the majority of transplanted patients return to insulin dependency within 5 years after transplantation.2,3
In data collected from pre-clinical and clinical studies, B lymphocytes appear to play a role in the pathogenesis of both islet allograft rejection4–7 and type 1 diabetes8–10 by means of pro-inflammatory cytokine release, antigen presentation and antibody production. For example, antibodies specific for donor alloantigens in patients that were transplanted with human islets serve as a marker for islet graft failure.5 Also, in human patients, high baseline B-cell counts are associated with poor islet transplantation outcome.4
B lymphocytes modulate immune responses by producing a vast array of pro- and anti-inflammatory mediators, including interleukin-4 (IL-4), IL-6, interferon-γ (IFN-γ), transforming growth factor-β and IL-10.11 B lymphocytes are activated by processes that are either T-cell-dependent or T-cell-independent. In both cases, B-lymphocyte activation leads to B-cell proliferation, as well as to an increase in cytokine release, up-regulation of co-stimulatory molecules (e.g. MHCII, CD86 and CD40), and the instigation of cell differentiation and antibody production.12
In the T-cell-independent pathway, lipopolysaccharide (LPS) can directly activate B lymphocytes through their membrane-bound Toll-like receptor 4.12 T-cell-dependent B-lymphocyte activation, on the other hand, occurs in secondary lymphoid organs and is considered the main alloimmune mechanism by which B lymphocytes mount specific antibody responses.13 This process involves antigen presentation on MHCII molecules and requires interaction between MHCII and CD40 molecules on B cells, with their corresponding T-cell receptor and CD40 ligand (CD40L), respectively.13 Following this interaction, in the presence of IL-4, B cells perform class switch recombination and, in addition, can differentiate into antibody-secreting cells.14
B-cell activation may also occur in the presence of B-cell activating factor (BAFF), released mainly by macrophages and dendritic cells. BAFF engages with BAFF receptor (BAFF-R) on B cells.15 Elevated levels of BAFF-R are found upon B-cell receptor activation.16 Elevated BAFF levels have been shown to increase IgM titres in vivo17,18 and to promote B-cell differentiation into plasma cells, which produce large amounts of IgM.19 Moreover, data suggest that levels of BAFF and BAFF-R may be associated with an allogeneic response in clinical transplantation studies.20
Lastly, a subpopulation of recently identified IL-10-producing B cells has been shown to be protective in the context of autoimmunity and, more specifically, to prevent diabetes in mice. Repeated transfusions of B-cell receptor-stimulated splenic B cells into non-obese diabetic (NOD) mice prevented disease development, while transfusion of activated B cells from IL-10-knockout mice failed to confer protection.21 Moreover, regulatory B cells were found to support regulatory T (Treg) cell differentiation in several proposed mechanisms.22,23 Hence, as far as the effect of B cells on Treg cells is concerned, the presence of intact B-cell populations and their positive modulation under particular circumstances may be of benefit to transplantation outcomes.
Human α1-antitrypsin (hAAT) is a circulating anti-inflammatory glycoprotein that exerts protective activities in the context of autoimmune diabetes and islet alloimmune responses both in vitro and in vivo.24–28 Specifically, our group has demonstrated that hAAT facilitates Treg cell expansion in various in vivo models.25,29,30 In addition, hAAT monotherapy improved diabetic parameters in the NOD mouse.26,27 Importantly, these observations are consistent across several orders of hAAT circulating levels; injections represent a clinically relevant range24,25,27 while transgenically expressed hAAT in both whole animal transfection29,31 and in a genetically engineered strain (hAAT+/+)30 represent low circulating hAAT levels, which all lead to an expansion of Treg cells. The mechanism of immunomodulation by hAAT has yet to be established.
The cellular targets of hAAT are a topic of recent interest. For example, it has been established that hAAT does not directly interfere with T-cell responses.25,27 Indeed, our findings indicate that the cellular targets of hAAT are predominantly antigen-presenting cells, such as macrophages and dendritic cells,32 which become tolerogenic in the presence of hAAT.25 Together with the lack of literature regarding hAAT and B lymphocytes, and the emerging role of these cells in important clinical conditions, the study of the effect of hAAT on B-lymphocyte responses is of great interest.
In the present study, we sought to establish whether the protective activities of hAAT might be directly related to B-lymphocyte modulation.
Materials and methods
Animals
Six- to eight–week-old BALB/c and C57BL/6 female mice were purchased from Harlan Laboratories Inc., Jerusalem, Israel, and were used as skin donors and skin recipients, respectively. hAAT transgenic mice that express hAAT under the surfactant promoter (background strain C57BL/6) were engineered as described previously.33 Interleukin-10-promoter-driven green fluorescent protein (GFP) transgenic mice and B-cell knockout mice were purchased from Jackson Laboratories, Inc. (Bar Harbor, ME) (B6.129S6-Il10tm1flv/J and B6.129S2-Ighmtm1Cgn/J, respectively). All mice were kept under specific pathogen-free conditions. Experiments were approved by the institutional animal care and use committee.
Cell isolation and culture
B220-positive and CD19-positive B-lymphocytes were isolated from spleens of C57BL/6 mice using magnetic beads, according to the manufacturer's instructions [B220 negative selection kit, EasySep®, STEMCELL (Vancouver, BC, Canada) and CD19 MicroBeads, MACS (Miltenyi Biotec, Bergisch Gladbach, Germany), respectively] resulting in > 95% purity. For IL-10 expression experiments, splenocytes were isolated from IL-10–GFP transgenic mice. Peritoneal macrophages were isolated from C57BL/6 mice by peritoneal lavage 72 hr after 3% thioglycolate (intraperitoneally). Isolated B cells, splenocytes and macrophages were cultured at 37° in 5% CO2 with complete RPMI-1640 medium containing 10% fetal calf serum, l-glutamine, penicillin and streptomycin. In all experiments, hAAT (0.5 mg/ml, Glassia, Kamada Ltd, Ness Ziona, Israel) was added 2 hr before stimulation. For IgM release assays, 1 × 105 B cells per well were cultured in 96-well round-bottom plates with LPS (1 μg/ml; Sigma-Aldrich, Rehovot, Israel) for 7 days. For activation marker expression, 1 × 106 B cells per well were cultured in 48-well plates and stimulated with LPS (100 ng/ml) or CD40 ligand combined with IL-4 (100 ng/ml; R&D Systems, Minneapolis, MN; 50 ng/ml; PeproTech, Rehovot, Israel, respectively). For IL-10-producing B-cell experiments, 3 × 106 splenocytes were cultured in six-well plates and stimulated with LPS (1 μg/ml) for 72 hr; 5 hr before harvest, PMA (50 ng/ml), ionomycin (500 ng/ml) both from Sigma-Aldrich and monensin (1 : 1000) from BioLegend® (San Diego, CA) were added. For the BAFF release assay, 0.5 × 106 peritoneal macrophages were cultured for 24, 48 and 72 hr in 48-well plates with IFN-γ (5 ng/ml, R&D Systems).
Assessment of proliferation marker expression, Ki-67
In vitro, B lymphocytes were isolated from spleens of C57BL/6 mice using a CD19-positive enrichment kit (CD19 MicroBeads, MACS, Miltenyi Biotec). Cells were cultured in 96-well round-bottom plates (2 × 105 B cells per well) and stimulated with LPS (1 μg/ml), CD40 ligand combined with IL-4 (100 ng/ml and 50 ng/ml, respectively) and recombinant BAFF (50 ng/ml; PeproTech) for 60 hr. hAAT (0.5 mg/ml) was added 2 hr before stimulation. Intracellular staining for Ki-67 was performed using an eBioscience staining kit (San Diego, CA) according to the manufacturer's instructions using Ki-67-allophycocyanin (APC). In vivo, B-lymphocyte proliferation marker expression in draining lymph nodes was evaluated according to surface expression of B-lymphocyte marker CD19 and intracellular Ki-67, 14 days after transplantation.
In vivo LPS stimulation
C57BL/6 and transgenic hAAT mice were injected intraperitoneally with LPS (1 mg/kg). Mice were harvested 72 hr after injection and splenocytes were evaluated by flow cytometry.
Skin transplantation
Allogeneic skin transplantation was performed as described.25 Briefly, donor BALB/c mice were harvested and shaved using surgical blade number 22. Skin was removed from the midline abdominal area directly into Petri plates containing sterile ice-cold PBS (BiologicalIndustries, Kibbutz Beit-Haemek, Israel). Blood vessels and hypodermis tissue were removed using a surgical blade. The skin was then cut under light stereomicroscope into 1 mm2 pieces. Skin was transplanted into the thigh region of recipient C57BL/6 mice through a 2-mm incision that was immediately closed with a 3-0 suture.
Immunization protocol
Wild-type and hAAT transgenic mice were immunized with BSA (Sigma-Aldrich), protocol and dosing, as described elsewhere.34,35 Briefly, solution containing 1 mg/ml BSA diluted in PBS, mixed with 100 μg/ml of complete Freund's adjuvant (CFA; Sigma-Aldrich) was emulsified using two glass syringes. Solution was injected intraperitoneally at a total volume of 200 μl per mouse. Mice were injected intraperitoneally for a second time 3 weeks later with 200 μl emulsified solution containing 1 mg/ml BSA and incomplete Freund's adjuvant (IFA; Sigma-Aldrich). Control mice were treated with CFA and IFA without BSA antigen. Serum was collected from the tail-vein at 1, 2, 3, 9 and 21 weeks after immunization and BSA-specific IgM and IgG titres were determined.
ELISA for BSA-specific IgM and IgG and for skin allo-antigen IgG
Antigen coating was performed in 96-well flat-bottom plates using 50 ng/ml BSA in PBS in a total volume of 100 μl, during 15 hr of incubation at room temperature. For detection of antibody response to skin-antigens, skin lysate was prepared by incubation with Tissue Protein Extraction Reagent (T-PER; Thermo Scientific, Waltham, MA) followed by an aggressive mechanical homogenizing protocol. Well-blocking was performed using 1% normal horse serum (Vector Laboratories, Inc., Burlingame, CA), diluted in PBS with 0.05% Tween-20 (TPBS) at a total volume of 300 μl. Plates were washed three times using TPBS. Sera were diluted in 100 μl PBS containing 1% normal horse serum (1 : 5000 for BSA-specific IgM ELISA, 1 : 125 000 for BSA-specific IgG ELISA, and 1 : 250 for skin-antigen ELISA, as optimized according to related calibration studies), and then loaded into wells and incubated for 3 hr at 4°. Wells were washed three times with TPBS before the addition of horseradish peroxidase-conjugated goat anti-mouse IgM or goat anti-mouse IgG (Invitrogen, Carlsbad, CA; 1 : 2000 and R&D Systems, 1 : 1000, respectively) in 100 μl PBS containing 1% normal horse serum. Samples were incubated for 2 hr. Plates were washed three times and then 100 μl TMB substrate (Southern Biotech, Birmingham, AL) was added. The reaction was stopped 10 min later using 100 μl HCl 0.5 m. Optical absorbance was determined by ELISA reader (BioTek instruments, Winooski, VT) at 450 nm wavelength. Total IgM was measured using total mouse IgM detection kit (R&D Systems).
RT-PCR
Total RNA from draining lymph nodes was extracted using PerfectPure RNA Tissue Kit (5 PRIME, Hamburg, Germany), according to the manufacturer's instructions. RNA concentration was determined using a spectrophotometer (NanoDrop, Thermo Scientific). Reverse transcription was performed using a Verso cDNA Kit (Thermo Scientific). PCR amplification was performed using XP cycler (BIOER, Hangzhou, China) using the following primers: mouse β-actin: forward 5′-GGGTCAGAAGGATTCCTATG-3′. reverse 5′-GGTCTCAAACATGATCTGGG-3′. mouse IL-4: forward 5′-ACGCCATGCACG GAGATGGA-3′. reverse 5′-CCAGGCATCGAAAAGCCCGA-3′. mouse BAFF: forward 5′-ACCCAGCCCTGCCATGCTCT-3′. reverse 5′-AGCGCGTCTGTTCCT GTGGC-3′.
Flow cytometry
Flow cytometry analysis was performed using FACSCalibur™ and FACSCanto™ II (Becton Dickinson) and analysed by CellQuest and Flowjo software. For in vitro experiments, 1 × 106 B220-positive and CD19-positive cells were stained with CD19-FITC, CD40-phycoerythrin (PE), CD86-PE, CD80-Peridinin chlorophyll protein-Cy5.5 (PerCP-Cy5.5) and MHCII-APC. For in vivo experiments 1 × 106 cells from inguinal draining lymph nodes or 1 × 106 splenocytes were stained with CD40-PE, B220-APC, CD19-FITC, CD86-PE, CD80-PerCP-Cy5.5 and MHCII-Pacific Blue. For intracellular staining of Ki-67 and foxp3, an eBioscience intracellular staining kit was used according to the manufacturer's instructions and staining was performed using Ki-67-APC and foxp3-FITC. All antibodies were purchased from Biolegend® and diluted according to the manufacturer's recommendations.
Assessment of BAFF and BAFF-R
Levels of BAFF were assessed by specific mouse BAFF ELISA (Quantikine®; R&D Systems). BAFF-R surface levels were induced by AffiniPure anti-F(ab')2 fragment donkey anti-mouse IgM (Jackson Immunoresearch, 25 μg/ml) and detected by FACS analysis using PE-labelled antibody at the recommended concentration.
Generation of chimeric mice
Transgenic hAAT mice were irradiated (1200 rad) and were introduced bone marrow from either B-cell knockout or transgenic hAAT mice. Animals with a restored immune system and below-detection B cells were used. Circulating levels of hAAT in transgenic hAAT mice and chimeric transgenic hAAT mice were determined by hAAT-specific ELISA (Immunology Consultants Laboratories, Portland, OR) and were uniform (not shown). Hence, the chimera represent mice that exhibit elevated circulating lung-derived hAAT and an immune system of a B-cell knockout strain.
Statistics
Comparisons between groups were performed using two-tailed Mann–Whitney U-test using GraphPad Prism 5.0 software (GraphPad Software, La Jolla, CA).
Results
hAAT modifies B lymphocyte responses to LPS in vitro and in vivo
The effect of hAAT on B lymphocyte responses was first addressed in vitro. Splenic B lymphocytes were isolated and cultured for several time periods in the presence of various concentrations of LPS. Before stimulation, cells were pre-incubated for 2 hr with the frequently employed concentration of hAAT (0.5 mg/ml).36 As shown in Fig. 1, responses evaluated at the end of the incubation period included assessment of Ki-67+ B cells, IgM release and surface expression of activation markers.
Figure 1.

Human α1-antitrypsin (hAAT) modifies B-lymphocyte responses to lipopolysaccharide (LPS). Spleen-derived bead-enriched B lymphocytes were isolated from C57BL/6 mice and incubated with LPS at indicated concentrations, with or without hAAT (0.5 mg/ml). CT represents untreated cells in all figure panels. (a) Intracellular Ki-67: cells were cultured in 96-well round-bottom plates (1 × 105/well, six replicates) and stimulated with LPS (1 μg/ml) for 60 hr. Intracellular Ki-67 staining gated for CD19+ B220+ cells. Results are presented as fold difference from CT. Right, representative FACS dot plot. (b) IgM release: cells were cultured in 96-well round-bottom plates (1 × 105/well, six replicates) and stimulated with LPS for 7 days. IgM levels were determined by ELISA. (c) Activation markers: cells were cultured in 48-well plates (1 × 106/well, four replicates) and stimulated with LPS (100 ng/ml) for 16 hr in the presence or absence of hAAT (dashed and solid lines, respectively). Untreated cells represent control group (shaded). Cells were stained for surface activation markers and gated for B220+ cells. Representative overlays are shown. (d) In vivo LPS stimulation: C57BL/6 and hAAT transgenic mice (n = 6 per group) were introduced intraperitoneally with LPS (1 mg/kg, solid and dashed lines, respectively). Control, untreated group (shaded). Splenocytes were isolated after 72 hr and analysed by FACS for activation markers. All panels are representative of two independent experiments. Mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001.
Lipopolysaccharide was introduced at the biologically effective concentration of 1 μg/ml. As shown in Fig. 1(a), the stimulated group displayed 6.34 ± 0.87-fold more Ki-67+ B cells than control non-stimulated cells (P = 0.016). However, cultures that were stimulated with LPS in the presence of hAAT exhibited near-background levels of Ki-67+ B cells.
Release of IgM antibodies was evoked using the same concentration of LPS (Fig. 1b). Stimulated cells exhibited a 1.49 ± 0.09-fold rise in total IgM compared with control cells (P < 0.01), whereas added hAAT resulted in near-background levels.
Cell surface activation markers were determined by FACS analysis in LPS-treated cells (100 ng/ml). As shown in Fig. 1(c), cultured B lymphocytes responded to LPS with elevated surface levels of CD40, CD86 and MHCII. However, LPS combined with hAAT exhibited a 1.45 ± 0.02-fold decrease in CD40HI expressing cells compared with LPS-stimulated cells (P < 0.05). CD86HI and MHCIIHI expression did not appear to change in the presence of hAAT.
We next examined in vivo changes in surface expression of the co-stimulatory molecules in response to systemic LPS. C57BL/6 and transgenic hAAT mice (n = 6 per each group) were injected with LPS (1 mg/kg). Expression of activation markers in splenic B lymphocytes was evaluated 72 hr later. As shown in Fig. 1(d), surface expression of CD40HI, CD86HI and MHCIIHI significantly increased after LPS treatment in comparison with the non-treated group (P < 0.01). On the other hand, surface expression on splenic B lymphocytes obtained from LPS-treated hAAT transgenic mice displayed significantly lower levels of surface expression of all markers examined, in comparison to LPS-treated control mice.
hAAT modifies CD40-related B-lymphocyte activation
As allograft rejection involves T-cell-dependent activation of B lymphocytes, we examined the effect of hAAT on the response of B lymphocytes to CD40 ligand. B lymphocytes were isolated and cultured with recombinant CD40 ligand combined with IL-4 (100 ng/ml and 50 ng/ml, respectively) in the presence or absence of hAAT. Ki-67 was assessed 72 hr after stimulation, while expression of activation markers CD86, CD80 and MHCII was evaluated 16 hr after stimulation.
As shown in Fig. 2(a), using Ki-67 intracellular staining we show that B lymphocytes displayed an increase in the Ki-67+ population in response to CD40 ligand. However, in the presence of hAAT, stimulated B lymphocytes exhibited near-background Ki-67 content. Similarly, as shown in Fig. 2(b), surface expression of CD86HI, CD80HI and MHCIIHI was significantly elevated upon stimulation, but partially responsive in the presence of hAAT (reduction in CD86HI, CD80HI and MHCIIHI-expressing cells of 1.07 ± 0.015-fold, 1.5 ± 0.04-fold and 1.14 ± 0.02-fold, respectively).
Figure 2.
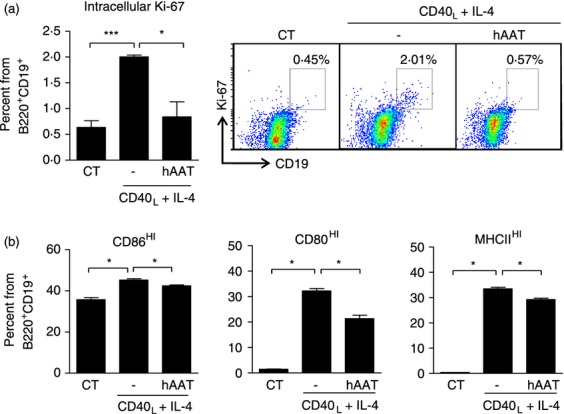
Human α1-antitrypsin (hAAT) modifies B-lymphocyte responses to CD40 stimulation. Spleen-derived bead-enriched B lymphocytes were isolated from C57BL/6 mice and incubated with CD40L combined with interleukin-4 (IL-4) (100 ng/ml and 50 ng/ml, respectively), with or without hAAT (0.5 mg/ml). CT represents untreated cells in all figure panels. (a) Intracellular Ki-67: cells were cultured in 96-well round-bottom plates (1 × 105/well, six replicates) and stimulated with CD40L combined with IL-4 for 60 hr. Intracellular Ki-67 staining gated for CD19+ B220+ cells. Right, representative FACS dot plots. (b) Activation markers: cells were cultured in 48-well plates (1 × 106/well, four replicates) and stimulated with CD40L combined with IL-4 for 16 hr. Cells were stained for surface activation markers and gated for CD19+ B220+ cells. All panels are representative of two independent experiments. Mean ± SEM, *P < 0.05, ***P < 0.001.
hAAT modifies B-lymphocyte responses in transplantation and vaccination models
The initial in vivo aspect that we sought to examine involves allograft transplantation (Fig. 3a–d). To evoke a potent and readily detectable immune response in allograft draining lymph nodes (DLN), skin transplantation was performed between major MHC disparate mouse strains. Both wild-type C57BL/6 and hAAT transgenic mice were grafted with 1 mm2 uni-size donor skin (BALB/c) in the subcutaneous compartment within the inner thigh region, and inguinal DLN were analysed 14 days later for Ki-67+ B lymphocytes, as well as for B-lymphocyte population size, IL-4 expression and B-lymphocyte activation markers.
Figure 3.

Human α1-antitrypsin (hAAT) modifies B-cell responses in vivo. (a–d) Skin allograft transplantation model and (e) vaccination model. Skin grafts from BALB/c donor mice (H-2d) were transplanted into the thigh region of wild-type C57BL/6 mice (H-2b) and hAAT transgenic mice (C57BL/6 backcrossed). Inguinal draining lymph nodes were removed 14 days after transplantation and analysed. (a) Intracellular Ki-67: expression of CD19 and Ki-67 was determined in draining lymph nodes (DLN) of transplanted wild-type and hAAT transgenic mice (n = 6 per group). Results are displayed as fold-change from non-treated wild-type mice. Right, representative FACS dot plots. (b) B-cell population size: percentage of B220+ CD19+ cells in DLN of non-treated and transplanted wild-type and transgenic hAAT mice (n = 8 per group). (c) IL-4 mRNA: β-actin-normalized IL-4 mRNA transcript levels in DLN of non-treated and transplanted wild-type and transgenic hAAT mice (n = 6 per group). (d) Activation markers: high expression of B-cell activation markers CD40 and MHCII in DLN of non-treated wild-type (CT) and transplanted wild-type and transgenic hAAT mice. (e) BSA-specific IgM and IgG ELISA in a vaccination model: wild-type and transgenic hAAT mice (n = 8 per group) were immunized with BSA/complete Freund's adjuvant (CFA) and 3 weeks later received a booster in the form of BSA/incomplete Freund's adjuvant (IFA). Non-immunized control group was injected with only CFA and IFA. Serum was collected at 1, 2, 3, 9 and 21 weeks after immunization. ELISA for BSA-specific IgM and IgG antibodies was performed in 96-well flat-bottom plate coated with 50 ng/ml of BSA. Samples were diluted (1 : 5000 for BSA-specific IgM and 1 : 125 000 for BSA-specific IgG) and incubated for 3 hr. Results displayed as fold change from non-immunized control. All graphs are representative out of at least two independent experiments. Mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001.
Skin graft survival was not measured in this model, because the model was used for the primary purpose of evoking a high-amplitude immune response with focus on B-lymphocyte responses. However, IgG antibody response to skin-antigen was evaluated, and found to display reduced IgG titres (1.29 ± 0.08-fold less anti-skin-Ag IgG in hAAT mice compared with wild-type mice at 6 weeks, P < 0.05). In addition, as shown in Fig. 3(a), the population of CD19+ Ki-67+ B lymphocytes increased 8.11 ± 2.06-fold in DLN obtained from transplanted wild-type mice, in comparison to the non-transplanted group, whereas CD19+ Ki-67+ B lymphocytes in DLN from transplanted transgenic hAAT mice were almost completely unaffected by the transplantation procedure, and displayed a decrease of 4.38 ± 1.1-fold from transplanted wild-type mice (P = 0.05). Representative FACS images are also shown.
The changes observed in Ki-67+ B lymphocytes are reflected in the B-cell population size in DLN, as shown in Fig. 3(b). Out of the total cell populations in the DLN, B220+ CD19+ population size in transplanted wild-type mice increased 1.36 ± 0.09-fold, compared with the wild-type non-transplanted mice (P = 0.0159). However, B-lymphocyte population size in the DLN of hAAT transplanted transgenic mice was near-normal. Of note, albeit without reaching statistical significance, non-transplanted hAAT transgenic mice (not shown) exhibited steady-state levels of B220+ CD19+ in DLN that were 1.25 ± 0.05-fold lower than in non-transplanted wild-type mice (P = 0.34).
To further depict B-lymphocyte downstream responses to the allogeneic skin transplant, that is, the T-cell-dependent aspect of activation, we examined DLN-related changes. DLN were examined by RT-PCR for changes in IL-4 transcript levels (Fig. 3c). As shown, graft recipient wild-type mice exhibited a 1.29 ± 0.08-fold increase in IL-4 transcript levels compared with non-grafted wild-type mice (P = 0.048). However, IL-4 transcript levels were in the non-stimulated range in DLN of transgenic hAAT recipient mice.
In a FACS analysis of the cellular content of DLN, changes in surface co-stimulation molecules were determined (Fig. 3d). As shown, we observed 1.19 ± 0.09-fold more MHCIIHI B220+ B lymphocytes and 1.29 ± 0.11-fold more CD40HI B220+ B lymphocytes in DLN of recipient wild-type mice than in DLN of non-grafted mice (P = 0.24 and P = 0.032, respectively). On the other hand, recipient hAAT transgenic mice displayed 1.39 ± 0.1-fold less MHCIIHI and 1.39 ± 0.11-fold less CD40HI B lymphocytes than recipient wild-type mice.
The effect of hAAT on downstream B-lymphocyte antigen-specific antibody production was addressed in a vaccination model (Fig. 3e). Wild-type and hAAT transgenic mice were introduced to antigenic BSA in a vaccination protocol; following the series of exposure and booster doses, circulating BSA-specific antibodies were determined at various time-points. As shown, 2 weeks after antigen introduction, BSA-specific IgM antibody levels increased 1.3 ± 0.12-fold in BSA-vaccinated animals compared with non-treated wild-type mice (open circles, P = 0.25), whereas BSA-specific IgG antibody levels increased 3 weeks after antigen introduction 22.58 ± 1.9-fold and 44.47 ± 6.85-fold after 9 weeks, in comparison to untreated wild-type mice (open circles, P < 0.001). On the other hand, BSA-specific IgM antibody levels were significantly elevated in hAAT transgenic mice (2.3 ± 0.36-fold, P < 0.001) 2 weeks after antigen introduction, in comparison to treated wild-type mice, whereas BSA-specific IgG antibody levels decreased in hAAT transgenic mice at 3 and 9 weeks after antigen introduction (5.0 ± 0.42-fold and 2.88 ± 0.44-fold from BSA-vaccinated wild-type mice, P = 0.03 and P = 0.11, respectively).
hAAT affects BAFF release and activity in vitro and in vivo
As our results indicate that hAAT interferes with B-lymphocyte activation both in culture and in an allogeneic skin transplantation model, we sought to establish the effect of hAAT on B-cell survival and activation factor, BAFF. Initially, we examined B-lymphocyte responsiveness to recombinant BAFF in culture (Fig. 4). Splenic B cells were cultured in the presence of recombinant BAFF (50 ng/ml), either alone or with hAAT (0.5 mg/ml). Ki-67 staining was performed 60 hr later. As shown in Fig. 4(a), Ki-67+ B lymphocytes increased 8.62 ± 0.25-fold in response to BAFF. However, stimulated culture in the presence of hAAT exhibited an intermediate response to BAFF, 2.04 ± 0.21-fold lower than that achieved in the presence of BAFF (P = 0.028). In vivo, allogeneic skin transplantation was performed and DLN were analysed for BAFF transcript levels 14 days later (Fig. 4b). Unexpectedly, BAFF transcript levels were grossly unchanged in DLN of recipient wild-type mice compared with non-grafted mice, but were 1.71 ± 0.08-fold greater in DLN from hAAT transgenic recipient animals compared with non-treated mice (P < 0.001).
Figure 4.

Human α1-antitrypsin (hAAT) affects B-cell activating factor (BAFF) pathway. (a) Intracellular Ki-67: 1 × 105 cells were cultured in 96-well round-bottom wells in six replicates and stimulated with recombinant BAFF (50 ng/ml) in the presence or absence of hAAT (0.5 mg/ml), for 60 hr. CT, non-treated cells. Ki-67+ CD19+ B220+ cells are shown as fold-change from control. (b) BAFF expression: skin allografts were transplanted into wild-type or transgenic hAAT mice. Inguinal draining lymph nodes were analysed for BAFF transcript levels 14 days later and normalized to β-actin. (c) BAFF release: peritoneal macrophages were stimulated with IFN-γ (5 ng/ml) and cultured in the presence or absence of hAAT. Accumulated BAFF in the supernatant was determined at 24, 48 and 72 hr after stimulation. (d) BAFF receptor expression: splenic B cells (2 × 106/well) were stimulated with anti-IgM for 16 hr in the presence or absence of hAAT pre-treatment (0.5 mg/ml). Top, pooled results; bottom, representative FACS analysis overlay histograms. Results are representative of at least two independent experiments. Mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001.
Macrophages are a major source of inducible BAFF; to study the effect of hAAT on macrophage-derived BAFF, IFN-γ was added to peritoneal macrophages in the presence or absence of hAAT (0.5 mg/ml). BAFF release was determined 24, 48 and 72 hr after stimulation. As shown in Fig. 4(c), BAFF accumulated in supernatants in a time-dependent manner, and displayed a significant increase in the presence of IFN-γ as early as 24 hr after stimulation. This trend was maintained throughout all incubation periods (circles compared with corresponding squares, P < 0.001 for all groups between control and stimulated cells at each time-point). On the other hand, BAFF release from cells stimulated with IFN-γ combined with hAAT (solid circles) resulted in 1.24 ± 0.04-fold more BAFF after 48 hr and in 1.34 ± 0.05-fold more BAFF 72 hr after stimulation.
The trend observed in steady-state non-stimulated BAFF (squares), was distinct from that achieved under the induction of IFN-γ. Hence, BAFF release from cultures treated with hAAT alone was lower compared with untreated cells (2.8 ± 0.46-fold, 2.14 ± 0.09-fold and 1.49 ± 0.12-fold at 24, 48 and 72 hr, respectively).
To examine whether hAAT affects BAFF-R expression on stimulated B cells, splenic B lymphocytes were isolated and then stimulated with anti-mouse IgM [anti-F(ab')2 fragment, 25 μg/ml], which promotes BCR signalling in the presence or absence of hAAT (0.5 mg/ml). As shown in Fig. 4(d), cultures stimulated with anti-mouse IgM exhibited a 3.52 ± 0.08-fold increase in BAFF-R expression, compared with non-treated control cells. On the other hand, the presence of hAAT in stimulated cultures resulted in lower BAFF-R levels, a 1.47 ± 0.05-fold decrease from those achieved in stimulated culture conditions (P < 0.01). Representative FACS histogram plots are shown in an overlay diagram.
hAAT increases IL-10-producing B-cell population size
As hAAT was found to increase IL-10 release in various cell types,25,32 we sought to investigate whether hAAT promotes IL-10-producing B cells in culture (Fig. 5a). Splenocytes were isolated from GFP-IL-10 mice and cultured with LPS (1 μg/ml) in the presence or absence of hAAT (0.5 mg/ml). Interleukin-10-producing B cells expressing CD1dHI, CD19 and B220 were identified by FACS analysis. As shown, stimulation of cells with LPS resulted in a 2.35 ± 0.05-fold increase in the proportion of IL-10-producing B cells compared with non-treated controls (CT, P < 0.001). On the other hand, cultures stimulated with LPS in the presence of hAAT resulted in a further significant increase in IL-10-producing B-cell population size, 1.5 ± 0.03-fold greater than levels observed in stimulated cells (P < 0.001).
Figure 5.
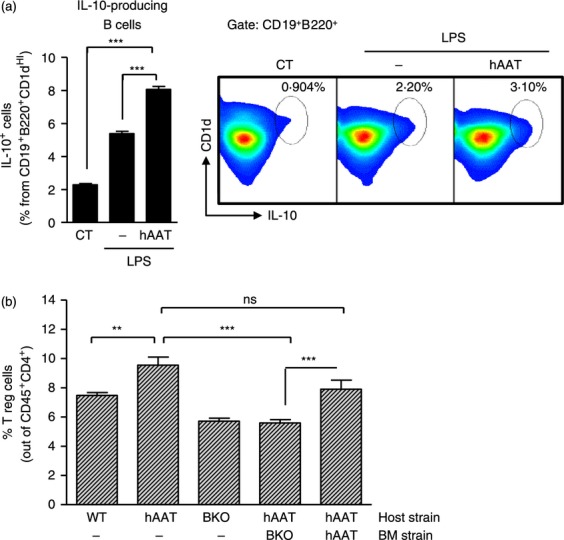
Human α1-antitrypsin (hAAT) promotes interleukin-10 (IL-10) -producing B cells and regulatory T (Treg) cells in a B-cell-dependent mechanism. (a) IL-10-producing B cells: splenocytes were isolated from IL-10-GFP transgenic mice, and cultured in 24-well plates at 3 × 106 cells/well in 10 replicates. Lipopolysaccharide (LPS) (1 μg/ml) was added in the presence or absence of hAAT (0.5 mg/ml). CT, untreated cells. Seventy-two hours later, IL-10-positive cells were identified by FACS gated for surface CD19, B220 and CD1d. Right, representative FACS images. (b) Regulatory T cells: leukocytes were obtained from peripheral blood of wild-type mice, hAAT transgenic mice, chimeric hAAT/hAAT mice, B-cell knockout mice (BKO) and chimeric BKO/hAAT mice (n > 5 per group). FACS analysis, regulatory T cells are shown as % foxp3+ cells out of CD45+ CD4+ lymphocytes. Representative results out of two independent repeats. Mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001.
hAAT increases Treg cell population size in a B-cell-dependent mechanism
Given that regulatory B cells were found to be involved in the induction of Treg cells, and that Treg cells were found to be associated with hAAT-mediated tolerance induction in allograft transplantation models, we sought to investigate whether the ability of hAAT to promote Treg cells might be B-cell-dependent. For this purpose, we generated chimeric hAAT transgenic mice that lack mature B lymphocytes by performing adoptive transfer of bone marrow from B-cell knockout (BKO) mice into irradiated hAAT transgenic mice. In addition, adoptive transfer from wild-type into irradiated hAAT transgenic mice was performed; however, this group was excluded in light of the ability of cells from the wild-type strain to mount an anti-hAAT antibody response with high animal mortality, as described previously.24 Irradiated hAAT transgenic mice were adoptively transferred with bone marrow from hAAT transgenic mice, as control. Treg cell population size in the circulation was determined by FACS analysis for CD4+ cells and for intracellular foxp3 (Fig. 5b). As shown, Treg cell population size in the circulation of hAAT transgenic mice was significantly greater than in WT mice, whereas BKO and BKO/hAAT mice displayed reduced levels of Treg cells.
Discussion
Human AAT displays a tolerogenic profile, particularly in the context of islet allograft transplantation; yet, besides the expansion of Treg cells and the alteration of dendritic cell responses, the complete cellular mechanism is unknown. In the current study we present data that support the involvement of B lymphocytes as cellular targets of hAAT in a manner that may take part in its tolerogenic activities during allogeneic transplantation. We show that LPS-stimulated cultures with hAAT added, displayed reduced B-lymphocyte activation and Ki-67+ B-cell population size, reduced IgM release, lower expression of activation markers CD40 and CD19, whereas, on the other hand, there was increased production and release of the anti-inflammatory cytokine, IL-10. Moreover, hAAT appears to affect T-cell-dependent B-cell activation pathways, characterized by lower expression of the co-stimulatory molecules CD80 and CD86 upon stimulation with CD40 ligand, a highly relevant stimulus for the engagement that occurs between B and T cells in the context of alloimmune responses. Lastly, hAAT positively affected IL-10-secreting B lymphocytes, characterized by expression of CD19, B220 and CD1d, and hAAT-driven Treg cells appeared to require an intact B-cell population.
The effect of hAAT on B lymphocytes was also illustrated in a skin allogeneic transplantation model, in which draining lymph nodes of recipient hAAT transgenic mice contained a smaller population of B lymphocytes, expressing less CD40 and MHC class II. These DLN also contained fewer mRNA transcripts for IL-4.
As we chose to focus on B-lymphocyte responses in a specific chosen time-point (14 days), skin graft survival was not measured in this experimental setup. However, antibody titres reflected a reduced IgG response, consistent with the vaccination model outcomes.
The effect of hAAT on events downstream to B-lymphocyte activation was specifically illustrated in the BSA-vaccination model. According to our results, transgenic hAAT BSA-vaccinated mice mounted a smaller BSA-specific IgG antibody response, but a greater BSA-specific IgM antibody response. This apparent inability to fulfill an isotype switch response is in agreement with a deficient CD40 pathway; together with IL-4, CD40 : CD40 ligand interaction is crucial for successful class switch recombination. In humans, hyper-IgM syndrome is a result of overt disruption in the CD40 pathway, resulting in excess circulating IgM levels.37 We suggest that the elevated levels of BSA-specific IgM isotype antibodies and reduced levels of BSA-specific IgG isotype antibodies are mostly the result of the ability of hAAT to interfere with the CD40 pathway.
Interestingly, when comparing the in vitro effects of hAAT on B cells and the in vivo changes in their response profile, an overlap is only partially obtained. Hence, while LPS-induced MHCII up-regulation is unaffected by hAAT in vitro, its levels on the surface of B cells is diminished significantly in the skin graft model. This may be attributed to the cellular responders to hAAT in the whole animal, such as dendritic cells,32 and the elaboration of the transplant setting as a whole under hAAT therapy, distinguishing it from cultured isolated enriched B cells.
The effect of hAAT on BAFF was unexpected. The increase in BAFF secretion in the presence of hAAT in vitro was supported by in vivo findings, according to which DLN of recipient hAAT transgenic mice contained more mRNA transcripts for BAFF than control recipient wild-type mice. We present two possible explanations for the observed rise in BAFF: (1) the cells are less responsive to BAFF in the presence of hAAT and (2) the cells express less BAFF receptor. According to our results, both options are viable; in vitro B lymphocytes stimulated with recombinant BAFF in the presence of hAAT displayed a reduced proportion of Ki-67+ cells, and the expression of anti-mouse IgM-induced BAFF receptor was diminished in the presence of hAAT. These findings are in line with recent evidence suggesting that abnormal BAFF signalling may be involved in several immunologically-driven mechanisms, such as rejection or loss of graft function.20
Although the effect of hAAT on B-lymphocyte activation appears potent, our results do show that essential properties that concern the ability of B lymphocytes to induce an effective response against bacterial antigens remain intact. In culture, CD86 and MHCII, important molecules for antigen presentation and activation of T lymphocytes, were unaffected by hAAT. Moreover, in vivo, in a non-sterile stimulation provoked by intestinal caecal puncture, hAAT transgenic mice displayed similar levels of circulating IgM isotype immunoglobulin in comparison to control wild-type (not shown). In this model, peritoneal fluids of wild-type mice contained elevated bacterial load upon harvest, while hAAT transgenic mice contained trace levels of bacteria, supporting the concept that the effect of hAAT does not compromise host response to bacterial infection. These data complement the findings of the ability of hAAT transgenic antigen-vaccinated mice to mount a higher antigen-specific IgM antibody response, which may compensate for the overall reduced activation profile. The aspect of protection from bacterial infection by hAAT is currently under investigation.
Lastly, our results suggest that the presence of an IL-10-producing B-cell population is elevated by hAAT. As the ability of hAAT to induce allograft tolerance was found to involve an expansion of Treg cells,25,27,29 we suggest that this phenomenon may in fact involve regulatory B cells. To establish whether the IL-10-producing B cells in the current study are indeed Breg cells and comply with functions and related markers (e.g. CD5/CD21/CD23/CD24), more studies are required. Nevertheless, we show in Fig. 5(b) that the rise of Treg cells in hAAT transgenic mice is abrogated in BKO/hAAT transgenic chimeric mice.
Further studies are required to fully elucidate the molecular mechanisms by which hAAT modifies B-cell responses, and, in light of its impressive clinical safety record, whether hAAT can modify other pathologies that involve excessive B-cell responses.
Acknowledgments
This study was supported by the Juvenile Diabetes Research Foundation (JDRF) and Israel Science Foundation (ISF) – JDRF Joint Program in Type I Diabetes Research. We thank Mrs Valeria Frischman and Mr Rafi Srebro for their excellent technical assistance.
Glossary
- APC
allophycocyanin
- BKO
B cell-knockout
- BAFF
B-cell activating factor
- CD40L
CD40 ligand
- CFA
complete Freund's adjuvant
- DLN
draining lymph node
- GFP
green fluorescent protein
- hAAT
human α1-antitrypsin
- IFA
incomplete Freund's adjuvant
- IFN
interferon
- IL-4
interleukin-4
- LPS
lipopolysaccharide
- NOD
non-obese diabetic
- PE
phycoerythrin
- PerCP
Peridinin chlorophyll protein
- TPBS
PBS/Tween
Disclosures
The authors have no potential conflict of interest.
References
- 1.Shapiro AM. Strategies toward single-donor islets of Langerhans transplantation. Curr Opin Organ Transplant. 2011;16:627–31. doi: 10.1097/MOT.0b013e32834cfb84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang X, Moore DJ, Ketchum RJ, Nunemaker CS, Kovatchev B, McCall AL, Brayman KL. Resolving the conundrum of islet transplantation by linking metabolic dysregulation, inflammation, and immune regulation. Endocr Rev. 2008;29:603–30. doi: 10.1210/er.2008-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leitao CB, Cure P, Tharavanij T, Baidal DA, Alejandro R. Current challenges in islet transplantation. Curr Diab Rep. 2008;8:324–31. doi: 10.1007/s11892-008-0057-3. [DOI] [PubMed] [Google Scholar]
- 4.Hilbrands R, Huurman VA, Gillard P, et al. Differences in baseline lymphocyte counts and autoreactivity are associated with differences in outcome of islet cell transplantation in type 1 diabetic patients. Diabetes. 2009;58:2267–76. doi: 10.2337/db09-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kessler L, Parissiadis A, Bayle F, et al. Evidence for humoral rejection of a pancreatic islet graft and rescue with rituximab and IV immunoglobulin therapy. Am J Transplant. 2009;9:1961–6. doi: 10.1111/j.1600-6143.2009.02711.x. [DOI] [PubMed] [Google Scholar]
- 6.Liu C, Noorchashm H, Sutter JA, et al. B lymphocyte-directed immunotherapy promotes long-term islet allograft survival in nonhuman primates. Nat Med. 2007;13:1295–8. doi: 10.1038/nm1673. [DOI] [PubMed] [Google Scholar]
- 7.Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med. 2009;361:2143–52. doi: 10.1056/NEJMoa0904452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noorchashm H, Lieu YK, Noorchashm N, et al. I-Ag7-mediated antigen presentation by B lymphocytes is critical in overcoming a checkpoint in T cell tolerance to islet beta cells of nonobese diabetic mice. J Immunol. 1999;163:743–50. [PubMed] [Google Scholar]
- 9.O'Neill SK, Liu E, Cambier JC. Change you can B(cell)eive in: recent progress confirms a critical role for B cells in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes. 2009;16:293–8. doi: 10.1097/MED.0b013e32832e06a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Silveira PA, Grey ST. B cells in the spotlight: innocent bystanders or major players in the pathogenesis of type 1 diabetes. Trends Endocrinol Metab. 2006;17:128–35. doi: 10.1016/j.tem.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 11.Lund FE. Cytokine-producing B lymphocytes-key regulators of immunity. Curr Opin Immunol. 2008;20:332–8. doi: 10.1016/j.coi.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pulendran B, Ahmed R. Translating innate immunity into immunological memory: implications for vaccine development. Cell. 2006;124:849–63. doi: 10.1016/j.cell.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 13.Clatworthy MR, Espeli M, Torpey N, Smith KG. The generation and maintenance of serum alloantibody. Curr Opin Immunol. 2010;22:669–81. doi: 10.1016/j.coi.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fairfax KA, Kallies A, Nutt SL, Tarlinton DM. Plasma cell development: from B-cell subsets to long-term survival niches. Semin Immunol. 2008;20:49–58. doi: 10.1016/j.smim.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, Cerutti A. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. 2002;3:822–9. doi: 10.1038/ni829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith SH, Cancro MP. Cutting edge: B cell receptor signals regulate BLyS receptor levels in mature B cells and their immediate progenitors. J Immunol. 2003;170:5820–3. doi: 10.4049/jimmunol.170.12.5820. [DOI] [PubMed] [Google Scholar]
- 17.Moore PA, Belvedere O, Orr A, et al. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science. 1999;285:260–3. doi: 10.1126/science.285.5425.260. [DOI] [PubMed] [Google Scholar]
- 18.Do RK, Hatada E, Lee H, Tourigny MR, Hilbert D, Chen-Kiang S. Attenuation of apoptosis underlies B lymphocyte stimulator enhancement of humoral immune response. J Exp Med. 2000;192:953–64. doi: 10.1084/jem.192.7.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darce JR, Arendt BK, Wu X, Jelinek DF. Regulated expression of BAFF-binding receptors during human B cell differentiation. J Immunol. 2007;179:7276–86. doi: 10.4049/jimmunol.179.11.7276. [DOI] [PubMed] [Google Scholar]
- 20.Thibault-Espitia A, Foucher Y, Danger R, et al. BAFF and BAFF-R levels are associated with risk of long-term kidney graft dysfunction and development of donor-specific antibodies. Am J Transplant. 2012;12:2754–62. doi: 10.1111/j.1600-6143.2012.04194.x. [DOI] [PubMed] [Google Scholar]
- 21.Hussain S, Delovitch TL. Intravenous transfusion of BCR-activated B cells protects NOD mice from type 1 diabetes in an IL-10-dependent manner. J Immunol. 2007;179:7225–32. doi: 10.4049/jimmunol.179.11.7225. [DOI] [PubMed] [Google Scholar]
- 22.Carter NA, Vasconcellos R, Rosser EC, et al. Mice lacking endogenous IL-10-producing regulatory B cells develop exacerbated disease and present with an increased frequency of Th1/Th17 but a decrease in regulatory T cells. J Immunol. 2011;186:5569–79. doi: 10.4049/jimmunol.1100284. [DOI] [PubMed] [Google Scholar]
- 23.Amu S, Saunders SP, Kronenberg M, Mangan NE, Atzberger A, Fallon PG. Regulatory B cells prevent and reverse allergic airway inflammation via FoxP3-positive T regulatory cells in a murine model. J Allergy Clin Immunol. 2010;125:1114–24. doi: 10.1016/j.jaci.2010.01.018. e8. [DOI] [PubMed] [Google Scholar]
- 24.Lewis EC, Shapiro L, Bowers OJ, Dinarello CA. α1-Antitrypsin monotherapy prolongs islet allograft survival in mice. Proc Natl Acad Sci U S A. 2005;102:12153–8. doi: 10.1073/pnas.0505579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lewis EC, Mizrahi M, Toledano M, Defelice N, Wright JL, Churg A, Shapiro L, Dinarello CA. α1-Antitrypsin monotherapy induces immune tolerance during islet allograft transplantation in mice. Proc Natl Acad Sci U S A. 2008;105:16236–41. doi: 10.1073/pnas.0807627105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pileggi A, Molano RD, Song S, et al. α-1 antitrypsin treatment of spontaneously diabetic nonobese diabetic mice receiving islet allografts. Transplant Proc. 2008;40:457–8. doi: 10.1016/j.transproceed.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 27.Koulmanda M, Bhasin M, Hoffman L, et al. Curative and β cell regenerative effects of α1-antitrypsin treatment in autoimmune diabetic NOD mice. Proc Natl Acad Sci U S A. 2008;105:16242–7. doi: 10.1073/pnas.0808031105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weir GC, Koulamnda M. Control of inflammation with α1-antitrypsin: a potential treatment for islet transplantation and new-onset type 1 diabetes. Curr Diab Rep. 2009;9:100–2. doi: 10.1007/s11892-009-0018-5. [DOI] [PubMed] [Google Scholar]
- 29.Shahaf G, Moser H, Ozeri E, Mizrahi M, Abecassis A, Lewis EC. α1-antitrypsin gene delivery reduces inflammation, increases T-regulatory cell population size and prevents islet allograft rejection. Mol Med. 2011;17:1000–11. doi: 10.2119/molmed.2011.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanian S, Shahaf G, Ozeri E, Miller LM, Vandenbark AA, Lewis EC, Offner H. Sustained expression of circulating human α1 antitrypsin reduces inflammation, increases CD4+ FoxP3+ Treg cell population and prevents signs of experimental autoimmune encephalomyelitis in mice. Metab Brain Dis. 2011;26:107–13. doi: 10.1007/s11011-011-9239-9. [DOI] [PubMed] [Google Scholar]
- 31.Lu Y, Tang M, Wasserfall C, et al. α1-antitrypsin gene therapy modulates cellular immunity and efficiently prevents type 1 diabetes in nonobese diabetic mice. Hum Gene Ther. 2006;17:625–34. doi: 10.1089/hum.2006.17.625. [DOI] [PubMed] [Google Scholar]
- 32.Ozeri E, Mizrahi M, Shahaf G, Lewis EC. α1 antitrypsin promotes semimature, IL-10-producing and readily migrating tolerogenic dendritic cells. J Immunol. 2012;189:146–53. doi: 10.4049/jimmunol.1101340. [DOI] [PubMed] [Google Scholar]
- 33.Dhami R, Zay K, Gilks B, Porter S, Wright JL, Churg A. Pulmonary epithelial expression of human α1-antitrypsin in transgenic mice results in delivery of α1-antitrypsin protein to the interstitium. J Mol Med (Berl) 1999;77:377–85. doi: 10.1007/s001090050364. [DOI] [PubMed] [Google Scholar]
- 34.Wang LF, Chen JS, Hsu CJ, Liu CY, Yu JS, Miaw SC. Antigen-driven bystander effect accelerates epicutaneous sensitization with a new protein allergen. J Biomed Sci. 2009;16:28. doi: 10.1186/1423-0127-16-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fuller SA, Takahashi M, Hurrell JG. Immunization of mice. Curr Protoc Mol Biol. 2001 doi: 10.1002/0471142727.mb1104s18. doi: 10.1002/0471142727.mb1104s18. [DOI] [PubMed] [Google Scholar]
- 36.Lewis EC. Expanding the clinical indications for α1-antitrypsin therapy. Mol Med. 2012;18:957–70. doi: 10.2119/molmed.2011.00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Winkelstein JA, Marino MC, Ochs H, Fuleihan R, Scholl PR, Geha R, Stiehm ER, Conley ME. The X-linked hyper-IgM syndrome: clinical and immunologic features of 79 patients. Medicine (Baltimore) 2003;82:373–84. doi: 10.1097/01.md.0000100046.06009.b0. [DOI] [PubMed] [Google Scholar]