

Abstract
We have previously described two types of protective B-cell epitopes in the O-antigen (OAg) of the Gram-negative bacterium Francisella tularensis: repeating internal epitopes targeted by the vast majority of anti-OAg monoclonal antibodies (mAbs), and a non-overlapping epitope at the non-reducing end targeted by the previously unique IgG2a mAb FB11. We have now generated and characterized three mAbs specific for the non-reducing end of F. tularensis OAg, partially encoded by the same variable region germline genes, indicating that they target the same epitope. Like FB11, the new mAbs, Ab63 (IgG3), N213 (IgG3) and N62 (IgG2b), had higher antigen-binding bivalent avidity than internally binding anti-OAg mAbs, and an oligosaccharide containing a single OAg repeat was sufficient for optimal inhibition of their antigen-binding. The X-ray crystal structure of N62 Fab showed that the antigen-binding site is lined mainly by aromatic amino acids that form a small cavity, which can accommodate no more than one and a third sugar residues, indicating that N62 binds mainly to the terminal Qui4NFm residue at the nonreducing end of OAg. In efficacy studies with mice infected intranasally with the highly virulent F. tularensis strain SchuS4, N62, N213 and Ab63 prolonged survival and reduced blood bacterial burden. These results yield insights into how antibodies to non-reducing ends of microbial polysaccharides can contribute to immune protection despite the smaller size of their target epitopes compared with antibodies to internal polysaccharide regions.
Keywords: bacteria/bacterial immunity, endotoxin/lipopolysaccharide, epitopes, immunoglobulins, vaccines
Introduction
Francisella tularensis, the Gram-negative facultative intracellular bacterium that causes tularaemia, has been classified by the Centers for Disease Control and Prevention as a category A priority pathogen, a likely bioterrorism agent.1–4 As few as 10 bacteria can cause respiratory tularaemia, the most severe form of the disease, with up to 30% mortality if untreated.1–5 Even when treated with antibiotics, respiratory tularaemia is still associated with considerable morbidity and up to 2% mortality.2–6 An attenuated type B live vaccine strain (LVS) partially protects against the highly virulent type A F. tularensis in humans but is not currently licensed because of safety concerns.6,7 Identification of protective F. tularensis antigens and epitopes will facilitate the development of potentially safer, subunit vaccines for tularaemia.
Lipopolysaccharide (LPS), the main component of the F. tularensis outer membrane, which is identical between type A and type B F. tularensis strains,8–12 is a main protective antigen in mice and circumstantially in humans.13–22 It is comprised of lipid A, a core oligosaccharide (C, mainly Hex4HexNAcKdo) and an O-polysaccharide [O-antigen (OAg)].8–12,23,24 The OAg consists of variable numbers of the tetrasaccharide repeat [2)-β-d-4,6- dideoxy-4-formamido-d-glucose(1→4)-α-d-2-acetamido-2 -deoxy-d-galacturonamide(1→4)-α-d-2-acetamido-2-deoxy-d-galacturonamide(1→3)-β-d-2-acetamido-2, 6-dideoxy-d-glucose(1→] [abbreviated as Qui4NFm-Gal NAcAN-GalNAcAN-QuiNAc or (oag)1], with Qui4NFm at the non-reducing end.8–12,23 Francisella tularensis capsular polysaccharide also consists of OAg.23,24
We have previously reported that anti-F. tularensis LPS mouse monoclonal antibodies (mAbs) can confer survival to BALB/c mice infected intranasally (i.n.) with an otherwise lethal dose of LVS, with the IgG2a isotype, the mouse analogue of human IgG1,25 being more effective than mouse IgG1 and IgG3.26 Subsequently, we found that the anti-LPS mAbs target OAg, and characterized the mouse IgG2a anti-OAg mAbs FB11 and Ab52,27 showing that both prolong survival of, and reduce blood bacterial burden in, BALB/c mice infected i.n. with the highly virulent F. tularensis type A strain SchuS4.28
We also showed that FB11 targets a terminal OAg epitope as exemplified by its even binding to both long and short chains of the LPS ladder on Western blots, whereas Ab52 targets an internal repeating OAg epitope as exemplified by the decrease in its binding intensity with decreasing LPS chain length.27 Using F. tularensis oligosaccharides of defined OAg-repeat length as molecular rulers in competition ELISA, the epitope targeted by FB11 was shown to span one tetrasaccharide repeat, whereas the epitope targeted by Ab52 was shown to span two tetrasaccharide repeats.28 The X-ray crystal structure of Ab52 Fab and computational studies revealed that the antigen-binding site of Ab52 has the shape of a large groove with a central pocket that accommodates a V-shaped epitope consisting of six sugar residues.29 The FB11 mAb, reported to have been obtained from BALB/c mice immunized with LVS or with F. tularensis LPS in complete Freund's adjuvant, using ‘different immunization schemes’,30 is a commercially sold hybridoma antibody whose X-ray crystal structure is unavailable. Furthermore, the anti-F. tularensis LPS mAbs reported by our group26,27 and others,31–35 all showed the Western blot binding pattern of Ab52, not FB11, suggesting that they target internal repeating OAg epitopes and that the repeating internal epitopes of F. tularensis OAg are much more immunogenic than the non-reducing OAg terminus. This is expected based on the higher multivalent binding between internal repeating OAg epitopes and B-cell receptor molecules during in vivo antigen stimulation of a B-cell, which was illustrated by the essentially irreversible multivalent binding between anti-immunoglobulin-captured Ab52 and OAgC (KD = 4·4 × 10–13 m) in surface plasmon resonance analysis.27 In contrast, the monovalent interaction between anti-immunoglobulin-captured FB11 and OAgC successfully measured the affinity of FB11 (KD = 4·0 × 10−7 m).27
As the antigen-binding affinity (i.e. the binding strength of a single binding site) of the intact Ab52 antibody could not be measured, the bivalent avidity of Ab52 and FB11 soluble antibodies for LPS was measured, which showed FB11 to have a 72-fold lower KD (higher bivalent avidity) than Ab52, the most avid of three IgG2a internally binding anti-OAg mAbs.27 This is presumably because greater complementarity can be achieved by head-on binding to a terminal epitope than by sideways binding to an internal epitope of a linear carbohydrate chain.
In an attempt to obtain additional terminally binding anti-OAg mAbs and study their antigen-binding characteristics, we generated hybridomas from BALB/c mice immunized with short LPS chains, capsule-enriched, or outer membrane-enriched F. tularensis preparations. We report here the binding characteristics and in vivo efficacy of one IgG2b and two IgG3 mAbs specific for the non-reducing end of F. tularensis OAg and the X-ray crystal structure of the IgG2b mAb.
Materials and methods
Bacterial strains and hybridoma antibodies
Francisella tularensis holarctica strain LVS was obtained from Dr J. Petersen (Centers for Disease Control and Prevention, Fort Collins, CO). Francisella tularensis strain SchuS4 was obtained from BEI Resources, Manassas, VA. Escherichia coli strain TG1 was purchased from Stratagene (La Jolla, CA). WbtIG191V (WbtI), an OAg-deficient LVS mutant,36 was obtained from Dr T. Inzana of Virginia Polytechnic Institute and State University, Blacksburg, VA. All strains were propagated and heat-inactivated as previously described.26
Protein G-purified mouse IgG2a mAb FB11, specific for F. tularensis OAg,30 and mouse IgG2a mAb GTX40330, specific for E. coli J5 LPS, were purchased from GeneTex® Inc. (Irvine, CA). Generation of anti-F. tularensis OAg mAbs Ab2 (IgG3)26 and Ab5227 was previously reported. Mouse hybridoma cell line CO17-1A, producing an IgG2a mAb specific for the human tumour-associated antigen EpCam,37,38 was obtained from Dr D. Herlyn of the Wistar Institute (Philadelphia, PA). Mouse hybridoma cell line TIB-114, producing an IgG3 mAb against sheep red blood cells, was obtained from ATCC (Manassas, VA).
The Ab63 mAb was generated in the current study by i.n. immunization of a 5-week-old BALB/cJ mouse (Jackson Laboratory, Bar Harbor, ME) with the sublethal dose of 2 × 103 colony-forming units (CFU) of LVS followed 17 days later by an intraperitoneal (i.p.) booster immunization with a mixture of LVS components containing short F. tularensis LPS chains (under a protocol approved by the Boston University Medical Center Institutional Animal Care and Use Committee). The mixture had been prepared by electrophoresis of an LVS lysate in a preparative 4–15% gradient SDS–polyacrylamide gel (BioRad, Hercules, CA), excision of the lower end components (7000–20 000 molecular weight range), and extraction by electroelution and dialysis against PBS to remove SDS with a D-tube™ Dialyzer (3500 molecular weight cutoff, Novagen, Madison, WI). Three days after the booster immunization, the spleen cells were harvested and used for fusion with Sp2/0-Ag14 mouse myeloma cells39 as previously described.26 Ab63 was determined to be an IgG3(κ) Ab by ELISA using the Mouse MonoAb ID Kit (HRP) from Zymed Laboratories (South San Francisco, CA).
Monoclonal antibodies N62 and N213 were also generated in the current study by intradermal immunization of a 24-week-old (N62) or a 13-week-old (N213) BALB/cJ mouse on day 0 with 2 × 107 or 2 × 105 CFU of LVS, i.p. booster immunization on day 56 or 66 with an outer membrane- or capsule-enriched LVS preparation, for N62 or N213, respectively, followed 3·75 days later by fusion of splenocytes with Sp2/0-Ag14 myeloma cells, as previously described.26 The LVS membrane preparation had been obtained by ultracentrifugation (200 000 g for 2 hr) of the supernatant of an LVS vortexate. The LVS capsule preparation had been obtained by high-salt extraction, as described by Hood.40 N62 and N213 were determined to be IgG2b(κ) and IgG3(κ), respectively, by IsoStrip (Mouse Monoclonal Antibody Isotyping Kit, Roche Diagnostics, Indianapolis, IN).
Purification of mAbs
Hybridoma cells were cultured in Iscove's modified Dulbecco's medium (GIBCO, Grand Island, NY) supplemented with 50 μg/ml gentamicin (IMDMg) supplemented with 10% fetal bovine serum, and grown to mass culture in IMDMg supplemented with 2% fetal bovine serum in 10-cm OPTILUX™ Petri dishes (Becton Dickinson Labware, Franklin Lakes, NJ) or in a CELLine classic 1000 two-compartment bioreactor (Wilson Wolf Manufacturing, New Brighton, MN) at 37° in a humidified environment of 5% CO2/95% air. The mAbs were separately purified from culture supernatants on PIERCE® Protein G (IgG1, IgG2a, IgG2b) Plus or Protein A (IgG3) Plus Agarose (Thermo Scientific, Rockford, IL) according to the manufacturer's instructions (modified to use 0·1 m sodium acetate pH 5·0 for elution of IgG3), and their purity and specificity were verified by SDS–PAGE and Western blot analysis on SchuS4, respectively.
Immunoassays and Biacore analysis
Direct and isotype-specific competition ELISA, Western blot analysis, and surface plasmon resonance (Biacore) analysis to determine equilibrium dissociation constants (KD) were performed as previously described.27 Bacterial microagglutination was performed as previously described,27 except for using less concentrated bacterial suspensions. Competition ELISA with purified F. tularensis LPS oligosaccharides23 was also performed as previously described.28 EIA/RIA plates were coated with 0·0625 μg/ml F. tularensis LPS when FB11 or Ab52 was used as reporter, with 0·15625 μg/ml F. tularensis LPS when N213 or N62 was used as reporter, or with 0·25 μg/ml F. tularensis LPS when Ab63 was used as reporter. The binding of a fixed concentration of reporter mAb (0·125 μg/ml for FB11, 5 μg/ml for Ab52, 1·2 μg/ml for N62, 0·158 μg/ml for N213 and 10 μg/ml for Ab63), in the presence of 10-fold serial dilutions of competitor oligosaccharides was determined using a secondary antibody specific for the isotype of the respective reporter mAb. Horseradish peroxidase-conjugated heavy-chain isotype-specific anti-mouse secondary antibodies (from SouthernBiotech, Birmingham, AL) were used for all ELISAs. For isotype-specific competition ELISA, EIA/RIA plates were coated as described above. The binding of a fixed concentration of reporter mAb (0·125 μg/ml for FB11, 5 μg/ml for Ab52, 1·2 μg/ml for N62 and 10 μg/ml for Ab63), in the presence of threefold serial dilutions of competitor mAb (starting at 500 or 800 μg/ml) were determined using secondary antibody specific for the isotype of the respective reporter mAb. The presence/binding of competitor mAbs to LPS were verified in a duplicate plate with secondary antibody specific for the isotype of the respective competitor mAb. Per cent inhibition was calculated (after subtracting the blank from all values) using the following formula: [(OD without competitor – OD with competitor)/(OD without competitor)] × 100; where OD is optical density. For Western blots, alkaline phosphatase-conjugated anti-mouse IgG (H + L) (Promega, Madison, WI) was used as secondary antibody, and the assay was developed with Western Blue stabilized substrate for alkaline phosphatase (Promega).
Nucleotide and deduced amino acid sequence determination
Variable region nucleotide sequences of mAbs were obtained from RT-PCR products as previously described for the Ab52 mAb.29 Homology to immunoglobulin germline genes was determined by IgBlast (http://www.ncbi.nlm.nih.gov/igblast/), and conversion to amino acid sequences was done by emboss Transeq (http://www.ebi.ac.uk/Tools/st/emboss_transeq/).
Fab X-ray crystal structure determination
N62 Fab was prepared from purified IgG using the Pierce® Fab Preparation Kit (Thermo, Rockford, IL). For crystallization trials, the Fab was dialysed and concentrated/diafiltered in a YM-10 Centricon Centrifugal Filter Device (Millipore, Bedford, MA) into 20 mm Tris–HCl pH 7·5, 150 mm NaCl, 0·02% NaN3, to 12 mg/ml as determined by absorbance at 280 nm. A sparse matrix screen was carried out using the reagents in the Index HT kit (Hampton Research, Aliso Viejo, CA) in a 96-well Corning Crystal EX microplate in sitting drops.
Initial screening resulted in clusters of crystals. These were optimized through microseeding in hanging drops by mixing 0·5 μl of Fab with 0·5 μl of reservoir solution [0·1 m bistris (pH 6·5), 0·2 m ammonium acetate, 25% weight/volume PEG 3350] on a siliconized coverslip, which was inverted over a well containing 0·5 ml of reservoir solution and sealed. After overnight equilibration at 17°, the drop was streak-seeded using a cat whisker that had been dipped into a seed stock mixture made by crushing a crystal cluster in 50 μl of reservoir solution. Before freezing in a nitrogen stream, the crystal used for diffraction studies was cryoprotected by soaking for 20 seconds in 10 μl of 4 : 1 reservoir solution and 85% volume/volume glycerol.
For ligand-soaking trials, crystals were transferred to a drop containing reservoir solution supplemented with 0·1 m N-acetylgalactosamine or purified oligosaccharide (oag)1C (3 μm estimated concentration; higher concentrations cracked the crystals) or (oag)1 (13 μm estimated concentration)23 for 10–60 min before soaking in the cryoprotectant solution. Diffraction data were collected on a RAXIS-IV imaging plate detector mounted on a Rigaku RU-300 rotating anode generator. Data indexing, integration and merging were carried out using the programs DENZO and Scalepack.41 The space group was determined to be either P212121 or P21212 (the alignment of the crystal during data collection prevented the determination of the third screw axis). To solve the N62 Fab structure, molecular replacement was employed as implemented in the AutoMR function in Phenix42 using as search models the heavy (H) chain of the mouse anti-lysozyme Fab HyHEL-10 (IgG2a)43 and the κ light (L) chain of the mouse anti-gp41 Fab NC-41 (PDB code 3OZ9; R.L. Stanfield, D.A. Calarese, S. Jiang, and I.A. Wilson, unpublished data). Each search model was divided into variable and constant domains for a total of four search models. In addition, the CDR3 loops of the variable domains were removed. In the first search, the variable domains of N62 Fab were placed, aided by the presence of pseudotranslational symmetry in the crystal. The constant domains were placed without accounting for the pseudotranslation. At this point, the space group was determined to be P212121. The molecular replacement solution was rebuilt with AutoBuild in Phenix, with the starting model not included in the building of the output structure so as to reduce phase bias.
Iterative cycles of manual rebuilding and refinement were performed using Coot44 and Phenix. For refinement of the final structure, individual atomic displacement parameters (ADPs) were refined using non-crystallographic symmetry (NCS) restraints and translation/libration/screw (TLS) parameters as determined by the programs in phenix.refine. The ligand-soaked crystals were solved using Difference Fourier in Phenix with the unliganded model as a starting structure.
In vivo efficacy studies
All animal procedures were approved by the Boston University Institutional Animal Care and Use Committee. BALB/cJ female mice were obtained from Jackson Laboratories (Bar Harbor, Maine) at 7–8 weeks of age, and inoculated i.n. with F. tularensis SchuS4 under ketamine/xylazine anaesthesia as previously described.26 For i.n. inoculation of mice, bacteria were serially diluted in PBS to the intended CFU/ml based on OD600 of the starting stock, and administered in 10 μl followed by 10 μl of PBS as described previously.22 The actual CFU inoculated per mouse was determined retrospectively after each experiment by plating serial dilutions of the bacterial preparation used for inoculation on chocolate agar plates. Two hours after SchuS4 inoculation, mice were injected i.p. with mAb or PBS (Vehicle). Survival was monitored every 12 hr. Kaplan–Meier survival curves were plotted using Graph-Pad Prism 5·0 (GraphPad Software, San Diego, CA) and the log-rank test was used to compare groups.
For determination of blood bacterial burden, blood was collected from the submandibular vein into a BD Microtainer® tube with Lithium Heparin additive (BD, Franklin Lakes, NJ) 3 days after bacterial inoculation. Undiluted blood and a fivefold serial dilution of the blood were plated on chocolate agar, and the plates were incubated at 35° for 2 days for CFU enumeration. Per cent CFU reduction compared with PBS was calculated from the median CFU obtained from the plate with ≥ 30 CFU or the undiluted-blood plate if both plates had < 30 CFU. The P-value was determined using the two-tailed Mann–Whitney U-test. P values of < 0·05 were considered statistically significant for both the log rank test and the two-tailed Mann–Whitney U-test.
Results
ELISA and Western blot analysis demonstrate the specificity of the new mAbs for both long and short F. tularensis OAg chains
In an attempt to obtain mAbs to non-immunodominant LPS epitopes, we infected BALB/c mice intradermally with a sublethal dose of LVS and boosted them i.p. either with a mixture of F. tularensis antigens enriched for short LPS chains or with enriched LVS outer membrane or capsule preparations, before using their splenocytes for generation of hybridomas. From a total of 26 hybridomas reactive with F. tularensis LPS in ELISA and with the F. tularensis LPS ladder in Western blot analysis, three showed the FB11 even-banding LPS ladder pattern and the rest showed the Ab52 pattern of decreasing band intensity with decreasing LPS chain length. One of the three FB11-like mAbs, Ab63 [IgG3(κ)], was obtained with the former immunization protocol, and the other two FB11-like mAbs, N213 [IgG3(κ)] and N62 [(IgG2b(κ)], were obtained from different mice using the latter immunization protocol.
All three new mAbs bound to F. tularensis SchuS4 but not to E. coli strain TG1 in ELISA (Fig. 1a), indicating their specificity for F. tularensis. Conversely, the anti-E. coli LPS mAb GTX40330 bound to E. coli TG1 but not to F. tularensis SchuS4 (Fig. 1a), confirming the presence of E. coli LPS on the plates and validating the specificity of the assay. The three new mAbs also agglutinated F. tularensis SchuS4 and LVS, but not the OAg-deficient LVS mutant WbtI (Fig. 1b), demonstrating their specificity for the OAg part of LPS. Furthermore, the new mAbs showed the same LPS ladder pattern as the terminally binding anti-F. tularensis OAg mAb FB11 on both SchuS4 lysate and purified F. tularensis LPS Western blots (Fig. 2). This ladder pattern is marked by relatively even intensity high molecular weight and low molecular weight bands, in contrast to the decreasing intensity of lower molecular weight bands displayed by internally binding anti-F. tularensis OAg mAbs, like Ab52 and Ab2 (Fig. 2). Although all three new mAbs have the same Western blot binding pattern, their binding potency varies, with Ab63 showing weaker binding than the other IgG3 mAb, N213, even at 10-fold higher concentration (Fig. 2). The previously characterized terminally binding anti-OAg mAb FB11 (IgG2a) appears to be at least 100-fold more potent than Ab63, however, this may be due at least in part to probable differences in the concentration of antibodies to different mouse immunoglobulin isotypes in the secondary antibody used to develop the Western blot (Fig. 2).
Figure 1.
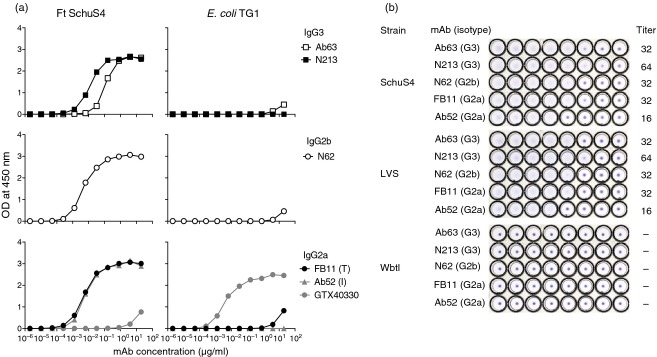
The three new monoclonal antibodies (mAbs) bind to Francisella tularensis SchuS4 or live vaccine strain (LVS), but not to Escherichia coli TG1 or the O-antigen (OAg) -deficient LVS mutant WbtI. (a) ELISA on heat-killed F. tularensis SchuS4 or E. coli TG1. The previously described terminally binding (T) and internally binding (I) anti-OAg mAbs, FB11 and Ab52, respectively, were included for reference. The anti-E. coli lipopolysaccharide (LPS) mAb GTX40330 was included as specificity control. The same isotype-specific secondary antibody was used for ELISAs of mAbs of the same isotype (indicated). (b) Microagglutination. Twofold serial dilutions of the indicated mAbs, from a starting concentration of 256 μg/ml in the first (left-most) well, were tested for ability to agglutinate the indicated F. tularensis strains. The titre was defined as the last dilution that showed agglutination.
Figure 2.
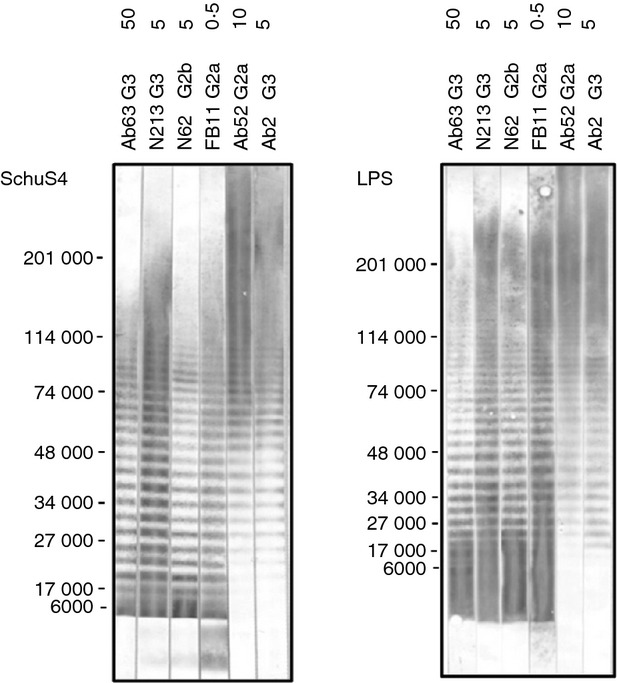
Ab63, N213 and N62, like FB11, bind strongly to both long and short lipopolysaccharide (LPS) chains on Western blots. Two hundred microlitres of SchuS4 lysate (equivalent to 0·05 OD600 of original bacterial culture) or Francisella tularensis LPS (20 μg) was electrophoresed in a preparative 4–15% polyacrylamide gel (8·6 cm wide by 6·8 cm long) under denaturing conditions. The positions of molecular weight markers are indicated. Strips were probed with the indicated monoclonal antibodies (mAbs) at the indicated μg/ml concentrations and AP-conjugated anti-mouse IgG (H + L) was used as secondary antibody. The terminally binding anti-OAg mAb FB11 (IgG2a), and the internally binding anti-OAg mAbs Ab52 (IgG2a) and Ab2 (IgG3) were included for comparison.
The antigen-binding affinities/bivalent avidities of the new mAbs are lower than that of FB11 but higher than those of internally binding anti-OAg mAbs
To assess whether the stronger Western blot reactivity of FB11 compared with the reactivity of the new mAbs reflects a higher affinity for antigen, we sought to determine the affinity of the new mAbs for F. tularensis OAgC by Biacore analysis, measuring the binding of graded concentrations of soluble OAgC to anti-immunoglobulin-captured mAb. As shown in Fig. 3(a), the KD of N213 and N62 were 1·37 × 10−6 m and 5·22 × 10−6 m, respectively. Hence, the affinity of FB11 for OAgC (4·01 × 10−7 m)27 is indeed 3·4-fold and 13·0-fold higher than those of N213 and N62, respectively. The Ab63 KD for this monovalent interaction with OAgC could not be determined at the OAgC concentrations used (up to 20 μm). Therefore, we determined the bivalent avidity of Ab63 by measuring the binding of graded concentrations of soluble mAb to chip-bound F. tularensis LPS, as was previously done to compare the binding potencies of internally binding anti-OAg mAbs with the binding potency of the terminally binding anti-OAg mAb FB11.27 The bivalent avidity of Ab63 was found to be 7·50 × 10−10 m (Fig. 3b), 4·8-fold higher than that of Ab52 (3·6 × 10−9 m), the most avid of three IgG2a internal OAg-binding mAbs, and 15-fold lower than the bivalent avidity of FB11 (5·0 × 10−11 m)27.
Figure 3.
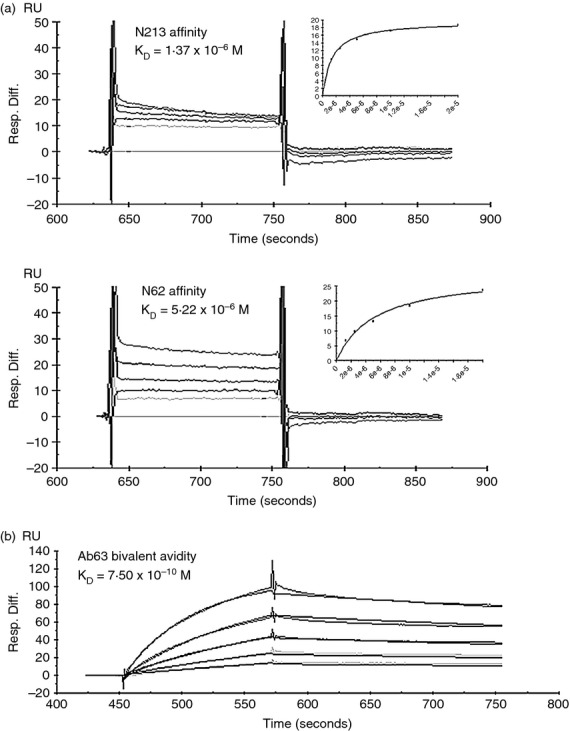
N213, N62 and Ab63 have antigen-binding affinities/bivalent avidities intermediate between FB11 and Ab52. (a) Biacore analysis of the binding of soluble Francisella tularensis O-antigen core (OAgC) to immobilized N213 and N62. Due to very fast off rates, the KDs were calculated based on the steady-state binding parameters. (b) Biacore analysis of the binding of soluble Ab63 to immobilized F. tularensis lipopolysaccharide (LPS). Ab52, captured on anti-mouse IgG-coated chips, was used to capture F. tularensis LPS, which was then probed with 0–13 nm of Ab63. Assays were performed by Precision Antibody (Columbia, MD). The axis labels on the original company graphs in (b) were relabelled for visibility.
Nucleotide sequencing reveals that all three new mAbs target the same epitope
To determine the amino acid sequence similarity, if any, among the three new mAbs, the nucleotide sequences of the expressed H and L chain variable (V) region genes that encode each mAb were determined, and the germline IGHV, IGHD, IGHJ and IGKV, IGKJ genes to which each has maximum identity were found. Despite originating from different mice, the Ab63, N213 and N62 variable regions are encoded by the same IGHV, IGHJ and IGKV germline genes (95–100% nucleotide identity, Table 1). Ab63 and N62 also share the IGKJ gene, and the different IGKJ gene of N213 has only two conservative amino acid differences (Gly → Ala and Ile → Leu, Fig. 4). N213 and N62 also share the same D gene and differ only by a single amino acid in CDR3 of the H chain (HCDR3). And although no D gene could be identified by the IgBlast program for Ab63 (Table 1), the HCDR3 sequences of Ab63 and N213 differ by a single amino acid, whereas those of N213 and N62 differ by two amino acids (Fig. 4). In contrast, none of the germline genes (except IGHJ, which is J3*01 for all the mAbs) are shared with those encoding the V regions of internally binding anti-OAg mAbs Ab52 and Ab2 (Table 1). The shared germline genes among Ab63, N213 and N62 indicate that the three new mAbs target the same epitope, and the number of somatic mutations (lower identity to the germline genes) correlates directly with the affinities/bivalent avidities of the three mAbs (Fig. 4, Table 1, and Fig. 3). The higher affinity mAbs, N213 and N62, share the H-FR2 and H-CDR2 somatic mutations Lys44Arg and Tyr58Phe, respectively, suggesting that these two mutations may be important for the higher affinity.
Table 1.
Nucleotide identity of anti-Francisella tularensis O-antigen monoclonal antibodies to the most homologous germline genes
| Germline gene (% nucleotide identity or number of contiguous nucleotide matches) | |||||
|---|---|---|---|---|---|
| mAb | IGHV | IGHD | IGHJ | IGKV | IGKJ |
| Ab63 | 3–8*02 (99·6) | –1 | 3*01 (100·0) | 4–80*01 (100·0) | 2*01 (100·0) |
| N213 | 3–8*02 (95·0) | 2–14*01 (9) | 3*01 (97·8) | 4–80*01 (97·1) | 5*01 (100·0) |
| N62 | 3–8*02 (97·7) | 2–14*01 (6) | 3*01 (100·0) | 4–80*01 (99·2) | 2*01 (100·0) |
| Ab52 | 1S130*01 (98·8) | –1 | 3*01 (100·0) | 8–24*01 (99·6) | 4*01 (100·0) |
| Ab2 | 9–2–1*01 (100·0) | 4–1*02 (5) | 3*01 (100·0) | 8–24*01 (100·0) | 4*01 (100·0) |
No D gene was identified by the IgBlast program (http://www.ncbi.nlm.nih.gov/igblast/) using the minimum requirement of five contiguous nucleotides.
Figure 4.

Variable region amino acid sequences indicate that Ab63, N213 and N62 target the same epitope and show a direct correlation between the number of somatic mutations and affinity. The amino acid sequences were deduced from the nucleotide sequences of the expressed heavy (H) and light (L) chains for each monoclonal antibody (mAb). Identities to the indicated germline genes are represented by dashes. The IGHD2-14*01 gene was not included because it could not be identified in all three mAbs. Amino acid differences from the germline genes or among mAbs are indicated (in one letter code), with differences from germline shown in bold type. The CDRs (and by exclusion the FRs) as defined by North et al.45 are indicated. There are three residues in H-FR3 following residue 82 that are labelled as 82A, 82B, and 82C, according to Kabat numbering as modified by Al-Lazikani et al.65 The residues that line the binding site in the N62 Fab crystal structure are also indicated. There is no amino acid at position 96 of the L chain in the three mAbs.
Ab63 and N62 cross-compete with FB11 but not with internally binding anti-OAg mAbs for binding to F. tularensis LPS
To ascertain that the new mAbs, like FB11, bind to the non-reducing end of F. tularensis OAg, the ability of two of the new mAbs, Ab63 and N62, to compete with FB11 or with internally binding anti-OAg mAb Ab52 for F. tularensis LPS-binding was determined in cross-blocking isotype-specific competition ELISA. As shown in Fig. 5(a) top row, Ab63 and FB11 cross-competed for binding to LPS, but neither mAb cross-competed for LPS-binding with the internally binding anti-OAg mAbs Ab52 or Ab2. Similarly, N62 cross-competed with FB11, but not with Ab52, for LPS-binding (Fig. 5b, top row) although, as expected, the inhibitory potencies of the two mAbs, especially of Ab63, were weaker than the inhibitory potency of FB11 (compare Fig. 5a and 5b, top left and middle panels). Conversely, the internally binding Ab2 but not Ab63 or N62 inhibited the binding of Ab52 to F. tularensis LPS (Fig. 5, right panels). The presence/binding of all competitors was confirmed in duplicate assays, using secondary antibodies specific for the respective competitors (Fig. 5a and 5b, bottom panels).
Figure 5.
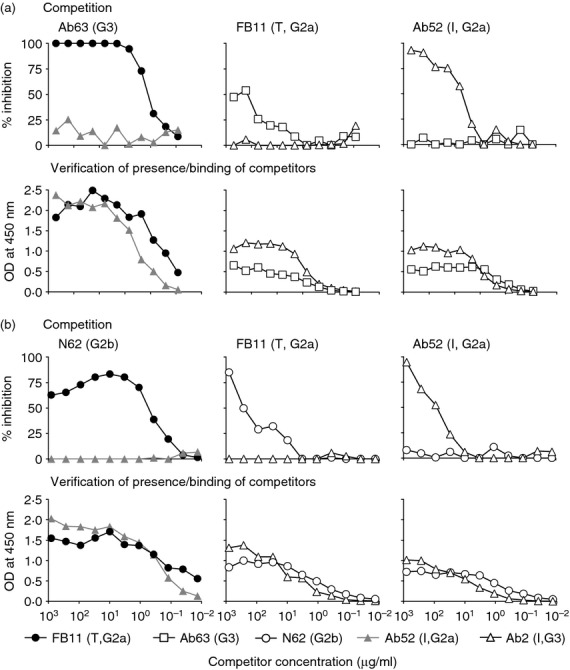
Ab63 or N62 and the terminally binding (T) anti-O-antigen (OAg) monoclonal antibody (mAb) FB11 cross-inhibit each other's binding to Francisella tularensis lipopolysaccharide (LPS), but no competition is observed between N62 and internally binding (I) anti-OAg mAbs. The internally binding anti-OAg mAbs Ab52 and Ab2 were included as specificity controls. (a,b upper panels) Competition. Horesradish peroxidase (HRP) -conjugated secondary antibody specific for the isotype of the reporter was used. (a,b lower panels) Verification that competitors are present and bind to F. tularensis LPS. HRP-conjugated secondary antibodies specific for the immunoglobulin isotypes of the competitors (indicated) were used. One of two experiments, with similar results, is shown.
Ab63, N213 and N62, like FB11, target an epitope contained within one OAg tetrasaccharide repeat
We had previously shown, using competition ELISA with oligosaccharides of graded numbers of OAg repeat units, that an OAgC oligosaccharide with two OAg repeat units is much better than an OAgC oligosaccharide with a single OAg repeat unit at inhibiting the binding of Ab52 to OAgC, whereas the two oligosaccharides are equally potent at inhibiting the binding of FB11 to OAgC.28 To test whether the new mAbs behave like FB11 in this assay and to define the maximum size of the targeted epitope, we tested the efficacy of three purified oligosaccharides containing core oligosaccharide (C), or one or two OAg repeats attached to core oligosaccharide [(oag)1C or (oag)2C]23 at inhibiting the binding of Ab63, N213 or N62 to F. tularensis LPS in competition ELISA. As shown in Fig. 6, core alone (C) was ineffective at inhibiting the binding of the three mAbs, but both OAg-containing oligosaccharides showed inhibition, confirming the specificity of Ab63, N213 and N62 for OAg. The binding of N213, like that of FB11, was equally inhibited by (oag)1C and (oag)2C, indicating that the epitope targeted by the two mAbs is contained within one OAg tetrasaccharide repeat. Although only (oag)1C, which was available in higher concentration than (oag)2C, reached 50% inhibition of Ab63 or N62 binding, the concentration required for 25% inhibition was similar for the two oligosaccharides, indicating that (oag)2C is not a more potent inhibitor than (oag)1C. Hence, the Ab63, N213 and N62 oligosaccharide inhibition profile resembles that of FB11 but differs from that of Ab52, where a 46-fold higher concentration of (oag)1C than of (oag)2C is required for 50% inhibition (Fig. 6).
Figure 6.

The epitope targeted by Ab63, N213 and N62 is contained within one O-antigen (OAg) tetrasaccharide repeat. Purified Francisella tularensis lipopolysaccharide (LPS) oligosaccharides were used to compete with the binding of Ab63, N213, N62, FB11 or Ab52 to F. tularensis LPS coated on ELISA plates. Bars denote standard deviation of the mean from two or three experiments. –, 50% inhibition was not reached at the highest available competitor concentrations.
The X-ray crystal structure of N62 Fab reveals an antigen-binding site lined by aromatic and basic amino acids that form a small cavity, which can accommodate one and a third sugar residues
To further localize the epitope targeted by the three in-house-generated terminally binding anti-OAg mAbs, the Fab of one of them, N62, was crystallized. The initial crystals of N62 Fab, obtained from sparse matrix screening, grew as clusters. Seeding was therefore used to generate large, single crystals, which grew as rectangular prisms and diffracted X-rays to 2·6-Å resolution. From analysis of the diffraction data and the molecular replacement solution (Table 2), the space group was determined to be P212121 with unit cell dimensions a = 68·874 Å, b = 87·076 Å, c = 153·589 Å, α = β = γ = 90·00° with two Fab molecules in the asymmetric unit.
Table 2.
X-ray crystal diffraction data collection and refinement statistics
| Item | Value |
|---|---|
| Data collection | |
| Number of reflections | 28 828 (2785)1 |
| Data cutoff | I ≤ −3σ |
| I/σ(I) | 20·1 (4·4) |
| Per cent completeness | 98·8 (98·0) |
| Redundancy | 3·6 (3·5) |
| Rmerge | 0·052 (0·262) |
| Refinement | |
| Rwork | 0·222 |
| Rfree | 0·268 |
| Number of atoms | 6255 |
| Deviations from Ideal values | |
| Bond lengths (Å) | 0·002 |
| Bond angles (°) | 0·67 |
| Ramachandran plot | |
| Favoured | 97% |
| Allowed | 3% |
| Outliers | 0·0% |
The resolution limits overall were 15–2·60 Å; the number in parentheses is the value for the highest resolution shell (2·69–2·60 Å).
After several cycles of refinement and manual rebuilding, the final model was constructed, including building in all residues of the CDR3 loops omitted during the molecular replacement search as well as altering the sequence of the H chain constant domain from the IgG2a isotype (of the molecular replacement search model) to the IgG2b isotype of N62. The model contains two Fab molecules comprising residues L1-211 and H2-212. Residues L148-157, H127-133, and H156-162 from one of the Fabs were not modelled because of poor electron density. Two N-terminal residues, one from an L chain and one from an H chain, were modelled as pyroglutamic acid, and H-Cys79 was modelled as cysteine sulphinic acid as indicated by the electron density. These modified residues are not located in the antigen-binding site and are unlikely to be essential for antigen-binding. The model also contained 31 water molecules and one acetate ion near L-Lys208 and H-Glu122 in the constant domains. The final structure has good geometry and agreement with the observed diffraction data in Table 2.
The CDR loops, with the exception of CDRH3, fall into conformational clusters found in other antibody structures.45 The clusters are L1-10-1, L2-8-1, L3-8-2, H1-13-7 and H2-9-1 for the CDRL1, CDRL2, CDRL3, CDRH1 and CDRH2 loops, respectively. The CDRH3 loop is short (seven residues) and does not fall into any cluster. The six CDRs form a relatively flat surface with a central cavity, the presumed antigen-binding site, which is 8 Å wide, 10 Å long and 9 Å deep (Fig. 7a). All the CDRs except L2 contribute to the central cavity, formed by residues L1-Tyr32, L1-His34, L3-His89, L3-Trp91, H1-Tyr33, H1-Asn35, H2-Tyr50, H3-Tyr95, H3-Arg96 and H3-Phe97 (Fig. 7b). The significance, if any, of the H-FR2 somatic mutation Lys44Arg, found in both of the higher affinity mAbs N213 and N62, is not apparent from the N62 crystal structure. However, the H-CDR2 somatic mutation Tyr58Phe in the two mAbs may positively fine-tune the antigen-binding cavity, because H-Phe58 is located in the VH-VL interface of the N62 crystal structure.
Figure 7.

The antigen-binding site of N62 is a cavity lined with aromatic and basic residues, which contacts mainly the terminal saccharide at the non-reducing end of the O-antigen (OAg) chain. (a) Space-filling model of the molecular surface of N62 VH and VL (head-on view, X-ray crystal structure), showing the central cavity, the presumed antigen-binding site. The surface-contributing areas from the complementarity-determining regions (CDRs) are coloured and indicated as H1, H2, H3, and L1, L2, L3 for HCDR1-3 and LCDR1-3, respectively. Non-CDR surface areas are coloured grey for VH and purple for VL. (b) Ribbon diagram of the N62 binding site in the same orientation as in (a). The amino acids lining the binding site are indicated and are coloured with grey carbons and ribbons for VH and purple for VL. (c) Stick diagram of the previously generated two-repeat model of F. tularensis OAg, obtained by docking a computational model in the binding site of the anti-F. tularensis OAg internally binding mAb Ab52.29 The four sugars in each of the first (non-reducing end) and second OAg repeat are colour-coded by sugar type and indicated as A, B, C, D, and A', B', C', D', respectively. The distance between the O5 oxygens of the A and A‘ sugars, and between the O1 oxygen of A and the C1 carbon of D‘, and the angle between the two repeats are indicated. (d) Cut-away side view (partial) of the N62 V regions, represented as ribbon diagram and wire-mesh molecular surface, with a manually docked model of F. tularensis OAg represented as a van der Waal's surface, which is colour-coded and in the same orientation as in (c). The orientation is rotated clockwise approximately 90° about the horizontal axis from panels (a) and (b).
Analysis of the N62 Fab crystal packing showed that the site is not blocked and ligand-soaking experiments were conducted in an attempt to solve the structure of an antibody–antigen complex. Two species of purified, single-repeat F. tularensis OAg, one with the core and one without, were used. Neither one resulted in successful complex formation after soaking. N-acetylgalactosamine was also tried, because it is an analogue of the GalNAcAN sugar that comprises two of the four sugars of the OAg repeat unit, but also failed to soak in.
In the absence of a mAb-OAg co-crystal structure, the feasibility of the N62 binding site accommodating the non-reducing end of F. tularensis OAg was tested by manual docking of a two-repeat (eight-sugar) model of F. tularensis OAg, obtained by docking a computational model in the binding site of the anti-F. tularensis OAg internally binding mAb Ab5229 (Fig. 7c). As shown in Fig. 7(d), the Qui4NFm (A) sugar at the non-reducing end can be fully accommodated in the central cavity, and the second sugar in the chain, GalNAcAN (B), can interact with the rim of the cavity. Due to the linear nature of the OAg, it is unlikely that any other sugars in the OAg chain are able to contribute to the binding. The snug fit of the OAg model in the N62 binding site (Fig. 7d) supports its placement, although an OAg conformation rotated 180° along the y-axis – albeit somewhat less well fitting – cannot be excluded.
The three new mAbs prolong survival in a mouse model of respiratory tularaemia
Ab63, N213 and N62 were tested for ability to prolong survival and/or reduce blood bacterial burden if administered to BALB/c mice after i.n. infection with F. tularensis SchuS4. Although all mice died, all three mAbs significantly prolonged survival at a dose of 200 μg, and N213 and N62 also significantly prolonged survival at a dose of 50 μg, with N62-treated mice surviving longest, an average of 36 hr longer compared with PBS (P = 0·0080) and 12 hr longer compared with N213 (P = 0·0326) (Fig. 8a). When blood SchuS4 burden was measured at 3 days post infection and 50 μg mAb treatment, all three mAbs showed significantly reduced levels, similar to the terminally binding anti-OAg mAb FB11 and the internally binding anti-OAg mAbs Ab52 and Ab2 (Fig. 8b). By comparison, the isotype-control mAbs CO17-1A (IgG2a) or TIB-114 (IgG3) had no effect on blood bacterial burden (Fig. 8b).
Figure 8.
Ab63, N213 and N62 prolong survival of SchuS4-infected BALB/c mice and reduce blood bacterial burden in a mouse model of respiratory tularaemia. (a) Survival. Mice (four or five per group) were inoculated with 24 colony-forming units (CFU) of SchuS4 intranasally (i.n.; in one experiment) or 36 CFU of SchuS4 i.n. (in a separate experiment), and injected intraperitoneally (i.p.) with the indicated monoclonal antibody (mAb) dose or with PBS 2 hr post infection. P values for mAb-treated groups compared with PBS-treated groups are indicated. (b) Blood bacterial burden. Mice were inoculated i.n. with 164 CFU of SchuS4 (left panel) or 109 CFU of SchuS4 (right panel), 2 hr post infection they were injected i.p. with 50 μg of the indicated mAbs or with PBS, then 3 days later they were bled and killed for blood CFU determination. Per cent CFU reduction compared with PBS was calculated from the median CFU numbers and the P-value was determined using the two-tailed Mann–Whitney U-test. The specificity of mAbs for terminal (T) or internal (I) O-antigen (OAg) epitope and their isotype are indicated.
Discussion
We generated three mAbs specific for the non-reducing end of F. tularensis OAg, as demonstrated by their binding to F. tularensis SchuS4 but not to E. coli TG1 in ELISA, their agglutination of F. tularensis SchuS4 and LVS but not of the OAg-deficient LVS mutant WbtI, their even reactivity with both long and short LPS chains on Western blots, and their ability to cross-compete for LPS-binding with the anti-F. tularensis OAg terminally binding mAb FB11 but not with internally binding mAbs. Furthermore, an oligosaccharide containing a single F. tularensis OAg tetrasaccharide repeat unit was sufficient for optimal inhibition of the binding of the new mAbs to LPS, as has been shown for FB11 previously27 and in the current study.
The three new mAbs, Ab63, N213 and N62, were derived from different BALB/c mice immunized with antigen preparations enriched for short F. tularensis LPS chains, F. tularensis outer membrane, or F. tularensis capsule. Ab63, N213 and N62 represent 11·5% of all anti-F. tularensis OAg mAbs obtained from these immunizations, confirming the relatively low immunogenicity of the OAg non-reducing end compared with repeating internal OAg epitopes.27 Remarkably, although the three mAbs were derived from three different mice, they are partially encoded by the same germline IGHV, IGHJ and IGKV genes, indicating that all three mAbs target the same epitope, and suggesting that their germline genes represent the main, if not only, combination that can yield terminally binding anti-F. tularensis OAg antibodies in BALB/c mice.
The three new mAbs are distinguished by somatic mutations, the number of which correlates directly with the antigen-binding affinity/avidity, exemplifying the maturation of the antibody response.25 Although N213, which has the highest affinity of the new mAbs, is still 3·4-fold less potent than FB11, all three new mAbs have higher affinities/bivalent avidities than previously characterized anti-F. tularensis OAg internally binding mAbs.27 Previous immunochemical and computational model-building studies had suggested that antibodies that target the non-reducing end of linear carbohydrate chains have cavity-type antigen-binding sites, which bind head-on to antigen, providing all-around complementarity, compared with antibodies that target repeating internal regions of linear carbohydrate chains that generally have groove-type sites, which bind sideways to antigen, providing only partial complementarity.46–48 The results of the current study are consistent with these predictions and with the higher affinity of the anti-(α→6) dextran terminally binding mAb W3129 – 5·9 × 10−6 m – compared with the affinities of 20 anti-(α→6) dextran internally binding mAbs, which range from 7·1 × 10−3 m to 1·3 × 10−5 m.46,49,50 The affinity of the anti-dextran terminally binding mAb W3129 is within a 15-fold range of the affinities of the anti-F. tularensis OAg terminally binding mAbs N62, N213 and FB1127 – 5·2 × 10−6 m, 1·4 × 10−6 m and 4·0 × 10−7 m,27 respectively, and of the 6·3 × 10−7 m affinity of the anti-Vibrio cholerae OAg terminally binding mAb S-20-4.51
The observed lower affinity of the internal binders compared with the terminally binding mAbs suggests that the expected positive enthalpic advantage of interacting with more of the sugar chain is outweighed by other energetic considerations. One such consideration is the loss of conformational entropy upon binding as a consequence of constraining flexible glycosidic bonds in the antigen-binding site. Constraining more glycosidic bonds would be expected to carry a larger entropic penalty. However, studies estimating the penalty, performed with tethered antigens that are restrained to the bound state conformation in solution, have resulted only in modest gains in affinity.52,53 Therefore, other factors, such as solvent reorganization, may have a larger effect on protein–carbohydrate binding than conformational restraint. Hence, the increased affinity of the terminally binding mAbs could be attributed to the complete sequestration of the epitope from solvent, whereas the internal binders would have much of the epitope and antibody still exposed to solvent.
The Fab X-ray crystal structure of N62 showed that the antigen-binding site is a cavity, 8 Å wide, 10 Å long and 9 Å deep, lined by 10 amino acid residues, eight of which are aromatic, including two histidines (which are both aromatic and basic). These 10 binding site residues are invariant in all three mAbs, suggesting that the binding sites of N213 and Ab63 have the same topologies as the N62 binding site.
Binding sites similar to that of N62 have been found in two other antibodies. The X-ray crystal structure of the Fab of the anti-OAg terminally binding mAb S-20-4 complexed with a terminal disaccharide from the Vibrio cholera Ogawa serotype, also shows the expected cavity-type binding site.51 The cavity of S-20-4 is somewhat smaller than that of N62 – 10 Å wide but only about 6 Å deep – and primarily binds the terminal sugar, 4-amino-4,6-dideoxy-d-mannose, which has a methyl group O-linked at the two position.51 In the crystal structure, the methyl group is buried in the cavity surrounded by aromatic residues (H-H99, L-Y34, L-W93, L-W98) and one alanine (H-A102), whereas the rest of the sugar is still partly solvent-exposed despite some contacts to the antibody; there are no contacts between the antibody and the penultimate sugar in the chain.51
A site even more similar to that of N62 can be seen in the X-ray crystal structures of complexes of Fab or single-chain Fv Se155-4 and the repeat unit of the OAg from Salmonella serogroup B.54,55 In contrast to the linear F. tularensis OAg, the Salmonella OAg has a branched repeat unit, with the structure [→3)-α-d-Gal(1→2)[α-d-Abe(1→3)]α-d-Man(1→4)-α-L-Rha(1→], where Rha is rhamnose and Abe is abequose. Se155-4 binds to the internal part of the OAg, however, in the crystal structures, the branching Abe non-reducing sugar and a conserved water molecule are located in a pocket (cavity) approximately 8 Å wide and 7 Å deep, which is formed by residues L-His32, L-Trp91, L-Trp96, H-Trp33, H-His35, H-Ala50, H-Phe58, H-Gly96, H-His97, H-Gly98 and H-Tyr99. The residues of the cavity primarily interact with the Abe, but also form interactions with the sugars of the main chain of the OAg, where L-Trp96 is hydrogen-bonded to the galactose sugar and H-His97 is hydrogen-bonded to the mannose sugar. The affinity of Se155-4 for its ligand (5 × 10−6 to 7·7 × 10−6 m) 54,55 is very similar to the affinity of N62 for F. tularensis OAg (5·2 × 10−6 m).
Since the Se155-4 antigen-binding cavity is similar in size to the one observed for N62 with multiple aromatic residues (8 out of 11 compared with 8 out of 10 for N62), it is reasonable to assume that the antigen-binding site in N62 serves a similar function to that in Se155-4 – to bind a single saccharide and optionally make surface contacts with the other sugars in the OAg chain. Because the F. tularensis OAg is linear, the only sugar capable of binding to the antibody pocket in this fashion is the terminal QuiNFm residue at the non-reducing end. The antibody could potentially contact the next sugar in the chain – GalNAcAN, but cannot completely surround it. This is consistent with the specificity of N62 for the non-reducing end of F. tularensis OAg.
Indeed, manual docking into the N62 X-ray crystal binding site of a model of F. tularensis OAg, obtained by docking a computational model in the binding site of the anti-F. tularensis OAg internally binding mAb Ab52,29 showed that the Qui4NFm sugar at the non-reducing end could be almost completely buried, whereas part of the next sugar, GalNacAN, could make contacts with the rim of the cavity. The burial of Qui4NFm in the binding cavity would be facilitated by interactions with the lining aromatic and polar amino acid residues, as has been observed in numerous protein–carbohydrate complexes.56–58 Many sugars have a hydrophobic face and a hydrophilic face as a consequence of chiral centres in the rigid pyranose ring, positioning CH groups on one face of the ring while OH and other polar groups reside on the opposite face of the sugar. Hence, binding of sugars face-to-face with aromatic residues allows for favourable CH-π interactions.56–58 In the Qui4NFm terminal sugar of F. tularensis OAg, the hydrophobic face is formed by the CH groups of carbons 1, 3 and 5. In the N62 antigen-binding site, residue L-W91 is ideally positioned for this type of interaction. The other aromatic residues could also participate in aromatic-carbohydrate interactions if they undergo an antigen-induced change in conformation. These residues could also create favourable interactions with the C6 methyl group of Qui4NFm. The polar residues lining the cavity, L-H34, L-H89, H-N35 and H-R96, could interact favourably with the polar groups of the Qui4NFm sugar – O2, O3 and the 4-N-formyl group. Hence, the observed binding site of N62 is well suited to accommodate all the functional groups present on the terminal sugar. Therefore, the X-ray crystal structure of N62 and the immunochemical data suggest that the epitope recognized by N62, (and by extension) N213 and Ab63 consists of the two sugars at the non-reducing end of the OAg – Qui4NFm(1→4)-α-d-GalNAcAN, with the primary contribution to the binding from the terminal saccharide.
The very high proportion of aromatic amino acids including histidine among presumed or known antigen-contacting residues in the binding cavities of N62, S-20-4 and Se155-4, is noteworthy. In general, the representation of aromatic residues among presumed59 or known60 contact residues in antibody binding sites is higher than that in an average protein loop – 25–30% versus 10%, respectively – due to an increase in Tyr and Trp (but not Phe or His).59,60 However, the percentage of aromatic contact residues (Tyr, Trp, Phe and His), calculated using the data of Collis et al.,59 does not differ between carbohydrate-binding and non-carbohydrate-binding antibodies, probably because the vast majority of anti-carbohydrate antibodies are directed against internal epitopes, which are much more immunogenic than terminal epitopes.27,49,61 In contrast, the percentages of aromatic contact residues in N62, S-20-4 and Se155-4 are 80, 80 and 73, respectively (current study and51,54,55). His is especially over-represented in these three antibodies (20–27% compared with 2·7% in other antibodies).59 This envelopment of the terminal sugar by aromatic residues, with His making polar interactions, may account for the higher affinity of antibodies to the non-reducing end, despite their smaller target epitope compared with antibodies to internal regions of carbohydrates.
All three new mAbs, like FB11 and the anti-F. tularensis OAg internally binding mAbs,28 prolonged survival and reduced blood bacterial burden in a mouse model of respiratory tularaemia with the virulent F. tularensis type A strain SchuS4. Although the lowest affinity mAb, Ab63 (IgG3), was least efficacious in these in vivo efficacy studies, prolonging survival when administered at 200 μg but not at 50 μg, the highest affinity mAb, N213 (IgG3) was not more efficacious than N62 (IgG2b). This is probably because the isotypes of the mAbs, as mouse antibodies of the IgG2a isotype, have been associated by our group26 and others62 with highest in vivo efficacy, whereas the IgG3 anti-OAg mAb Ab2 has been associated with the lowest in vivo efficacy compared with IgG2a and IgG1 mAbs of the same specificity.26 Since the IgG2a and IgG2b isotypes have been shown to be equally potent at mediating effector functions,63,64 N62 may have more affect in the mouse model of respiratory tularaemia than N213, despite the higher antigen-binding affinity of N213. Hence, the contribution of antibodies to non-reducing ends of microbial polysaccharides to immune protection probably depends both on a relatively high affinity for antigen due to the aromatic envelopment of the terminal sugar and on the antibody isotype.
Acknowledgments
ZL, MJR, CYY, GM, HMP and JS performed the experiments. QW, CEC and JZ prepared the purified oligosaccharides used in some of the studies. JS, ZL, MJR and BAS designed the study. GM, JZ, CEC and QW provided suggestions and critical discussion. JS, MJR and ZL wrote the paper. This work was supported in its entirety with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. HHSN272200900054C.
Glossary
- (oag)1
Qui4NFm-GalNAcAN-GalNAcAN-QuiNAc
- C
core oligosaccharide = Hex4HexNAcKdo
- CDR
complementarity-determining region
- CFU
colony-forming units
- FR
framework region
- GalNAcAN
2-acetamido-2-deoxy-d-galacturonamide
- i.n.
intranasal(ly)
- i.p.
intraperitoneal(ly)
- LPS
lipopolysaccharide
- LVS
live vaccine strain
- mAb
monoclonal antibody
- OAgC
OAg-core
- OAg
O-antigen (O-polysaccharide)
- Qui4NFm
4,6-dideoxy-4-formamido-d-glucose
- QuiNAc
2-acetamido-2,6-dideoxy-d-glucose
- V region
variable region
Disclosures
The authors have no potential conflicts of interest.
References
- 1.McLendon MK, Apicella MA, Allen LA. Francisella tularensis: taxonomy, genetics, and immunopathogenesis of a potential agent of biowarfare. Annu Rev Microbiol. 2006;60:167–85. doi: 10.1146/annurev.micro.60.080805.142126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sjostedt A. Tularemia: history, epidemiology, pathogen physiology, and clinical manifestations. Ann N Y Acad Sci. 2007;1105:1–29. doi: 10.1196/annals.1409.009. [DOI] [PubMed] [Google Scholar]
- 3.Thomas LD, Schaffner W. Tularemia pneumonia. Infect Dis Clin North Am. 2010;24:43–55. doi: 10.1016/j.idc.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 4.Tarnvik A, Chu MC. New approaches to diagnosis and therapy of tularemia. Ann N Y Acad Sci. 2007;1105:378–404. doi: 10.1196/annals.1409.017. [DOI] [PubMed] [Google Scholar]
- 5.Dennis DT, Inglesby TV, Henderson DA, et al. Tularemia as a biological weapon: medical and public health management. JAMA. 2001;285:2763–73. doi: 10.1001/jama.285.21.2763. [DOI] [PubMed] [Google Scholar]
- 6.Conlan JW. Tularemia vaccines: recent developments and remaining hurdles. Future Microbiol. 2011;6:391–405. doi: 10.2217/fmb.11.22. [DOI] [PubMed] [Google Scholar]
- 7.Oyston PC. Francisella tularensis vaccines. Vaccine. 2009;27(Suppl 4):D48–51. doi: 10.1016/j.vaccine.2009.07.090. [DOI] [PubMed] [Google Scholar]
- 8.Conlan JW, Shen H, Webb A, Perry MB. Mice vaccinated with the O-antigen of Francisella tularensis LVS lipopolysaccharide conjugated to bovine serum albumin develop varying degrees of protective immunity against systemic or aerosol challenge with virulent type A and type B strains of the pathogen. Vaccine. 2002;20:3465–71. doi: 10.1016/s0264-410x(02)00345-6. [DOI] [PubMed] [Google Scholar]
- 9.Gunn JS, Ernst RK. The structure and function of Francisella lipopolysaccharide. Ann N Y Acad Sci. 2007;1105:202–18. doi: 10.1196/annals.1409.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prior JL, Prior RG, Hitchen PG, Diaper H, Griffin KF, Morris HR, Dell A, Titball RW. Characterization of the O antigen gene cluster and structural analysis of the O antigen of Francisella tularensis subsp. tularensis. J Med Microbiol. 2003;52:845–51. doi: 10.1099/jmm.0.05184-0. [DOI] [PubMed] [Google Scholar]
- 11.Thirumalapura NR, Goad DW, Mort A, Morton RJ, Clarke J, Malayer J. Structural analysis of the O-antigen of Francisella tularensis subspecies tularensis strain OSU 10. J Med Microbiol. 2005;54:693–5. doi: 10.1099/jmm.0.45931-0. [DOI] [PubMed] [Google Scholar]
- 12.Vinogradov EV, Shashkov AS, Knirel YA, Kochetkov NK, Tochtamysheva NV, Averin SF, Goncharova OV, Khlebnikov VS. Structure of the O-antigen of Francisella tularensis strain 15. Carbohydr Res. 1991;214:289–97. doi: 10.1016/0008-6215(91)80036-m. [DOI] [PubMed] [Google Scholar]
- 13.Drabick JJ, Narayanan RB, Williams JC, Leduc JW, Nacy CA. Passive protection of mice against lethal Francisella tularensis (live tularemia vaccine strain) infection by the sera of human recipients of the live tularemia vaccine. Am J Med Sci. 1994;308:83–7. doi: 10.1097/00000441-199408000-00003. [DOI] [PubMed] [Google Scholar]
- 14.Fortier AH, Slayter MV, Ziemba R, Meltzer MS, Nacy CA. Live vaccine strain of Francisella tularensis: infection and immunity in mice. Infect Immun. 1991;59:2922–8. doi: 10.1128/iai.59.9.2922-2928.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foshay L. Tularemia: a summary of certain aspects of disease including methods for early diagnosis and the results of serum treatment in 600 patients. Medicine. 1940;19:1–83. [Google Scholar]
- 16.Fulop M, Mastroeni P, Green M, Titball RW. Role of antibody to lipopolysaccharide in protection against low- and high-virulence strains of Francisella tularensis. Vaccine. 2001;19:4465–72. doi: 10.1016/s0264-410x(01)00189-x. [DOI] [PubMed] [Google Scholar]
- 17.Kirimanjeswara GS, Golden JM, Bakshi CS, Metzger DW. Prophylactic and therapeutic use of antibodies for protection against respiratory infection with Francisella tularensis. J Immunol. 2007;179:532–9. doi: 10.4049/jimmunol.179.1.532. [DOI] [PubMed] [Google Scholar]
- 18.Rhinehart-Jones TR, Fortier AH, Elkins KL. Transfer of immunity against lethal murine Francisella infection by specific antibody depends on host γ interferon and T cells. Infect Immun. 1994;62:3129–37. doi: 10.1128/iai.62.8.3129-3137.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stenmark S, Lindgren H, Tarnvik A, Sjostedt A. Specific antibodies contribute to the host protection against strains of Francisella tularensis subspecies holarctica. Microb Pathog. 2003;35:73–80. doi: 10.1016/s0882-4010(03)00095-0. [DOI] [PubMed] [Google Scholar]
- 20.Sebastian S, Dillon ST, Lynch JG, et al. A defined O-antigen polysaccharide mutant of Francisella tularensis live vaccine strain has attenuated virulence while retaining its protective capacity. Infect Immun. 2007;75:2591–602. doi: 10.1128/IAI.01789-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirimanjeswara GS, Olmos S, Bakshi CS, Metzger DW. Humoral and cell-mediated immunity to the intracellular pathogen Francisella tularensis. Immunol Rev. 2008;225:244–55. doi: 10.1111/j.1600-065X.2008.00689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klimpel GR, Eaves-Pyles T, Moen ST, et al. Levofloxacin rescues mice from lethal intra-nasal infections with virulent Francisella tularensis and induces immunity and production of protective antibody. Vaccine. 2008;26:6874–82. doi: 10.1016/j.vaccine.2008.09.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Q, Shi X, Leymarie N, Madico G, Sharon J, Costello CE, Zaia J. A typical preparation of Francisella tularensis O-antigen yields a mixture of three types of saccharides. Biochemistry. 2011;50:10941–50. doi: 10.1021/bi201450v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Apicella MA, Post DM, Fowler AC, et al. Identification, characterization and immunogenicity of an O-antigen capsular polysaccharide of Francisella tularensis. PLoS ONE. 2010;5:e11060. doi: 10.1371/journal.pone.0011060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murphy K, Travers P, Walport M, Janeway C. Janeway's Immunobiology. 7th edn. New York: Garland Science; 2008. [Google Scholar]
- 26.Lu Z, Roche MI, Hui JH, Unal B, Felgner PL, Gulati S, Madico G, Sharon J. Generation and characterization of hybridoma antibodies for immunotherapy of tularemia. Immunol Lett. 2007;112:92–103. doi: 10.1016/j.imlet.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roche MI, Lu Z, Hui JH, Sharon J. Characterization of monoclonal antibodies to terminal and internal O-antigen epitopes of Francisella tularensis lipopolysaccharide. Hybridoma (Larchmt) 2011;30:19–28. doi: 10.1089/hyb.2010.0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu Z, Madico G, Roche MI, et al. Protective B-cell epitopes of Francisella tularensis O-polysaccharide in a mouse model of respiratory tularaemia. Immunology. 2012;136:352–60. doi: 10.1111/j.1365-2567.2012.03589.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rynkiewicz MJ, Lu Z, Hui JH, Sharon J, Seaton BA. Structural analysis of a protective epitope of the Francisella tularensis O-polysaccharide. Biochemistry. 2012;51:5684–94. doi: 10.1021/bi201711m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khlebnikov VS, Golovlev IR, Tokhtamysheva NV, Averin SF, Kulevatskii DP, Grechko GK, Averina AA, Vetchinin SS. The determination of the antigenic determinant of protective monoclonal antibodies specific to the Francisella tularensis lipopolysaccharide. Zh Mikrobiol Epidemiol Immunobiol. 1993;1:83–8. [PubMed] [Google Scholar]
- 31.Fulop MJ, Webber T, Manchee RJ, Kelly DC. Production and characterization of monoclonal antibodies directed against the lipopolysaccharide of Francisella tularensis. J Clin Microbiol. 1991;29:1407–12. doi: 10.1128/jcm.29.7.1407-1412.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gubbins MJ, Berry JD, Schmidt L, Cabral T, Kabani A, Tsang RS. Production and characterization of a monoclonal antibody to Francisella tularensis lipopolysaccharide. Hybridoma (Larchmt) 2007;26:98–103. doi: 10.1089/hyb.2006.049. [DOI] [PubMed] [Google Scholar]
- 33.Hotta A, Uda A, Fujita O, Tanabayashi K, Yamada A. Preparation of monoclonal antibodies for detection and identification of Francisella tularensis. Clin Vaccine Immunol. 2007;14:81–4. doi: 10.1128/CVI.00057-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Narayanan RB, Drabick JJ, Williams JC, Fortier AH, Meltzer MS, Sadoff JC, Bolt CR, Nacy CA. Immunotherapy of tularemia: characterization of a monoclonal antibody reactive with Francisella tularensis. J Leukoc Biol. 1993;53:112–16. doi: 10.1002/jlb.53.1.112. [DOI] [PubMed] [Google Scholar]
- 35.Savitt AG, Mena-Taboada P, Monsalve G, Benach JL. Francisella tularensis infection-derived monoclonal antibodies provide detection, protection, and therapy. Clin Vaccine Immunol. 2009;16:414–22. doi: 10.1128/CVI.00362-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J, Ryder C, Mandal M, et al. Attenuation and protective efficacy of an O-antigen-deficient mutant of Francisella tularensis LVS. Microbiology. 2007;153:3141–53. doi: 10.1099/mic.0.2007/006460-0. [DOI] [PubMed] [Google Scholar]
- 37.Herlyn DM, Steplewski Z, Herlyn MF, Koprowski H. Inhibition of growth of colorectal carcinoma in nude mice by monoclonal antibody. Cancer Res. 1980;40:717–21. [PubMed] [Google Scholar]
- 38.Schlimok G, Gottlinger H, Funke I, Swierkot S, Hauser H, Riethmuller G. In vivo and in vitro labelling of epithelial tumor cells with anti 17-1A monoclonal antibodies in bone marrow of cancer patients. Hybridoma. 1986;5(Suppl 1):S163–70. [PubMed] [Google Scholar]
- 39.Shulman M, Wilde CD, Kohler G. A better cell line for making hybridomas secreting specific antibodies. Nature. 1978;276:269–70. doi: 10.1038/276269a0. [DOI] [PubMed] [Google Scholar]
- 40.Hood AM. Virulence factors of Francisella tularensis. J Hyg (Lond) 1977;79:47–60. doi: 10.1017/s0022172400052840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–26. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 42.Adams PD, Afonine PV, Bunkoczi G, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Acchione M, Lipschultz CA, DeSantis ME, Shanmuganathan A, Li M, Wlodawer A, Tarasov S, Smith-Gill SJ. Light chain somatic mutations change thermodynamics of binding and water coordination in the HyHEL-10 family of antibodies. Mol Immunol. 2009;47:457–64. doi: 10.1016/j.molimm.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 45.North B, Lehmann A, Dunbrack RL., Jr A new clustering of antibody CDR loop conformations. J Mol Biol. 2011;406:228–56. doi: 10.1016/j.jmb.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cisar J, Kabat EA, Dorner MM, Liao J. Binding properties of immunoglobulin combining sites specific for terminal or nonterminal antigenic determinants in dextran. J Exp Med. 1975;142:435–59. doi: 10.1084/jem.142.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kabat EA. The nature of an antigenic determinant. J Immunol. 1966;97:1–11. [PubMed] [Google Scholar]
- 48.Padlan EA, Kabat EA. Model-building study of the combining sites of two antibodies to α(1–6)dextran. Proc Natl Acad Sci USA. 1988;85:6885–9. doi: 10.1073/pnas.85.18.6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Newman BA, Kabat EA. An immunochemical study of the combining site specificities of C57BL/6J monoclonal antibodies to α(1–6)-linked dextran B512. J Immunol. 1985;135:1220–31. [PubMed] [Google Scholar]
- 50.Sharon J, Kabat EA, Morrison SL. Association constants of hybridoma antibodies specific for α (1 leads to 6) linked dextran determined by affinity electrophoresis. Mol Immunol. 1982;19:389–97. doi: 10.1016/0161-5890(82)90204-8. [DOI] [PubMed] [Google Scholar]
- 51.Villeneuve S, Souchon H, Riottot MM, et al. Crystal structure of an anti-carbohydrate antibody directed against Vibrio cholerae O1 in complex with antigen: molecular basis for serotype specificity. Proc Natl Acad Sci USA. 2000;97:8433–8. doi: 10.1073/pnas.060022997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McGavin RS, Gagne RA, Chervenak MC, Bundle DR. The design, synthesis and evaluation of high affinity macrocyclic carbohydrate inhibitors. Org Biomol Chem. 2005;3:2723–32. doi: 10.1039/b416105j. [DOI] [PubMed] [Google Scholar]
- 53.Bundle DR, Alibés R, Nilar S, Otter A, Warwas M, Zhang P. Thermodynamic and conformational implications of glycosidic rotamers preorganized for binding. J Am Chem Soc. 1998;120:5317–18. [Google Scholar]
- 54.Cygler M, Wu S, Zdanov A, Bundle DR, Rose DR. Recognition of a carbohydrate antigenic determinant of Salmonella by an antibody. Biochem Soc Trans. 1993;21:437–41. doi: 10.1042/bst0210437. [DOI] [PubMed] [Google Scholar]
- 55.Zdanov A, Li Y, Bundle DR, Deng SJ, MacKenzie CR, Narang SA, Young NM, Cygler M. Structure of a single-chain antibody variable domain (Fv) fragment complexed with a carbohydrate antigen at 1.7-A resolution. Proc Natl Acad Sci USA. 1994;91:6423–7. doi: 10.1073/pnas.91.14.6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muraki M. The importance of CH/pi interactions to the function of carbohydrate binding proteins. Protein Pept Lett. 2002;9:195–209. doi: 10.2174/0929866023408751. [DOI] [PubMed] [Google Scholar]
- 57.Weis WI, Drickamer K. Structural basis of lectin-carbohydrate recognition. Annu Rev Biochem. 1996;65:441–73. doi: 10.1146/annurev.bi.65.070196.002301. [DOI] [PubMed] [Google Scholar]
- 58.Laughrey ZR, Kiehna SE, Riemen AJ, Waters ML. Carbohydrate–π interactions: what are they worth? J Am Chem Soc. 2008;130:14625–33. doi: 10.1021/ja803960x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Collis AV, Brouwer AP, Martin AC. Analysis of the antigen combining site: correlations between length and sequence composition of the hypervariable loops and the nature of the antigen. J Mol Biol. 2003;325:337–54. doi: 10.1016/s0022-2836(02)01222-6. [DOI] [PubMed] [Google Scholar]
- 60.Ramaraj T, Angel T, Dratz EA, Jesaitis AJ, Mumey B. Antigen–antibody interface properties: composition, residue interactions, and features of 53 non-redundant structures. Biochim Biophys Acta. 2012;1824:520–32. doi: 10.1016/j.bbapap.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sharon J, Kabat EA, Morrison SL. Immunochemical characterization of binding sites of hybridoma antibodies specific for α (1 leads to 6) linked dextran. Mol Immunol. 1982;19:375–88. doi: 10.1016/0161-5890(82)90203-6. [DOI] [PubMed] [Google Scholar]
- 62.Eyles JE, Unal B, Hartley MG, et al. Immunodominant Francisella tularensis antigens identified using proteome microarray. Proteomics. 2007;7:2172–83. doi: 10.1002/pmic.200600985. [DOI] [PubMed] [Google Scholar]
- 63.Dangl JL, Wensel TG, Morrison SL, Stryer L, Herzenberg LA, Oi VT. Segmental flexibility and complement fixation of genetically engineered chimeric human, rabbit and mouse antibodies. EMBO J. 1988;7:1989–94. doi: 10.1002/j.1460-2075.1988.tb03037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rashid A, Auchincloss H, Jr, Sharon J. Comparison of GK1.5 and chimeric rat/mouse GK1.5 anti-CD4 antibodies for prolongation of skin allograft survival and suppression of alloantibody production in mice. J Immunol. 1992;148:1382–8. [PubMed] [Google Scholar]
- 65.Al-Lazikani B, Lesk AM, Chothia C. Standard conformations for the canonical structures of immunoglobulins. J Mol Biol. 1997;273:927–48. doi: 10.1006/jmbi.1997.1354. [DOI] [PubMed] [Google Scholar]