

Abstract
We recently described a new form of neural integration and firing in a subset of interneurons, in which evoking hundreds of action potentials over tens of seconds to minutes produces a sudden barrage of action potentials lasting about a minute beyond the inciting stimulation. During this persistent firing, action potentials are generated in the distal axon and propagate retrogradely to the soma. To distinguish this from other forms of persistent firing, we refer to it here as ‘retroaxonal barrage firing’, or ‘barrage firing’ for short. Its induction is blocked by chemical inhibitors of gap junctions and curiously, stimulation of one interneuron in some cases triggers barrage firing in a nearby, unstimulated interneuron. Beyond these clues, the mechanisms of barrage firing are unknown. Here we report new results related to these mechanisms. Induction of barrage firing was blocked by lowering extracellular calcium, as long as normal action potential threshold was maintained, and it was inhibited by blocking L-type voltage-gated calcium channels. Despite its calcium dependence, barrage firing was not prevented by inhibiting chemical synaptic transmission. Furthermore, loading the stimulated/recorded interneuron with BAPTA did not block barrage firing, suggesting that the required calcium entry occurs in other cells. Finally, barrage firing was normal in mice with deletion of the primary gene for neuronal gap junctions (connexin36), suggesting that non-neuronal gap junctions may be involved. Together, these findings suggest that barrage firing is probably triggered by a multicellular mechanism involving calcium signalling and gap junctions, but operating independently of chemical synaptic transmission.
Key points
Persistent firing can be triggered in a population of inhibitory interneurons found in the hippocampus and neocortex. Repeated stimulation eventually triggers an autonomous barrage of spikes that is generated and maintained in the axon, followed by antidromic propagation to the soma.
This barrage of spikes is generated and maintained in the axon, followed by antidromic propagation to the soma. The mechanisms underlying this ‘retroaxonal barrage firing’ are unknown.
We find that retroaxonal barrage firing is Ca2+ dependent, is inhibited by the L-type Ca2+ channel blockers cadmium, nifedipine and verapamil, and does not require synaptic transmission. Loading the stimulated interneuron with BAPTA did not block barrage firing, suggesting that the required Ca2+ entry may occur in other cells.
Retroaxonal barrage firing was observed in mice lacking the Cx36 isoform (most common neuronal isoform), indicating that this particular isoform is not required.
Introduction
The conventional view of synaptic inhibition is that inhibitory interneurons are activated by excitatory synaptic input onto their dendritic trees, leading to action potential firing and inhibition of either the same neurons that provided excitation (i.e. feedback inhibition) or other neurons in the circuit (i.e. feedforward or lateral inhibition). Alternatively, some inhibitory neurons (e.g. cerebellar Purkinje neurons) fire action potentials spontaneously, thus providing nearly continuous inhibition, except when their firing pauses in response to synaptic input (Häusser et al. 2004). In each of these scenarios, action potentials in the inhibitory neuron are initiated in the axon hillock or initial segment (Palmer et al. 2010) and firing is modulated by synaptic inputs to the soma and dendrites, which act on a time scale of tens or hundreds of milliseconds.
We previously described, in a subset of interneurons of the rodent hippocampus and neocortex, an unusual and novel form of signalling that operates on a much longer time scale (Sheffield et al. 2011). In these cells, repeated stimulation eventually triggers a barrage of autonomous action potential firing that outlasts the stimulus by over a minute. We noted a number of unique characteristics of this persistent firing, including a slow time scale of signal integration (minutes), sudden switching into persistent firing, and action potential initiation in the distal axon far from the initial segment. To distinguish this form of persistent firing from other forms (Major & Tank, 2004), we refer to this phenomenon as “retroaxonal barrage firing”, or simply “barrage firing”, for short. This form of persistent firing was also recently reported in neuropeptide-Y-expressing neurons, where it was blocked by activation of μ-opioid receptors (Krook-Magnuson et al. 2011).
Several aspects of retroaxonal barrage firing deviate from the conventional view of action potential initiation in most cortical neurons, including inhibitory neurons, as described above. Most curiously, in a few paired recordings (n= 3/19), stimulation of one cell was able to induce barrage firing in another cell that was not stimulated. This result suggested that some aspect of the barrage firing mechanistic cascade (integration, switching, or readout) is able to spread transcellularly, directly into the distal axons of the interneurons.
Here we describe a series of experiments in which we further investigated the mechanisms underlying retroaxonal barrage firing in interneurons of the hippocampus. We report that barrage firing relies on voltage-gated calcium signalling outside of the stimulated cell and depends on gap junctions (most likely non-neuronal), but not chemical synapses. These data suggest a novel form of intercellular signalling responsible for the slow integration and/or abrupt onset of barrage firing in hippocampal interneurons.
Methods
All experiments were performed under the approval of the Northwestern University Animal Care and Use Committee, and conform to the principles of UK legislation.
Hippocampal slice preparation
Parasagittal hippocampal slices (∼300 μm) were prepared from postnatal day 14–30 mice anaesthetized with isofluorane (inhalation). Htr5b-EGFP BAC transgenic (GENSAT; Heintz, 2004), Cx36 knockout (KO) (line acquired from the laboratory of Dr David Paul, Harvard University; Deans et al. 2001) and wild-type (WT; C57BL/6) mice were used as indicated in the Results. Briefly, animals were decapitated and the brain was rapidly removed and placed under ice-cold sucrose-rich slicing solution containing (in mm): 85 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 75 sucrose, 0.5 CaCl2 and 4 MgCl2 bubbled with 95% O2/5% CO2. Slices were then transferred to a warmed (30°C) incubation chamber for 20 min with bubbled artificial cerebrospinal fluid (ACSF) consisting of (in mm): 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, 25 dextrose. Slices were then maintained in bubbled ACSF at room temperature until placed in the recording chamber.
Hippocampal slice electrophysiology
During recording slices were bathed in bubbled ACSF maintained at a constant temperature (34–37°C). Somatic whole-cell current clamp recordings were made using patch-clamp electrodes pulled from borosilicate glass and filled with intracellular solution containing (in mm): 135 potassium gluconate, 7.5 KCl, 10 sodium phosphocreatine, 10 Hepes, 2 MgATP, 0.3 NaGTP, and 0.5% biocytin. Recordings were made using a bridge amplifier (BVC-700; Dagan, Minneapolis, MN, USA). Electrode resistance in the bath was 3–7 MΩ and series resistance was 15–35 MΩ; the resultant errors were minimized using bridge balance and capacitance compensation. Electrophysiological traces were digitized using an ITC-16 board (HEKA Instruments, Bellmore, NY, USA) under control of custom macros programmed using IGOR Pro software (WaveMetrics, Portland, OR, USA). During the stimulation protocol, cells were maintained at a potential between −65 and −70 mV with holding current, when needed (from 0 to ±100 pA; negative in most recordings). Current pulses of 1 s were delivered at the beginning of 3 s sweeps using one of two stimulation protocols: either current pulses were delivered starting at 40 or 50 pA and incremented by 20 pA up to a maximum of 800 pA, or 200 pA pulses were repeatedly delivered with no increment. If the current stimulus initiated barrage firing (>8 Hz firing for at least 1 s), stimulation was stopped and the cell was allowed to fire for up to 4 min before the protocol was repeated after a recovery interval of several minutes.
For experiments requiring synaptic stimulation, we placed a stimulating electrode in the stratum radiatum approximately equidistant from the soma of the recorded cell in each case. Stimulation intensity was initially adjusted to evoke an appropriately-sized EPSP, and the same stimulation intensity was used when comparing control with drug conditions. For bafilomycin experiments, a 400 μA stimulus intensity was selected to stimulate synaptic release in all control and bafilomycin treated cells. This intensity was chosen because it evoked large EPSPs in all control cells.
Pharmacology
All chemicals were obtained from Sigma (St Louis, MO, USA). For the experiments conducted in Ca2+-buffered solution we replaced 2 mm CaCl2 in the external ACSF solution with 6 mm MgCl2 and 1 mm EGTA. For Ca2+-free experiments we replaced 2 mM CaCl2 with 2 mM MgCl2 in the ACSF. Cadmium (Cd2+), nifedipine, verapamil, carbenoxolone and mefloquine were bath applied at the concentrations indicated in the figures after inducing three episodes of barrage firing in ACSF to establish a baseline. For the experiments with bafilomycin, slices were pre-incubated for 1.5 h in either ACSF alone (control), or ACSF containing 1 μm bafilomycin. Slices were then transferred to the recording chamber. For BAPTA experiments, we replaced equimolar potassium gluconate with 10 mm BAPTA in the internal solution in order to maintain osmolarity. To determine whether the reduction of potassium gluconate had any effects on barrage firing, we also made an internal solution containing sucrose instead of BAPTA.
Data analysis and statistics
Analyses of electrophysiological data were performed using custom programs designed with IGOR Pro software (WaveMetrics) and statistical analyses were performed using the Prism 4 software (GraphPad, La Jolla, CA, USA). Pooled data from multiple cells were tested for statistically significant differences using either paired or unpaired Student's t tests or a one-way ANOVA with Tukey's post hoc comparisons. For all statistical tests, significance was defined as P < 0.05. All measurements are presented as mean ± SEM unless otherwise indicated.
Results
We performed patch-clamp recordings from interneurons that expressed enhanced green fluorescent protein (EGFP) near the border of stratum radiatum (SR) and stratum lacunosum-moleculare of hippocampal area CA1 in acute brain slices prepared from serotonin 5b receptor (Htr5b) BAC transgenic mice (see Methods). For all of the experiments described here, 1 s current injections were delivered at the beginning of 3 s sweeps and repeated until barrage firing began, at which time the stimulus was stopped (see Methods). Cells that did not express barrage firing in response to this stimulus were not analysed further.
Barrage firing is calcium dependent
We first tested the role of voltage-gated calcium channels (VGCCs) in retroaxonal barrage firing in these cells. We examined the role of VGCCs directly by attempting to evoke barrage firing in the presence of the non-selective VGCC antagonist cadmium (Cd2+). Bath application of 100 μm Cd2+ caused complete block of barrage firing (Fig. 1A and B). Each attempt to induce barrage firing (i.e. several 1 s stimuli constituting one trial) was separated by approximately 5 min (exact times varied depending on how long it took to evoke barrage firing in each cell); complete block took approximately 20 min (Fig. 1B). This time course is in line with Cd2+ effects on cellular excitability as determined by the number of action potentials evoked by a 100 pA, 1 s current injection (Fig. 1C).
Figure 1. Cd2+ completely blocks retroaxonal barrage (RaB) firing.
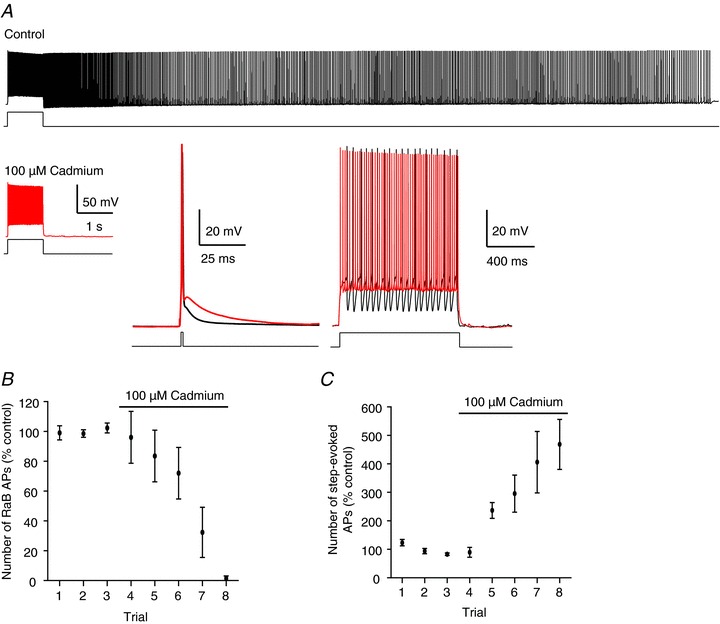
A, example recording showing RaB firing in the absence (top), and lack of RaB firing in the presence (bottom), of 100 μm Cd2+ in the same cell. Insets: Cd2+ (red traces) reduces post-spike membrane potential following an evoked spike elicited with a 1 ms, 1 nA pulse, and inhibits AHPs in spike trains during a 1 s, 200 pA step. Note: the number of action potentials (APs) is greatly increased in Cd2+ in response to the same amplitude step. Lines under each trace depict periods of stimulation. B, grouped data from 5 cells plotted on consecutive trials showing the effect of Cd2+ on the number of RaB APs. Control is calculated as the mean from 3 consecutive trials taken prior to Cd2+ application. All points are normalized to the control in each cell. C, the number of APs required to evoke RaB firing with a 1 s, 100 pA step progressively increased in the presence of Cd2+.
The fact that barrage firing can be completely blocked by Cd2+ suggests a calcium-dependent mechanism. However, we previously reported that removing calcium from the extracellular solution did not inhibit barrage firing. To reconcile these observations, we revisited our previous results, in which we found that bath application of artificial cerebrospinal fluid (ACSF) containing no added calcium (‘Ca2+-free’ ASCF: 2 mm Ca2+ replaced with 2 mm Mg2+) actually increased the duration of barrage firing (Sheffield et al. 2011). Under these conditions, the threshold for evoked action potentials was significantly hyperpolarized (Fig. 2A, left), presumably due to the difference in the charge screening effects of Mg2+ versus Ca2+ (Madeja, 2000). This change in cellular excitability has the potential to confound our measurements of barrage firing. We therefore adjusted the Mg2+ concentration such that evoked action potential threshold was maintained. We also added 1 mm EGTA to further reduce free Ca2+ (Fig. 2A, right; ‘Ca2+-buffered’ ACSF: 2 mm Ca2+ replaced with 6 mm Mg2+ and 1 mm EGTA). Under these conditions Ca2+-buffered ACSF inhibited barrage firing but did not block it completely, even as overall cellular excitability increased due to a reduction in after-hyperpolarization (AHP) amplitude (Fig. 2B–E). These data are consistent with a Ca2+-dependent mechanism for barrage firing, but they also suggest that even low levels of Ca2+ may be sufficient to induce barrage firing when the action potential threshold is lowered.
Figure 2. Ca2+ effects on evoked firing and RaB firing.
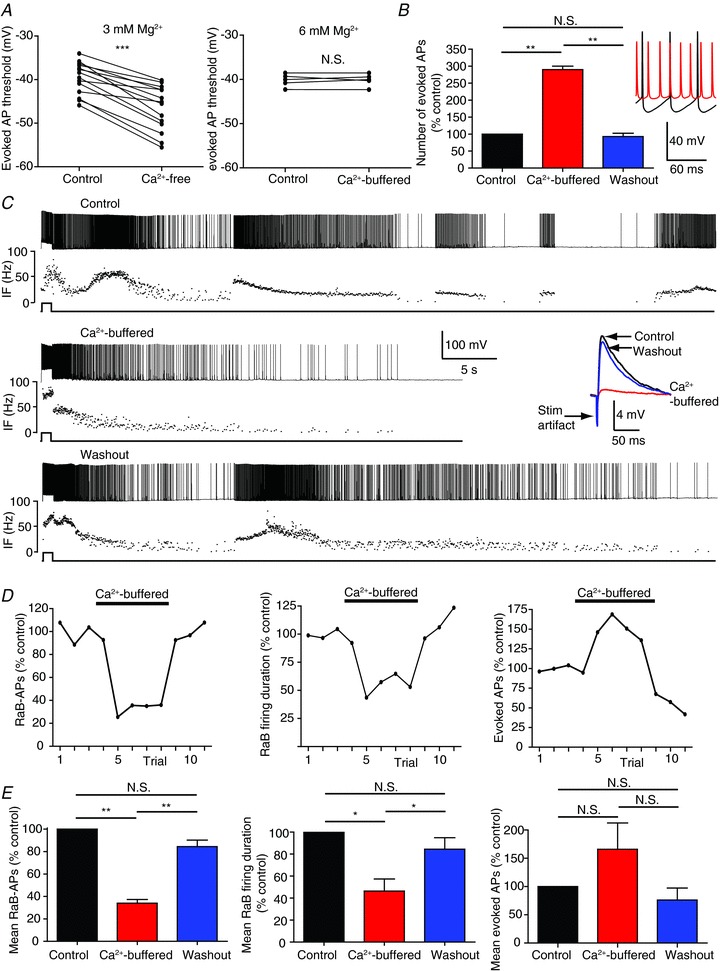
A, replacing 2 mm Ca2+ with 2 mm Mg2+ (3 mm total Mg2+; n= 14) significantly decreased evoked AP threshold from −39.6 ± 1 mV to −46.6 ± 1.4 mV in Htr5b-EGFP-positive hippocampal interneurons, but threshold remained constant when 2 mm Ca2+ was replaced with 6 mm Mg2+ (n= 6). B, Ca2+-buffered ACSF with 6 mm Mg2+ significantly increased the number of evoked APs in response to a 200 pA, 1 s current injection (n= 3). Inset: evoked AP trains in 2 mm Ca2+ (black) and Ca2+-buffered ACSF (red). Note the lack of AHPs in Ca2+-buffered ACSF. C, whole-cell current-clamp recordings of RaB firing in control (top: 2 mm Ca2+), Ca2+-buffered ACSF (middle), and after washout (bottom: 2 mm Ca2+). Underneath each spike train is the instantaneous firing frequency (IF). Note the first second in the train is evoked firing followed by RaB firing. Inset: EPSPs evoked by a stimulating electrode placed in the SR. Black, blue and red show traces from control, washout, and Ca2+-buffered ACSF conditions, respectively. D, the number of RaB APs (left), the duration of RaB firing (middle) and the number of evoked APs required to trigger RaB firing (right) measured on consecutive trials taken from the cell in C. ‘Control’ was calculated as the mean from the first 3 trials. All subsequent trials were normalized to this mean. E, grouped data from 5 cells. All statistics are paired-sample comparisons in the same cell, *P < 0.05; **P < 0.001; ***P < 0.0001; N.S., not significant. For grouped control data, mean measurements were taken from 3 consecutive repeats before Ca2+-buffered ACSF exchange. For grouped Ca2+-buffered ACSF data, mean measurements were taken from at least 2 consecutive trials after a plateau was reached. For grouped washout data, at least 2 consecutive trials were measured following the switch back to 2 mm Ca2+ (ignoring the first trial after the switch to allow time for bath exchange).
We next tested whether Ca2+ flux through L-type Ca2+ channels was involved. The dihydropyridine VGCC blocker nifedipine (30 μm) induced a reversible inhibition of barrage firing (Fig. 3A–C), reducing both the number of action potentials (to 7.5 ± 2.9% of control) and the duration of barrage firing (to 13.1 ± 5.0% of control). Another L-type channel blocker, verapamil (phenylalkylamine), which inhibits channels via a mechanism distinct from nifedipine (Rampe & Triggle, 1990), rapidly and completely abolished barrage firing (Fig. 3D). These data are consistent with a specific role for L-type Ca2+ channels in barrage firing, but since these channels are not essential for synaptic transmission (Poncer et al. 1997; Fisher & Bourque, 2001; Golding et al. 2002), they must act via a different mechanism.
Figure 3. L-type calcium channel antagonists inhibit RaB firing.
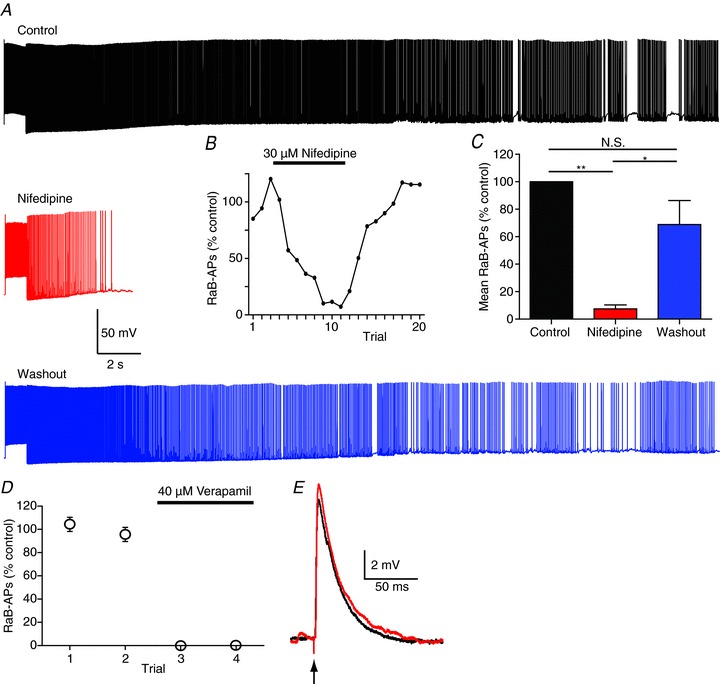
A, example recordings showing control (black), 30 μm nifedipine (red) and washout (blue) in the same cell. B, number of RaB APs measured as % control (control calculated as the mean of the first 3 trials; data are from cell in A). Nifedipine applied after trial 3 and washed out after trial 11. C, grouped data from 5 cells showing number of RaB APs before, during and after nifedipine. D, grouped data from 3 cells before and during application of verapamil. Verapamil was applied after trial 2 and caused complete block of RaB firing. E, evoked EPSPs are unchanged in verapamil (black, control; red, verapamil). N.S., not significant.
To test whether the L-type Ca2+ channels trigger barrage firing via an increase in Ca2+ in the stimulated interneuron, we added the fast Ca2+ chelator BAPTA (10 mm) to the pipette solution. To ensure complete intracellular dialysis such that free calcium would be chelated by BAPTA throughout the recorded cell, including in the distal axon, before attempting to trigger barrage firing, we conducted whole-cell recordings 30–60 min after membrane rupture. BAPTA has been shown to diffuse into the axon terminals of cortical interneurons in 15–20 min (Blatow et al. 2003). Barrage firing was unaffected by the presence of intracellular BAPTA compared to cells recorded using control pipette solution (Fig. 4). As a control experiment, we used a pipette solution in which BAPTA was replaced with equimolar sucrose; this was also found to have no effect on barrage firing.
Figure 4. RaB firing remains unchanged by intracellular Ca2+ chelation.
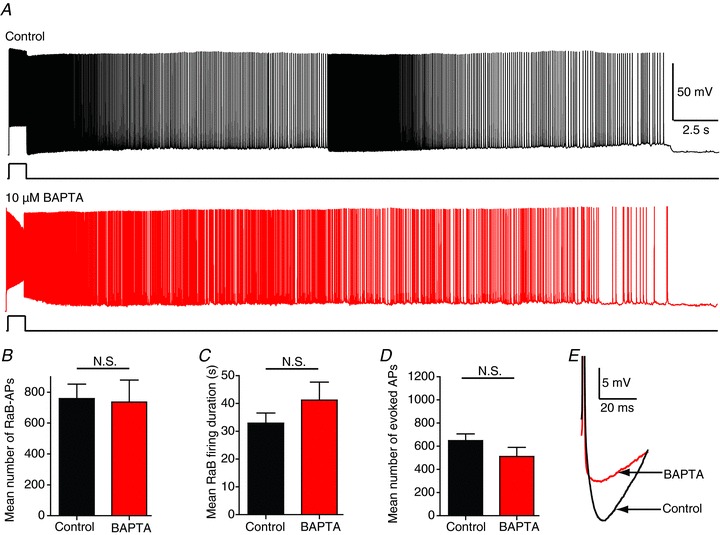
A, recordings showing RaB firing with different internal solutions: control (top; black) and 10 mm BAPTA (middle; red) from different cells. BAPTA measurements were taken between 30 and 60 min following rupture of the membrane for whole-cell recordings to allow for complete cellular dialysis. B–D, population data (n= 14 control; n= 9 BAPTA) showing number of RaB APs (B); RAB firing duration (C); and mean number of evoked APs required to trigger RaB firing in control (black) and BAPTA (red) (D). E, recordings showing the reduced AHP in BAPTA. All statistics are unpaired sample comparisons from different cell groups. N.S., not significant.
Barrage firing does not depend on synaptic vesicle release
Although the involvement of Ca2+ in barrage firing suggests a possible role for synaptic transmission, the specific role of L-type Ca2+ channels and the lack of effect of BAPTA would both suggest otherwise. To more directly test whether barrage firing requires vesicular release, we performed experiments using bafilomycin A1, a potent and specific blocker of the vacuolar type (V-type) H+-ATPase. It acts by eliminating the driving force for uptake of neurotransmitters into synaptic vesicles in both neurons (Zhou et al. 2000) and glia (Araque et al. 2000), thus effectively inhibiting neurotransmitter release from all V-type synaptic vesicles. We pre-incubated slices in 1 μm bafilomycin for 1.5 h. Following this treatment both miniature excitatory postsynaptic potential (mEPSP) frequency and evoked EPSP amplitude were significantly reduced, indicating successful block of synaptic transmission in the slice (Fig. 5A and B). Under these conditions we were still able to evoke barrage firing (Fig. 5C) and found no significant differences in the total number of evoked action potentials required to elicit barrage firing, the duration of barrage firing, or the number of action potentials recorded during barrage firing (Fig. 5D–F). These data indicate that barrage firing does not require active synaptic transmission in the slice.
Figure 5. Depletion of synaptic vesicles blocks synaptic transmission but not RaB firing.
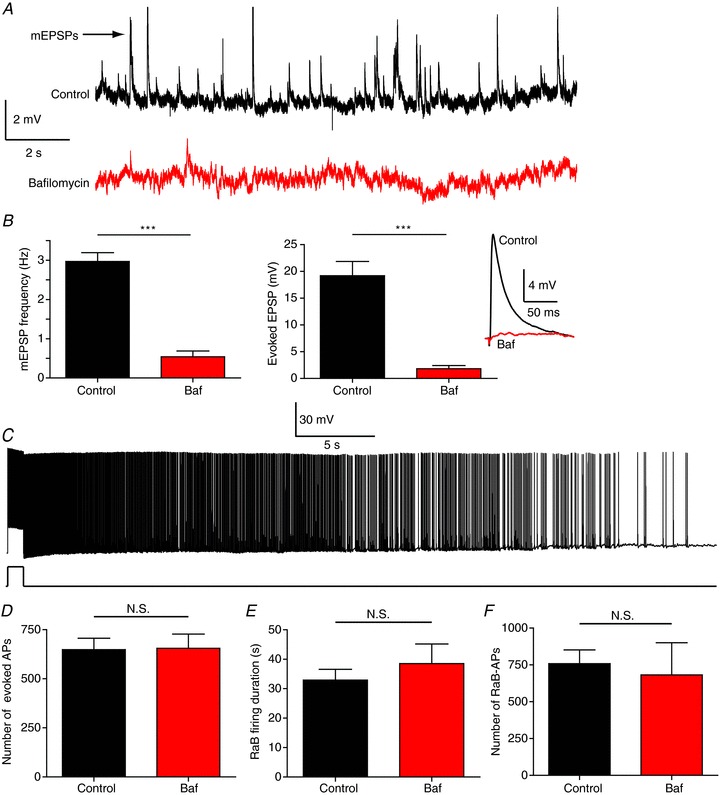
A, traces from control (black) and bafilomycin A1-treated (Baf; red) cells. B, mEPSP frequency (left; 15 s recording, no stimulation; n= 9 control; n= 14 Baf) and evoked EPSP amplitude (right; 400 μA pulse from stimulating electrode placed in the SR ∼500 μm away from recorded cell; n= 9 control; n= 11 Baf). Inset: response to Schaffer collateral stimulation in control (black) and Baf-treated (red) cells. C, example recording showing the presence of normal RaB firing after pre-incubation in Baf. D–F, population analysis of the number of evoked APs required to trigger RaB firing (D), RaB firing duration (E) and the number of RaB APs (F) (n= 14 control; n= 8 Baf). N.S., not significant.
The role of gap junctions
Our previous work indicated that inter-axonal communication can occur during barrage firing (Sheffield et al. 2011). Specifically, stimulation of one interneuron can result in barrage firing in a nearby cell that is not directly stimulated. This observation, together with the L-type Ca2+ channel and BAPTA results described above, raises the interesting possibility that the signal initiating barrage firing may not rely on mechanisms occurring within the stimulated cell itself. Collectively, our data suggest that the mechanism of barrage firing in these cells involves intercellular communication with Ca2+ signalling occurring outside of the stimulated cell, and independently of the effects of Ca2+ on synaptic transmission. Gap junctions are another plausible pathway for intercellular communication of the barrage firing signal that does not require synaptic transmission. Our previous work supported a role for gap junctions in barrage firing. Two commonly used gap junction blockers, carbenoxolone and mefloquine, both inhibited barrage firing (Sheffield et al. 2011); however, neither carbenoxolone nor mefloquine block specific connexin isoforms selectively, making it impossible to determine from these data whether the important gap junctions were those connecting neurons or glial cells.
To address this question, we took advantage of an available mouse line lacking Cx36 (Deans et al. 2001), the most common connexin isoform found in neurons (Söhl et al. 2004). We attempted to generate barrage firing in hippocampal interneurons in these animals using the same approach described for both WT and Htr5b-EGFP animals. Barrage firing occurred in approximately 30% (23/65) of recorded cells. This is comparable to the frequency with which barrage firing was observed in WT animals (13/66). Furthermore, we observed no differences in the number of evoked action potentials required to trigger barrage firing, average duration of barrage firing, or number of barrage firing action potentials (Fig. 6B and C). Thus, the neuronal isoform Cx36 is not required for barrage firing.
Figure 6. Gap junction blockers inhibit RaB firing independently of Cx36.
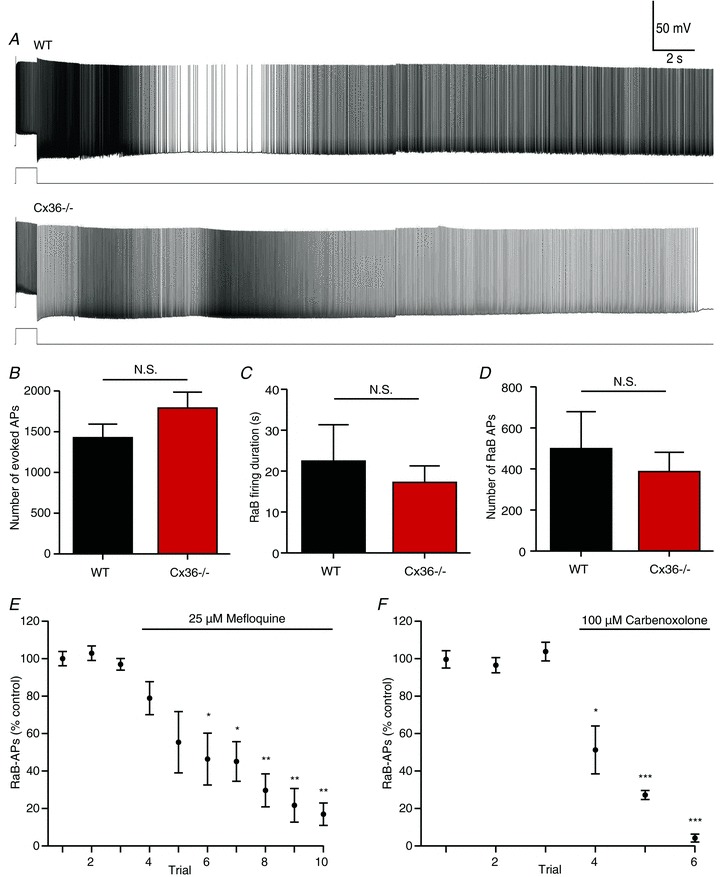
Example recordings (A) and population data (B–E) from WT (n= 13) and Cx36−/− (n= 23) mice. There were no significant differences in the number of evoked APs required to trigger RaB firing (B), duration of RaB firing (C), or number of RaB APs (D). E and F, grouped data from KO showing the effect of 25 μm mefloquine (E; n= 7) and 100 μm carbenoxolone (F; n= 4) on consecutive RaB firing trials. For each cell, AP counts were normalized to the mean of 2–3 control trials before drug application. *P < 0.05, **P < 0.001, ***P < 0.0001 relative to each of the 3 control groups, one-way ANOVA with Tukey's post test. N.S., not significant.
To confirm that the effects of carbenoxolone and mefloquine on barrage firing are independent of its action on Cx36, we repeated these experiments using the Cx36 KO animals. As was observed in Htr5b-EGFP animals, mefloquine (25 μm) inhibited barrage firing in Cx36 KO animals (n= 7 cells; Fig. 6E). The number of barrage firing spikes decreased steadily for each firing episode following mefloquine application, falling to 50% of pre-application levels within 4 episodes and reaching a plateau of approximately 80% block after 9–10 episodes. The effect of carbenoxolone (100 μm) was more dramatic; barrage firing was nearly completely blocked within 3 firing episodes following drug application (n= 4 cells; Fig. 6F). This discrepancy may reflect the reduced potency of mefloquine for blocking isoforms other than Cx36 (Cruikshank et al. 2004), whereas carbenoxolone is very non-selective (Juszczak & Swiergiel, 2009). It is therefore likely that the inhibition of barrage firing by these gap junction blockers is via their action on connexins other than Cx36.
Discussion
In our initial description of this form of persistent firing in hippocampal and neocortical interneurons we detailed a novel integration and firing pattern in which the slow integration of stimulus-evoked firing eventually triggered a barrage of spontaneous action potentials initiated in the distal axon. Here we have shown that barrage firing requires the activation of Ca2+ signalling but is not dependent on vesicular release or intracellular Ca2+ elevation within the stimulated cell. Furthermore, in Ca2+-buffered ACSF, barrage firing had a reduced duration and fewer action potentials. This could be due to a small amount of remaining Ca2+ being sufficient to evoke barrage firing. Alternatively, there could be two distinct mechanisms involved in barrage firing: a Ca2+-independent mechanism required to generate barrage firing, and a Ca2+-dependent mechanism that determines the barrage firing duration and number of action potentials fired during barrage firing. It is also worth noting that the inhibition of barrage firing in Ca2+-buffered ACSF is complicated by the effect of Ca2+-buffered ACSF on excitability. Even though we were able to maintain action potential threshold by increasing extracellular magnesium, under these conditions AHPs were reduced, leading to an increase in the number of evoked action potentials. This is also likely to affect action potentials during barrage firing, which may partially mask the inhibitory effects of eliminating extracellular Ca2+ on barrage firing.
Despite the incomplete block of barrage firing in Ca2+-buffered ACSF, our pharmacological data support a role for VGCCs in the expression of barrage firing. In particular we found that 30 μm nifedipine significantly inhibited the duration of barrage firing in these cells. At this concentration, nifedipine is selective for L-type channels over other types of VGCCs, and blocks both Cav1.2 and Cav1.3, the two L-type channel isoforms expressed ubiquitously in neurons in the mammalian brain (Lipscombe et al. 2004; Marcantoni et al. 2010), and in hippocampal neurons specifically (Xu et al. 2007). Verapamil, a blocker of L-type calcium channels that is mechanistically distinct from nifedipine (Ramp & Triggle, 1990), completely blocked barrage firing in our experiments. However, it is possible that some of its effects can be attributed to inhibition of other VGCC subtypes (Dobrev et al. 1999; Freeze et al. 2006) or potassium channels (Hogg et al. 1999). Nevertheless, the effects of both Ca2+ channel blockers and Ca2+-buffered ACSF strongly support an important role for Ca2+ in the induction, triggering, and/or maintenance of barrage firing.
Several features distinguish the retroaxonal barrage firing observed in hippocampal interneurons from forms of persistent firing that have been observed in other cell types. In many cases, persistent firing is thought to be the result of recurrent activity among networks of synaptically connected neurons (Major & Tank, 2004). The barrage firing we studied, however, was unaffected by the depletion of synaptic vesicles, suggesting it is not the result of synaptically mediated reverberations within neuronal networks.
Some pyramidal cells in layer 5 of the entorhinal cortex and cells in the lateral amygdala show a form of persistent firing that does not require intact synaptic connectivity (Egorov et al. 2002, 2006; Fransén et al. 2006). Like the barrage firing we studied in interneurons, persistent firing in pyramidal neurons is dependent on calcium and blocked by inhibitors of L-type VGCCs. However, there are a number of differences between persistent firing in pyramidal neurons and barrage firing in interneurons. In pyramidal neurons, persistent firing was dependent on cholinergic activation and the ‘readout’ is graded and continuous: the firing rate increases in frequency with additional depolarizing stimuli and decreases with hyperpolarizing stimuli. Moreover, BAPTA applied intracellularly blocked graded persistent firing in pyramidal neurons suggesting a cell-autonomous mechanism. These features are not observed in barrage firing in hippocampal interneurons.
Importantly, a number of observations suggest that multicellular mechanisms may contribute to retroaxonal barrage firing in interneurons. We previously showed that stimulation of one interneuron could trigger barrage firing in another interneuron, despite a lack of evident chemical synaptic transmission or electrical coupling between the two neurons (Sheffield et al. 2011). The lack of observed electrical coupling did not rule out the possibility that intercellular signalling relevant to barrage firing could occur through axo-axonal gap junctions that are too far from the soma to be observable in somatic recordings. Direct electrical coupling via gap junctions is known to occur between dendrites of hippocampal interneurons (Hestrin & Galarreta, 2005), and a few reports suggest that the same types of connections can be formed between axons (Schmitz et al. 2001; Hamzei-Sichani et al. 2007).
Although inhibitors of gap junctions blocked interneuron barrage firing, our results from the Cx36 knockout mice suggest that the presence of Cx36, the predominant neuronal isoform, is not required for barrage firing. While we cannot rule out the possibility that these interneurons express other connexin isoforms that may be involved, to our knowledge only one other isoform, Cx30.2, has been localized to hippocampal interneurons (Kreuzberg et al. 2008). Expression of Cx30.2 was confined to interneurons of the stratum pyramidale and stratum oriens, whereas we targeted interneurons at the border of stratum radiatum and stratum lacunosum-moleculare, making it unlikely that Cx30.2 could have a role in barrage firing. Thus, although barrage firing in interneurons appears to depend on gap junctions, it is unlikely that the relevant gap junctions are formed between the axons of interneurons.
The unlikely role of direct axo-axonal coupling leaves two possibilities: either the ability of carbenoxolone and mefloquine to inhibit barrage firing is due to off-target effects of these drugs or the gap junctions involved in barrage firing consist of other, non-neuronal connexin isoforms. Carbenoxolone is a non-specific connexin blocker, but it can also act on a number of other targets, including VGCCs and P2X7 receptors (Juszczak & Swiergiel, 2009). At low concentrations in vitro (<5 μm), mefloquine selectively blocks Cx36 (Cruikshank et al. 2004). However, at the concentrations required to effectively inhibit coupling in brain slices (≥25 μm), mefloquine significantly affects several other connexin isoforms, including the major astrocytic isoform Cx43. Like carbenoxolone, mefloquine has been shown to cause other physiological changes, including inhibition of inositol 1,4,5-trisphosphate (IP3)-induced Ca2+ release, acetylcholinesterase and P2X7 receptors (Juszczak & Swiergiel, 2009). Although any of these other targets could plausibly be involved in barrage firing, the fact that both carbenoxolone and mefloquine block barrage firing increases the likelihood that gap junctions are the relevant target. Furthermore, our finding that intracellular BAPTA does not affect barrage firing argues against a role for IP3-induced Ca2+ release in the recorded interneuron.
Taken together, our results provide intriguing new constraints on the molecular mechanisms underlying this unusual form of signalling. The ability of gap junction inhibitors to block barrage firing even in Cx36 KO mice suggests the possibility that glial cells may participate in the integration or spread of barrage firing. Astrocytes and oligodendrocytes are extensively coupled by gap junctions, forming an elaborate syncytium that provides metabolic support to adjacent neurons (Söhl et al. 2004). In addition to this supportive role, a growing body of evidence has shown that astrocytes dynamically monitor and modulate neuronal activity by releasing the ‘gliotransmitters’ glutamate, d-serine and ATP onto synapses and axons (Bullock et al. 2005; Fields, 2008; Halassa & Haydon, 2010; Sasaki et al. 2011; Di Castro et al. 2011; Navarrete et al. 2012). These processes are mediated by Ca2+-signalling pathways within astrocytes (Agulhon et al. 2008), which include L-type VGCCs (Parri et al. 2001), consistent with our results demonstrating that barrage firing depends on Ca2+ extrinsic to the stimulated neuron. Furthermore, Ca2+ transients initiated in one astrocyte can spread broadly throughout the astrocytic network via gap junctions (Kuga et al. 2011), providing a potential explanation for the ability of barrage firing to spread from one interneuron to another (Sheffield et al. 2011). Future studies will directly address the hypothesis that neuron–astrocyte interactions underlie this unusual form of integration and firing in hippocampal and other cortical interneurons.
Acknowledgments
The authors thank Bill Kath and Austin Graves for valuable feedback and discussions and to Dr. David Paul for providing connexin 36 knockout mice.
Glossary
- AHP
after-hyperpolarization
- AP
action potential
- EGFP
enhanced green fluorescent protein
- IF
instantaneous firing frequency
- KO
knockout
- mEPSP
miniature excitatory postsynaptic potential
- RaB
retroaxonal barrage
- SR
stratum radiatum
- VGCC
voltage-gated calcium channel
- WT
wild-type
Additional information
Competing interests
None declared.
Author contributions
Experiments were performed in the laboratory of N. Spruston at Northwestern University and the HHMI Janelia Farm Research Campus. N.S. was involved in the conception and design of the experiments as well as the interpretation of the results. M.E.J.S. was involved in the conception and design of the experiments and performed the majority of data collection and analysis. G.B.E. participated in the discussions of experimental design and data analysis and interpretation. G.B.E. was also responsible for drafting and revising the article with input from N.S. and B.D.M. R.J.H. participated in discussions of experimental design and data analysis and interpretation. R.J.H. was also responsible for data collected using the Cx36 KO animals. T.D. contributed data to the Cx36 KO experiments. B.D.M. participated in discussions of data analysis and interpretation and was involved in writing and revising the article. All authors approved the final version of the manuscript.
Funding
Financial support was provided by the US National Institutes of Health (NS-046064 to N.S.) and the Howard Hughes Medical Institute. M.E.J.S. was supported by a Presidential Fellowship from Northwestern University.
References
- Agulhon C, Petravicz J, McMullen AB, Sweger EJ, Minton SK, Taves SR, Casper KB, Fiacco TA, McCarthy KD. What is the role of astrocyte calcium in neurophysiology. Neuron. 2008;59:932–946. doi: 10.1016/j.neuron.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Li N, Doyle RT, Haydon PG. SNARE protein-dependent glutamate release from astrocytes. J Neurosci. 2000;20:666–673. doi: 10.1523/JNEUROSCI.20-02-00666.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatow M, Caputi A, Burnashev N, Monyer H, Rozov A. Ca2+ buffer saturation underlies paired pulse facilitation in calbindin-D28k-containing terminals. Neuron. 2003;38:79–88. doi: 10.1016/s0896-6273(03)00196-x. [DOI] [PubMed] [Google Scholar]
- Bullock TH, Bennett MVL, Johnston D, Josephson R, Marder E, Fields RD. Neuroscience. The neuron doctrine, redux. Science. 2005;310:791–793. doi: 10.1126/science.1114394. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Hopperstad M, Younger M, Connors BW, Spray DC, Srinivas M. Potent block of Cx36 and Cx50 gap junction channels by mefloquine. Proc Natl Acad Sci U S A. 2004;101:12364–12369. doi: 10.1073/pnas.0402044101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans MR, Gibson JR, Sellitto C, Connors BW, Paul DL. Synchronous activity of inhibitory networks in neocortex requires electrical synapses containing connexin36. Neuron. 2001;31:477–485. doi: 10.1016/s0896-6273(01)00373-7. [DOI] [PubMed] [Google Scholar]
- Di Castro MA, Chuquet J, Liaudet N, Bhaukaurally K, Santello M, Bouvier D, Tiret P, Volterra A. Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat Neurosci. 2011;14:1276–1284. doi: 10.1038/nn.2929. [DOI] [PubMed] [Google Scholar]
- Dobrev D, Milde AS, Andreas K, Ravens U. The effects of verapamil and diltiazem on N-, P- and Q-type calcium channels mediating dopamine release in rat striatum. Br J Pharmacol. 1999;127:576–582. doi: 10.1038/sj.bjp.0702574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egorov AV, Hamam BN, Fransen E, Hasselmo ME, Alonso AA. Graded persistent activity in entorhinal cortex neurons. Nature. 2002;420:173–178. doi: 10.1038/nature01171. [DOI] [PubMed] [Google Scholar]
- Egorov AV, Unsicker K, von Bohlen und Halbach O. Muscarinic control of graded persistent activity in lateral amygdala neurons. Eur J Neurosci. 2006;24:3183–3194. doi: 10.1111/j.1460-9568.2006.05200.x. [DOI] [PubMed] [Google Scholar]
- Fields RD. Oligodendrocytes changing the rules: action potentials in glia and oligodendrocytes controlling action potentials. Neuroscientist. 2008;14:540–543. doi: 10.1177/1073858408320294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher TE, Bourque CW. The function of Ca2+ channel subtypes in exocytotic secretion: new perspectives from synaptic and non-synaptic release. Prog Biophys Mol Biol. 2001;77:269–303. doi: 10.1016/s0079-6107(01)00017-7. [DOI] [PubMed] [Google Scholar]
- Fransén E, Tahvildari B, Egorov AV, Hasselmo ME, Alonso AA. Mechanism of graded persistent cellular activity of entorhinal cortex layer V neurons. Neuron. 2006;49:735–746. doi: 10.1016/j.neuron.2006.01.036. [DOI] [PubMed] [Google Scholar]
- Freeze BS, McNulty MM, Hanck DA. State-dependent verapamil block of the cloned human Cav3.1 T-type Ca2+ channel. Mol Pharmacol. 2006;70:718–726. doi: 10.1124/mol.106.023473. [DOI] [PubMed] [Google Scholar]
- Golding NL, Staff NP, Spruston N. Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature. 2002;418:326–331. doi: 10.1038/nature00854. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behaviour. Annu Rev Physiol. 2010;72:335–355. doi: 10.1146/annurev-physiol-021909-135843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamzei-Sichani F, Kamasawa N, Janssen WG, Yasumura T, Davidson KG, Hof PR, Wearne SL, Stewart MG, Young SR, Whittington MA, Rash JE, Traub RD. Gap junctions on hippocampal mossy fibre axons demonstrated by thin- section electron microscopy and freeze fracture replica imm- unogold labelling. Proc Natl Acad Sci USA. 2007;104:12548–12553. doi: 10.1073/pnas.0705281104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häusser M, Raman IM, Otis T, Smith SL, Nelson A, du Lac S, Loewenstein Y, Mahon S, Pennartz C, Cohen I, Yarom Y. The beat goes on: spontaneous firing in mammalian neuronal microcircuits. J Neurosci. 2004;24:9215–9219. doi: 10.1523/JNEUROSCI.3375-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz N. Gene expression nervous system atlas (GENSAT) Nat Neurosci. 2004;7:483. doi: 10.1038/nn0504-483. [DOI] [PubMed] [Google Scholar]
- Hestrin S, Galarreta M. Electrical synapses define networks of neocortical GABAergic neurons. Trends Neurosci. 2005;28:304–309. doi: 10.1016/j.tins.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Hogg RC, Trequattrini C, Catacuzzeno L, Petris A, Franciolini F, Adams DJ. Mechanisms of verapamil inhibition of action potential firing in rat intracardiac ganglion neurons. J Pharmacol Exp Ther. 1999;289:1502–1508. [PubMed] [Google Scholar]
- Juszczak GR, Swiergiel AH. Properties of gap junction blockers and their behavioural, cognitive and electrophysiological effects: animal and human studies. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:181–198. doi: 10.1016/j.pnpbp.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Kreuzberg M, Deuchars J, Weiss E, Schober A, Sonntag S, Wellershaus K, Draguhn A, Willecke K. Expression of connexin30.2 in interneurons of the central nervous system in the mouse. Mol Cell Neurosci. 2008;37:119–134. doi: 10.1016/j.mcn.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Krook-Magnuson E, Luu L, Lee S-H, Varga C, Soltesz I. Ivy and neurogliaform interneurons are a major target of μ-opioid receptor modulation. J Neurosci. 2011;31:14861–14870. doi: 10.1523/JNEUROSCI.2269-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuga N, Sasaki T, Takahara Y, Matsuki N, Ikegaya Y. Large-scale calcium waves traveling through astrocytic networks in vivo. J Neurosci. 2011;31:2607–2614. doi: 10.1523/JNEUROSCI.5319-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscombe D, Helton TD, Xu W. L-type calcium channels: the low down. J Neurophysiol. 2004;92:2633–2641. doi: 10.1152/jn.00486.2004. [DOI] [PubMed] [Google Scholar]
- Madeja M. Extracellular surface charges in voltage-gated ion channels. News Physiol Sci. 2000;15:15–19. doi: 10.1152/physiologyonline.2000.15.1.15. [DOI] [PubMed] [Google Scholar]
- Major G, Tank D. Persistent neural activity: prevalence and mechanisms. Curr Opin Neurobiol. 2004;14:675–684. doi: 10.1016/j.conb.2004.10.017. [DOI] [PubMed] [Google Scholar]
- Marcantoni A, Vandael DH, Mahapatra S, Carabelli V, Sinnegger-Brauns MJ, Striessnig J, Carbone E. Loss of Cav1.3 channels reveals the critical role of L-type and BK channel coupling in pacemaking mouse adrenal chromaffin cells. J Neurosci. 2010;30:491–504. doi: 10.1523/JNEUROSCI.4961-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete M, Perea G, Fernandez de Sevilla D, Gómez-Gonzalo M, Núñez A, Martín ED, Araque A. Astrocytes mediate in vivo cholinergic-induced synaptic plasticity. PLoS Biol. 2012;10:e1001259. doi: 10.1371/journal.pbio.1001259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LM, Clark BA, Gründemann J, Roth A, Stuart GJ, Häusser M. Initiation of simple and complex spikes in cerebellar Purkinje cells. J Physiol. 2010;588:1709–1717. doi: 10.1113/jphysiol.2010.188300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parri HR, Gould TM, Crunelli V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat Neurosci. 2001;4:803–812. doi: 10.1038/90507. [DOI] [PubMed] [Google Scholar]
- Poncer JC, McKinney RA, Gähwiler BH, Thompson SM. Either N- or P-type calcium channels mediate GABA release at distinct hippocampal inhibitory synapses. Neuron. 1997;18:463–472. doi: 10.1016/s0896-6273(00)81246-5. [DOI] [PubMed] [Google Scholar]
- Rampe D, Triggle D. New ligands for L-type Ca2+ channels. Trends Pharmacol Sci. 1990;11:112–115. doi: 10.1016/0165-6147(90)90196-f. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Matsuki N, Ikegaya Y. Action-potential modulation during axonal conduction. Science. 2011;331:599–601. doi: 10.1126/science.1197598. [DOI] [PubMed] [Google Scholar]
- Schmitz D, Schuchmann S, Fisahn A, Draguhn A, Buhl EH, Petrasch-Parwez E, Dermietzel R, Heinemann U, Traub RD. Axo-axonal coupling: a novel mechanism for ultrafast neuronal communication. Neuron. 2001;31:831–840. doi: 10.1016/s0896-6273(01)00410-x. [DOI] [PubMed] [Google Scholar]
- Sheffield M, Best T, Mensh B, Kath W, Spruston N. Slow integration leads to persistent action potential firing in distal axons of coupled interneurons. Nat Neurosci. 2011;14:200–207. doi: 10.1038/nn.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söhl G, Odermatt B, Maxeiner S, Degen J, Willecke K. New insights into the expression and function of neural connexins with transgenic mouse mutants. Brain Res Rev. 2004;47:245–259. doi: 10.1016/j.brainresrev.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Xu JH, Long L, Tang YC, Hu HT, Tang FR. Cav1.2, Cav1.3, and Cav2.1 in the mouse hippocampus during and after pilocarpine-induced status epilepticus. Hippocampus. 2007;17:235–251. doi: 10.1002/hipo.20263. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Petersen CC, Nicoll RA. Effects of reduced vesicular filling on synaptic transmission in rat hippocampal neurones. J Physiol. 2000;525:195–206. doi: 10.1111/j.1469-7793.2000.t01-1-00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]