

Abstract
Synaptic projections from the ventral tegmental area (VTA) to the nucleus accumbens (NAc) make up the backbone of the brain reward pathway, a neural circuit that mediates behavioural responses elicited by natural rewards as well as by cocaine and other drugs of abuse. In addition to the well-known modulatory dopaminergic projection, the VTA also provides fast excitatory and inhibitory synaptic input to the NAc, directly regulating NAc medium spiny neurons (MSNs). However, the cellular nature of VTA-to-NAc fast synaptic transmission and its roles in drug-induced adaptations are not well understood. Using viral-mediated in vivo expression of channelrhodopsin 2, the present study dissected fast excitatory and inhibitory synaptic transmission from the VTA to NAc MSNs in rats. Our results suggest that, following repeated exposure to cocaine (15 mg kg−1 day−1× 5 days, i.p., 1 or 21 day withdrawal), a presynaptic enhancement of excitatory transmission and suppression of inhibitory transmission occurred at different withdrawal time points at VTA-to-NAc core synapses. In contrast, no postsynaptic alterations were detected at either type of synapse. These results suggest that changes in VTA-to-NAc fast excitatory and inhibitory synaptic transmissions may contribute to cocaine-induced alteration of the brain reward circuitry.
Key points
Synaptic projections from the ventral tegmental area (VTA) to the nucleus accumbens (NAc) make up the backbone of the brain reward pathway; in addition to the well-known modulatory dopaminergic projection, the VTA also provides fast excitatory and inhibitory synaptic input to the NAc, but the cellular nature of VTA-to-NAc fast synaptic transmission and its roles in drug-induced adaptations are not well understood.
Using optogenetic approaches, the present study profiled fast excitatory synaptic projection from dopaminergic neurons and inhibitory synaptic projection from GABAergic neurons in the VTA to NAc core (NAcCo) medium spiny neurons.
We further identified that, following repeated non-contingent exposure to cocaine, VTA-to-NAcCo inhibitory synaptic transmission appears to be enhanced by an increase in the presynaptic release probability.
No postsynaptic alterations were detected at either excitatory or inhibitory synapses within the VTA-to-NAcCo projection.
Introduction
Emotional and motivational responses are critically regulated by the brain reward system, which comprises reciprocal projections among the ventral tegmental area (VTA), the nucleus accumbens (NAc) and other related brain regions (Phillips, 1984; Wise, 1987). Pathophysiological alterations of these projections may critically contribute to a variety of emotional and motivational disorders, such as drug addiction and depression (Wise, 1996; Hyman et al. 2006).
Within the complex neural network of the brain reward system, the synaptic projection from VTA dopamine (DA) neurons to NAc medium spiny neurons (MSNs) is relatively well defined for its role in the acute rewarding effect and emotion/motivation-associated learning (Kelley, 2004; Hyman et al. 2006). However, DA neurons and possibly other neuronal types within the VTA also project excitatory glutamatergic axons onto NAc MSNs (Sulzer et al. 1998; Joyce & Rayport, 2000; Chuhma et al. 2004; Lavin et al. 2005; Nair-Roberts et al. 2008; Hnasko et al. 2010; Stuber et al. 2010; Tecuapetla et al. 2010). Furthermore, GABAergic neurons in the VTA, which are intermixed with DA neurons, receive synaptic inputs from the same brain regions, and project to NAc MSNs in parallel with DA neurons (Nagai et al. 1983; Steffensen et al. 1998; Carr & Sesack, 2000). Compared with the modulatory role of DA, fast excitatory and inhibitory VTA-to-NAc synaptic transmissions may exert more direct and timing-specific control over NAc MSNs and their functional output. However, the cellular properties of these fast VTA-to-NAc synaptic transmissions and their cellular responses to drugs of abuse have not been well characterized.
The NAc contains two subregions, the shell (NAcSh) and core (NAcCo), receiving projections from the VTA with a medial-to-lateral topographical pattern. Specifically, dopaminergic neurons that project to the ventromedial NAcSh are primarily located in the posteromedial VTA, including the paranigral nucleus (PN), the central linear nucleus and the medial part of the parabrachial pigmented area (PBP), whereas the dopaminergic cell bodies projecting to the NAcCo are primarily located in the anterolateral part of the VTA, in particular the lateral part of the PBP (Ikemoto, 2007). However, from the medial to lateral VTA–nigra complex, the co-expression of vesicular glutamate transporter 2 (VGLUT2) with tyrosine hydroxylase (TH) gradually decreases (Kawano et al. 2006). These topographical features suggest that the NAcSh receives more VTA glutamate/DA co-release than the NAcCo. The differences in these innervation patterns probably contribute to the differential roles of the NAcSh and NAcCo in cocaine-induced behaviours. One hypothesis is that the NAcSh is preferentially involved in the emotional and motivational aspects of the additive state, whereas the NAcCo is preferentially involved in the habitual learning aspect (Kelley, 2004). Thus far, a number of previous studies have been devoted to the characterization of the VTA-to-NAcSh excitatory synapses (Stuber et al. 2010; Huang et al. 2011), leaving the VTA-to-NAcCo excitatory synapses underexplored. The present study examines and compares VTA glutamatergic projections to the NAcSh and NAcCo, with more detailed characterization on VTA-to-NAcCo synapses.
Compared with glutamatergic projections, VTA-to-NAc GABAergic projections have been less well characterized. A recent study has reported that activation of VTA-to-NAc GABAergic projection fails to elicit responses in 96.6% of NAc MSNs (without discriminating between NAcSh and NAcCo) (Brown et al. 2012). However, our subsequent study detected VTA-to-NAc GABAergic synaptic responses in the majority of NAcCo MSNs (Ishikawa et al. 2013). Although the cellular and circuitry basis underlying these discrepant results has not been characterized (see Discussion), it is clear that at least NAcCo MSNs receive reliable VTA GABAergic projections (Ishikawa et al. 2013). Our current study thus focuses on NAcCo MSNs to examine VTA-to-NAc GABAergic projection.
Using viral-mediated in vivo expression of channelrhodopsin 2 (ChR2) combined with pharmacological and electrophysiological manipulations, we dissected and characterized the fast excitatory and inhibitory synaptic transmission from the VTA to the NAc. Furthermore, following repeated exposure to cocaine (15 mg kg−1 i.p.× 5 days, 1 day withdrawal), we did not detect postsynaptic alterations at VTA-to-NAc excitatory and inhibitory synapses, but observed differential alterations in presynaptic activity at VTA-to-NAcCo excitatory and inhibitory synapses. These results may provide insights into the role of fast VTA-to-NAc synaptic transmission for cocaine-induced circuitry and behavioural alterations.
Methods
Viral vectors
ChR2 fused to Venus (ChR2Y; Addgene plasmid 20071) or mCherry (ChR2R) was expressed from an adeno-associated viral vector 2 (AAV2) with AAV2 internal repeats (Atasoy et al. 2008; Petreanu et al. 2009; Suska et al. 2013). AAV2-flexed-ChR2R expression was induced specifically in Cre recombinase-expressing neurons, where the inverted expressing cassette was flipped. In this study, AAV2-flexed-ChR2R was used in Gad-cre or TH-cre mouse lines, and AAV2-ChR2Y was used for rats. AAV2 stereotype AAV vectors were generated in HEK293T cells and purified by discontinuous iodixanol gradient centrifugation.
Animal use, stereotaxic injections and cocaine administration
Male Sprague-Dawley rats were purchased from the Simonsen (CA, USA) and Harlan Laboratories (MD, USA). Intra-VTA or intra-NAcSh viral injections were performed when animals were at a postnatal age of 26–34 days. For in vivo delivery of viral vectors, animals were anaesthetized with a mixture of ketamine/xylazine (100/10 mg kg−1, 0.1 ml) and placed in a stereotaxic apparatus (Stoelting, Wood Dale, IL, USA), as described previously (Huang et al. 2011; Ishikawa et al. 2013). Briefly, a 28-gauge injection needle was used to bilaterally inject 1 μl (0.2 μl min−1) of the AAV-ChR2Y solution via a Hamilton syringe into the VTA (AP −5.00, ML ±0.90, DV −7.65) or NAcSh (AP +1.55, ML ±0.80, DV −6.50) using a Thermo Orion M365 pump (Thermo Scientific, Barrington, IL, USA). Injection needles were left in place for 5 min following injections. After the recovery period (∼2 weeks), rats received repeated i.p. injections of cocaine (15 mg kg−1 day in 0.1 ml saline × 5 days) or saline (0.1 ml × 5 days) in a novel environment, as described previously (Ishikawa et al. 2009; Mu et al. 2010; Brown et al. 2011). Rats were placed back to the home cage for withdrawal. This timeline allowed 3 weeks to pass following the viral injections, allowing for adequate viral-mediated protein expression.
In a set of experiments, the Gad2-IRES-Cre (Gad2tm2(cre)zjh; Jackson, Laboratories; ME, USA) and TH-IRES-Cre (B6.129 × 1-Thtm1(cre)Te/Kieg, The European Mouse Mutant Archive, Munich, Germany) (Lindeberg et al. 2004) mouse lines were used. For these mice, a 28-gauge needle was used to bilaterally inject 1 μl (0.2 μl min−1) of the virus solution into the VTA (−3.44/±0.48/−4.4).
NAc slice preparation and electrophysiology
Similar to that described previously (Ishikawa et al. 2009, 2013; Mu et al. 2010; Huang et al. 2011), the animal was decapitated following isoflurane anaesthesia. The brain was removed and glued to a block and sliced with a vibratome in 4°C cutting solution containing (in mm) N-methyl-D-glucamine (135), KCl (1), KH2PO4 (1.2), CaCl2 (0.5), MgCl2 (1.5), choline-HCO3 (20) and glucose (10), saturated with 95% O2/5% CO2, with the pH adjusted to 7.4 with HCl. The slices were incubated in artificial cerebrospinal fluid (aCSF) containing (in mm) NaCl (119), KCl (2.5), CaCl2 (2.5), MgCl2 (1.3), NaH2PO4 (1), NaHCO3 (26.2) and glucose (11), saturated with 95% O2/5% CO2 at 37°C for 30 min and then allowed to recover for at least 30 min at room temperature before experimentation. Signature anatomical landmarks (e.g. the anterior commissure) were used to delineate the NAcSh and NAcCo.
During recordings, slices were superfused with aCSF that was heated to 31–33°C by passing the solution through a feedback-controlled in-line heater (Warner Instruments, Hamden, CT, USA) before entering the chamber. Recordings were made under visual guidance (40×, differential interference contrast optics) with electrodes (3–5 MΩ) filled with (in mm) CsCH3O3S (140), tetraethylammonium chloride (TEA-Cl) (5), EGTA (0.4), Hepes (20), Mg-ATP (2.5), Na-GTP (0.25) and QX-314 (1), pH 7.3. To record excitatory postsynaptic currents (EPSCs), picrotoxin (PTX) (100 μm) was included in the external perfusion aCSF to block GABAA. The N-methyl-d-aspartate receptor (NMDAR)-mediated component was recorded at +40 mV, and the amplitude was operationally defined as the amplitude of the current 30 ms after the onset of the evoked current.
To evoke VTA-to-NAc synaptic transmission, axons expressing ChR2 were stimulated by 473 nm DPSS laser (IkeCool, Los Angeles, CA, USA) coupled to a 62.5 μm optic fibre. An optical stimulation of <1 ms duration was used to stimulate the ChR2-expressing axons at 0.1 Hz. EPSCs were recorded at –70 mV, which was around the reversal potential of inhibitory postsynaptic currents (IPSCs). IPSCs were recorded at +10 mV, which was around the reversal potential of EPSCs. Liquid junction potentials were not corrected.
To measure the α-amino-3-hydroxy-5-methyl-4-isoxa-zolepropionic acid receptor (AMPAR)/NMDAR ratio, the NMDAR-mediated components were measured at +50 mV and the AMPAR-mediated EPSCs were measured at –70 mV. Operationally, the amplitude of AMPAR EPSCs was measured at the peak of EPSCs at –70 mV, and the amplitude of NMDAR EPSCs was measured at +50 mV, 30 ms after the onset of EPSCs; at this time point AMPAR EPSCs were largely inactivated (Fig. 2). This measurement is similar to some (Dong et al. 2004) but different from other (Kourrich et al. 2007; Mameli et al. 2009) previous studies measuring the AMPAR/NMDAR ratio. Specifically, the peak amplitudes of AMPAR-mediated EPSCs were larger at –70 mV than at +40 or +50 mV, resulting in larger absolute values of the AMPAR/NMDAR ratio. Because the AMPAR/NMDAR ratio is a relative measurement, the absolute values of this ratio do not carry physiological significance, whereas the changes in this ratio under the same recording conditions reflect synaptic alterations.
Figure 2. VTA-to-NAc fast excitatory synaptic transmission following cocaine administration.
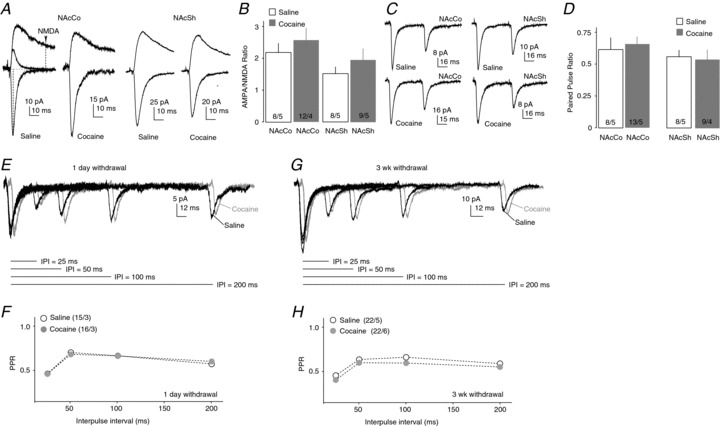
A, Example EPSCs recorded at −70 and +50 mV from VTA-to-NAcCo (left) and VTA-to-NAcSh (right) synapses from saline- and cocaine-treated rats. In some recordings, AMPAR-mediated EPSCs were pharmacologically isolated at +50 mV (left), which decayed rapidly to the baseline. Operationally, the amplitude of AMPAR EPSCs was measured at the peak of EPSCs at –70 mV, and the amplitude of NMDAR EPSCs was measured at +50 mV at 30 ms after the onset of EPSCs, at which time point AMPAR EPSCs had largely returned to baseline. B, Summarized results showing that, after 1 day of withdrawal from repeated i.p. injections of cocaine, the AMPAR/NMDAR ratio was not altered within the VTA-to-NAcCo/Sh projection. C, Example EPSCs elicited by paired-pulse optical stimulations in NAcCo (left) and NAcSh (right) MSNs from saline-treated (top) and cocaine-treated (bottom) rats. D, Summarized results showing that the PPR (IPI = 50 ms) at VTA-to-NAcCo or VTA-to-NAcSh excitatory synapses was not altered 1 day after repeated exposure to cocaine (15 mg kg−1 day−1× 5 days, i.p. injection). E, Example EPSCs elicited from rat VTA-to-NAcCo synapses after 1 day of withdrawal by paired-pulse optical stimulations with IPI of 25, 50, 100 or 200 ms. F, Summarized results showing that the PPRs of EPSCs from VTA-to-NAcCo synapses were not affected after 1 day of withdrawal from cocaine exposure. G, Example EPSCs elicited from rat VTA-to-NAcCo synapses after 21 days of withdrawal by paired-pulse optical stimulations with IPI of 25, 50, 100 or 200 ms. H, Summarized results showing that the PPRs of EPSCs from VTA-to-NAcCo synapses were modestly but significantly enhanced after 21 days of withdrawal from cocaine exposure. n/m, number of cells/number of animals.
To measure the decay kinetics of IPSCs, the time elapsed from the peak amplitude of IPSCs to one-third of the peak amplitude (T1/3) was used. The value of T1/3 is an operational estimate of the time constant (i.e. T36.8%) assuming that the decay of IPSCs follows single-exponential decay kinetics.
All chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA) unless specified otherwise. For all recordings, the series resistance was 8–14 MΩ and was left uncompensated. The series resistance was monitored continuously during all recordings, and a change beyond 20% was not accepted for data analysis. Synaptic currents were recorded with a MultiClamp 700A amplifier (Molecular Devices), filtered at 3 kHz, amplified five times and then digitized at 20 kHz with a Digidata 1322A analogue-to-digital converter (Molecular Devices, CA, USA).
All procedures were performed by strictly following the standard procedures approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh or European Neuroscience Institute.
Data analysis and statistics
All results are shown as the mean ± SEM. Statistical significance was assessed using one- or two-factor ANOVA with Bonferroni post-tests or two-tailed t test. One to four cells were recorded from each animal. The total number of cells is presented as ‘n’ and the total number of animals as ‘m’. Thus, the sample size is presented as n/m. Cell-based statistics were performed and presented for all results.
Results
To electrophysiologically dissect fast excitatory and inhibitory synaptic transmission from the VTA to NAc, we injected AAV2-ChR2Y into the VTA of rats (Petreanu et al. 2009). To selectively target VTA dopaminergic or GABAergic neurons, we used the TH-cre or GAD-cre mice with intra-VTA injections of AAV2-flexed-ChR2R. Three weeks following virus injection, naïve animals and animals treated with repeated i.p. injections of saline or cocaine (15 mg kg day−1× 5 days, 24 h withdrawal) were sacrificed to obtain horizontal brain slices. Within these slices, strong fluorescence was observed in the VTA, indicating the expression of ChR2 (Fig. 1A). In addition, a large number of ChR2-positive neural fibres were observed projecting from the VTA to the forebrain including the NAc (Fig. 1A). By laser-mediated activation of ChR2, the VTA-to-NAc synaptic transmission was selectively activated and examined.
Figure 1. VTA-to-NAc excitatory synaptic transmission.
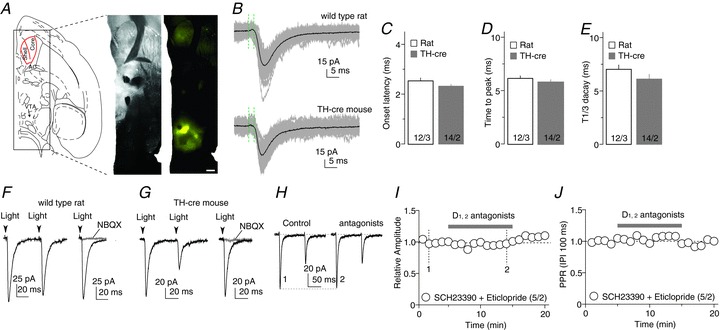
A, Diagram (left) and images (right) showing that intra-VTA injection of ChR2-expressing AAV2 resulted in ChR2-YFP expression in the VTA and its projections. Images were taken from the differential interference contrast microscope used for electrophysiological recordings of this study. B, Optogenetically evoked synaptic currents in example NAcCo neurons from a rat (top) with intra-VTA injection of AAV-ChR2Y and a TH-cre mouse (bottom) with intra-VTA injection of AAV-flexed-ChR2R. Recordings were made by holding the membrane potential at –70 mV, which was close to the reversal potential of IPSCs, resulting in minimal IPSCs. The time elapsed from the artifacts by optogenetic stimulation (indicated by the first green vertical dashed line) to the initiation of the evoked synaptic currents (indicated by the second dashed line) was operationally used to measure the delay of synaptic transmission. C, Summarized result showing that the delays of optogenetically evoked VTA-to-NAcCo synaptic transmission were short, consistent with monosynaptic transmission; they were not different between rats and TH-cre mice. D, Summarized results showing that the activation kinetics (operationally measured as the time elapsed from the initiation to the peak of evoked synaptic currents) of optically evoked synaptic currents were fast and not different between rats and TH-cre mice with intra-VTA injections of AAV-flexed-ChR2R. E, Summarized results showing that the decay kinetics (operationally measured as the decay time from the peak to one-third of the peak) of optically evoked synaptic currents were fast and were not different between rats and TH-cre mice. F, Examples showing that, in rats, optogenetic stimulations evoked synaptic currents from the VTA-to-NAcCo afferent (left), which were completely inhibited by the AMPAR-selective antagonist NBQX (right), indicating that they were EPSCs. G, Examples showing that, in TH-cre mice, optogenetic stimulations evoked synaptic currents from the VTA-to-NAcCo afferent (left), which were completely inhibited by the AMPAR-selective antagonist NBQX (right), indicating that they were EPSCs. H, Example EPSCs from VTA-to-NAcCo synapses in a rat before and after perfusion of the dopamine D1 and D2 class receptor antagonists SCH23390 and eticlopride. I, Summarized results showing that the inhibition of D1 and D2 receptors did not affect the EPSC amplitude from VTA-to-NAcCo synapses. J, Summarized results showing that the inhibition of D1 and D2 receptors did not affect the PPR (IPI = 100 ms) of EPSCs from VTA-to-NAcCo synapses.
Fast excitatory synaptic transmission
In wild-type rats and TH-cre mice with intra-VTA injection of ChR2-expressing viruses, fast postsynaptic currents were consistently elicited in NAcCo neurons by brief laser exposures (<1 ms, λ= 473 nm) directed towards the ChR2-expressing fibres (in the presence of 100 μm PTX to inhibit GABAA receptors) (Fig. 1B). These synaptic currents exhibited a short delay from optical stimulation to the onset of the current (rats, 2.54 ± 0.12 ms; mice, 2.33 ± 0.08 ms; Fig. 1C), fast activation (time to peak current: rats, 6.17 ± 0.24 ms; mice, 5.77 ± 0.25 ms; Fig. 1D) and inactivation (time to decay to one-third of peak amplitude: rats, 7.03 ± 0.42 ms; mice, 6.10 ± 0.46 ms; Fig. 1E). These synaptic parameters are consistent with those of monosynaptic transmission evoked using optogenetic stimulation in the striatum (Tritsch et al. 2012). Furthermore, these postsynaptic currents could be blocked by the AMPAR-selective antagonist 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline (NBQX) (5 μm) in both rat (n= 5 of 5 cells; Fig. 1F) and TH-cre mouse (n= 5 of 5 cells; Fig. 1G) preparations, indicating that they were AMPAR EPSCs. In our experimental preparations, in addition to triggering glutamatergic transmission, optical activation of VTA-to-NAcCo projections was also likely to trigger DA release, which may, in turn, tonically regulate VTA-to-NAcCo synaptic transmission. However, co-perfusion of the DA D1 and D2 class receptor antagonists SCH23390 (1 μm) and eticlopride (3 μm) did not affect either the peak amplitude (P= 0.38 before vs. during perfusion) or the paired-pulse ratio (PPR; the ratio of the peak amplitude of the second EPSC over the peak amplitude of the first EPSC) (P= 0.57 before vs. during perfusion) of the evoked EPSCs at the stimulation frequency of 0.1 Hz (Fig. 1I, J). Taken together, this set of results shows that, similar to the optogenetic activation of VTA-to-NAcCo dopaminergic projection in transgenic mice, activation of the VTA-to-NAcCo projection triggered monosynaptic EPSCs, which were not affected by concurrent dopaminergic signalling under our experimental conditions.
After establishing the VTA-to-NAc excitatory transmission in slices, we examined whether it was affected after 1 day of withdrawal from repeated cocaine exposure. We first measured the AMPAR/NMDAR ratio (see Methods) as an indicator of potential postsynaptic alterations. Because AMPAR and NMDAR EPSCs were simultaneously elicited by the same presynaptic stimulation, changes in the AMPAR/NMDAR ratio should be largely attributable to altered postsynaptic receptors (i.e. AMPARs, NMDARs or both). In this experiment, both VTA-to-NAcCo and VTA-to-NAcSh excitatory synaptic transmissions were examined using the rat preparation. AMPAR-mediated EPSCs were measured at –70 mV and NMDAR-mediated components were measured at +50 mV (see Methods). Cocaine treatment did not alter the AMPAR/NMDAR ratio at either the VTA-to-NAcCo or VTA-to-NAcSh synapse (saline-Co, 2.14 ± 0.25, n= 8/5; saline-Sh, 1.41 ± 0.13, n= 12/6; cocaine-Co, 2.60 ± 0.43, n= 8/5; cocaine-Sh, 1.94 ± 0.36, n= 9/5; F = 2.70, P= 0.06, one-way ANOVA; Fig. 2A, B). Notably, the AMPAR/NMDAR ratio appeared to be lower at VTA-to-NAcSh synapses relative to VTA-to-NAcCo synapses in both saline- and cocaine-exposed groups, but no statistical significance was detected.
We next examined the potential presynaptic effects of cocaine exposure on VTA-to-NAcCo excitatory synapses by recording paired EPSCs (interpulse interval, IPI = 50 ms) (Fig. 2C, D). The PPR was similar at VTA-to-NAcCo excitatory synapses between saline- and cocaine-treated rats (saline, 0.60 ± 0.03, n= 8/5; cocaine, 0.69 ± 0.06, n= 13/5; P= 0.8, t test; Fig. 2C, D). It is possible that presynaptic alterations cannot be detected using PPR analysis at a single IPI. As such, we next examined the PPR at four different IPIs (25, 50, 100 and 200 ms), and our results again showed no significant difference at VTA-to-NAcCo excitatory synapses between saline- and cocaine-treated animals after 1 day of withdrawal (F1,116= 0.005, P= 0.944, treatment effect; F = 0.314, P= 0.815, IPI × treatment; two-factor ANOVA; Fig. 2E, F). However, a modest but significant decrease in the PPR was detected after 21 days of withdrawal (F1,158= 6.77, P= 0.01, treatment effect; F = 0.146, P= 0.932, IPI × treatment; two-factor ANOVA; Fig. 2G, H). Thus, similar to the prefrontal cortex-to-NAc afferent (Suska et al. 2013), exposure to cocaine may induce an increase in the presynaptic release of neurotransmitters at VTA-to-NAcCo synapses. It is worth noting that, in our experimental conditions, the PPR returned to the basal level (i.e. 1) after ∼1 s, a recovery slower than that analysed using electrical stimulation (Kourrich et al. 2007).
In addition to the VTA-to-NAc projection, the NAc receives extensive excitatory projections from a large number of limbic and paralimbic brain regions, including the prefrontal cortex, amygdala, thalamus and hypothalamus (Phillipson & Griffiths, 1985; Sesack & Grace, 2010). By random sampling of NAc excitatory synapses, extensive previous studies have demonstrated dynamic cocaine-induced alterations in the overall excitatory transmission onto NAc MSNs. The lack of cocaine-induced effects on VTA-to-NAc excitatory synapses led us to speculate that VTA-to-NAc excitatory synapses respond differently to cocaine treatment. To test this speculation, we sequentially recorded light-evoked VTA-to-NAc excitatory transmission and electrically evoked, randomly sampled excitatory transmission from the same NAcCo neurons. We found that light-evoked EPSCs readily reached a plateau amplitude as the stimulation intensity was increased (46.0 ± 6.2 pA, n= 37). In addition, light-evoked EPSCs at VTA-to-NAcSh synapses were larger than those at VTA-to-NAcCo synapses (182.0 ± 35.7 pA, n= 7; P < 0.001, t test). However, the peak amplitude of electrically induced (randomly sampled) excitatory monosynaptic transmission exhibited a wide range and could be as large as several thousands of pA upon strong stimulations (Fig. 3A). Furthermore, the postsynaptic properties, measured by the AMPAR/NMDAR ratio, were also different between VTA-to-NAc and randomly sampled excitatory synapses. Specifically, the AMPAR/NMDAR ratio was significantly higher at randomly sampled excitatory synapses than at VTA-to-NAcCo synapses in both saline-exposed (AMPAR/NMDAR ratio from electrical stimulation relative to optical stimulation, 2.40 ± 0.25; P < 0.01, n= 8/5; paired t test) and cocaine-exposed (1 day of withdrawal) (AMPAR/NMDAR ratio from electrical stimulation relative to optical stimulation, 1.92 ± 0.34, n= 8/5; P < 0.05, paired t test; Fig. 3B, C) rats. Further analysis revealed that this synapse type-based difference in the AMPAR/NMDAR ratio was not affected by exposure to cocaine (P= 0.25, t test). These results suggest that VTA-to-NAc excitatory synaptic transmission is not likely to be a major contributor to the overall excitatory inputs onto NAc MSNs, exhibits different compositions of postsynaptic receptor populations compared with other excitatory synapses, and is probably resistant to changes upon cocaine exposure.
Figure 3. Different synaptic properties between VTA-to-NAcCo excitatory synapses and randomly sampled excitatory synapses on NAcCo MSNs.
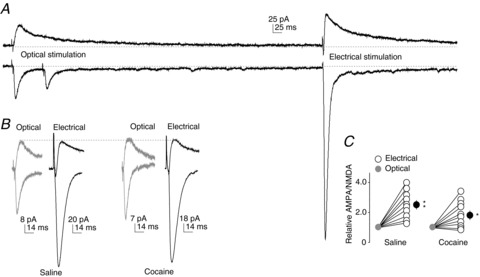
A, Example AMPAR- and NMDAR-mediated EPSCs recorded at +40 mV (top) and –70 mV (bottom) from VTA-to-NAcCo synapses by optical stimulation (left) and randomly sampled synapses by electrical stimulation (right) from the same NAcCo neuron in a saline-exposed rat. Large EPSCs could be evoked from randomly sampled synapses upon strong stimulation, whereas EPSCs from VTA-to-NAcCo synapses were typically small and insensitive to the intensity of optical stimulation. B, Examples of normalized EPSCs (to optically evoked NMDAR-mediated components) from VTA-to-NAcCo synapses and randomly sampled NAcCo synapses in saline- and cocaine-exposed rats. C, Summarized results showing that the AMPAR/NMDAR ratio at VTA-to-NAcCo excitatory synapses is lower than that in randomly sampled excitatory NAcCo synapses, and this difference was not altered after 1 day of withdrawal from exposure to cocaine.
Fast inhibitory synaptic transmission
Compared with glutamatergic projections, VTA-to-NAc GABAergic projections have been characterized less consistently. A recent study has shown that the VTA-to-NAc GABAergic projection primarily synapses on cholinergic interneurons, and spares the majority of NAc MSNs (NAcSh or NAcCo, not specified) (Brown et al. 2012). However, our subsequent study consistently detected VTA-to-NAc GABAergic synaptic responses in the majority of NAcCo MSNs (Ishikawa et al. 2013). Our current study thus focuses on NAcCo MSNs to examine the VTA-to-NAc GABAergic projection.
Using similar approaches, we recorded optically evoked fast synaptic currents from VTA-to-NAcCo synapses at the holding potential of +10 mV, which was close to the reversal potential of AMPAR and NMDAR EPSCs, thus minimizing the potential contamination from EPSCs. Some recordings were also made at +10 mV in the presence of NBQX, and the recorded IPSCs were no different in amplitude or kinetics from those recorded without the application of NBQX. Data were thus pooled for the following analysis.
In wild-type rats and GAD-cre mice with intra-VTA injections of ChR2-expressing viruses, light-evoked IPSC-like currents were recorded from NAcCo MSNs (Fig. 4A, B). These synaptic currents exhibited short-delay onset (rats, 2.30 ± 0.16 ms; mice, 1.90 ± 0.14 ms; Fig. 4C), fast activation (time to peak: rats, 6.60 ± 0.44 ms; mice, 6.10 ± 0.41 ms; Fig. 4D) and inactivation (time to decay to one-third of peak amplitude: rats, 15.90 ± 1.64 ms; mice, 15.20 ± 0.67 ms; Fig. 4E). These synaptic parameters are consistent with those of monosynaptic transmission evoked using optogenetic stimulation in the striatum (Tritsch et al. 2012). Furthermore, these postsynaptic currents could be consistently evoked by optical stimulations and could be completely inhibited by the GABAA receptor-selective antagonist PTX (100 μm) in both the rat (n= 5 of 5 cells; Fig. 4F) and GAD-cre mouse (n= 5 of 5 cells; Fig. 4G) preparations, indicating that they were GABAA receptor IPSCs. In addition, co-perfusion of the DA D1 and D2 class receptor antagonists SCH23390 (1 μm) and eticlopride (3 μm) did not affect either the peak amplitude (P= 0.57 before vs. during perfusion) or the PPR (P= 0.38 before vs. during perfusion) of evoked IPSCs, suggesting that VTA-to-NAcCo co-release of DA does not tonically regulate the GABAergic transmission under the current experimental conditions (i.e. at 0.1 Hz) (Fig. 4I, J). Taken together, this set of results shows that, similar to the optogenetic activation of VTA-to-NAcCo dopaminergic projection in transgenic mice, activation of the VTA-to-NAcCo projection triggers monosynaptic IPSCs, which were not affected by concurrent dopaminergic signalling under our experimental conditions.
Figure 4. VTA-to-NAcCo inhibitory synaptic transmission.
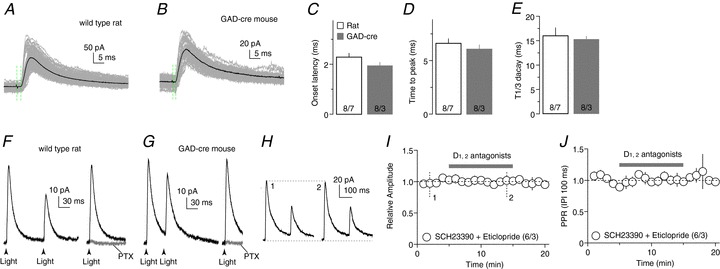
A and B, Optogenetically evoked synaptic currents in example NAcCo neurons from a rat with intra-VTA injection of AAV2-ChR2Y (A) and a GAD-cre mouse with intra-VTA injection of AAV2-flexed-ChR2R (B). Recordings were made by holding the membrane potential at +10 mV, which was close to the reversal potential of EPSCs, resulting in minimal EPSCs. The time elapsed from the artifacts by optogenetic stimulation (indicated by the first green vertical dashed line) to the initiation of the evoked synaptic currents (indicated by the second dashed line) was operationally used to measure the delay of synaptic transmission. C, Summarized result showing that the delay of optogenetically evoked VTA-to-NAcCo synaptic transmission was short, which was consistent with monosynaptic transmission; the onset latency was not different between wild-type rats and GAD-cre mice. D, Summarized results showing that the activation kinetics (operationally measured as the time elapsed from the initiation to the peak of evoked synaptic currents) of optically evoked synaptic currents were fast and not different between rats and GAD-cre mice. E, Summarized results showing that the decay kinetics (peak to one-third peak decay time) of optically evoked synaptic currents were fast and not different between rats and GAD-cre mice. F, Examples showing that, in the rat, optogenetic stimulations evoked synaptic currents from the VTA-to-NAcCo afferent (left), which were completely inhibited by the GAGAA receptor-selective antagonist PTX (right), indicating that they were GABA receptor IPSCs. G, Examples showing that, in the GAD-cre mouse, optogenetic stimulations evoked synaptic currents from the VTA-to-NAcCo afferent (left), which were completely inhibited by the GABAA receptor-selective antagonist PTX (right), indicating that they were GABAR IPSCs. H, Example EPSCs from VTA-to-NAcCo synapses in a rat before and after application of the dopamine D1 and D2 class receptor antagonists SCH23390 (1 μm) and eticlopride (3 μm). I, Summarized results showing that the inhibition of D1 and D2 receptors did not affect the IPSC amplitude from VTA-to-NAcCo synapses. J, Summarized results showing that the inhibition of D1 and D2 receptors did not affect the PPR (IPI = 100 ms) of IPSCs from VTA-to-NAcCo synapses.
After verifying the above experimental approach, we examined whether exposure to cocaine altered VTA-to-NAcCo inhibitory synaptic transmission. On withdrawal day 1 from repeated exposure to cocaine, a significant increase in the PPR was observed at VTA-to-NAcCo synapses (F1,144= 34.57, P < 0.01, treatment effect; F = 0.19, P= 0.90, IPI × treatment; two-factor ANOVA; Fig. 5A, B), suggesting an inhibition of presynaptic release. In parallel, we also examined the inhibitory projection from the NAcSh to NAcCo, which provides important lateral inhibitory control of NAcCo neurons (Morgan et al. 2011). This was achieved by intra-NAcSh injection of AAV-ChR2Y (Fig. 5C). In saline-treated animals, NAcSh-to-NAcCo inhibitory synapses exhibited a significantly higher PPR relative to VTA-to-NAcCo inhibitory synapses at all four tested IPIs (25, 50, 100 and 200 ms) (F1,100= 194.4, P < 0.001, treatment effect; F = 0.72, P= 0.54, IPI × treatment; two-factor ANOVA), suggesting a higher presynaptic release probability at VTA-to-NAcCo inhibitory synapses. Furthermore, exposure to cocaine (examined on withdrawal day 1) did not affect the PPR at NAcSh-to-NAcCo synapses (F1,156= 0.08, P= 0.77, treatment effect; F = 0.09, P= 0.97, IPI × treatment; two-factor ANOVA; Fig. 5D, E). However, the effect of cocaine on the PPR of VTA-to-NAcCo inhibitory synapses was no longer detectable after 21 days of withdrawal, suggesting that this effect was relatively transient (F1,136= 2.08, P= 0.152, treatment effect; F = 0.34, P= 0.80, IPI × treatment; two-factor ANOVA; Fig. 5F, G). Thus, presynaptic release of VTA to NAcCo GABAergic transmission was probably targeted by exposure to cocaine, and the resulting inhibition may transiently reduce the tonic inhibitory influence of the VTA input on NAcCo neurons. Notably, the PPR of VTA-to-NAcCo inhibitory synapses appeared to exhibit developmental regulation; it increased when the animal became older (saline withdrawal day 1 vs. day 21).
Figure 5. Presynaptic inhibition was detected at VTA-to-NAcCo inhibitory synapses after cocaine administration.
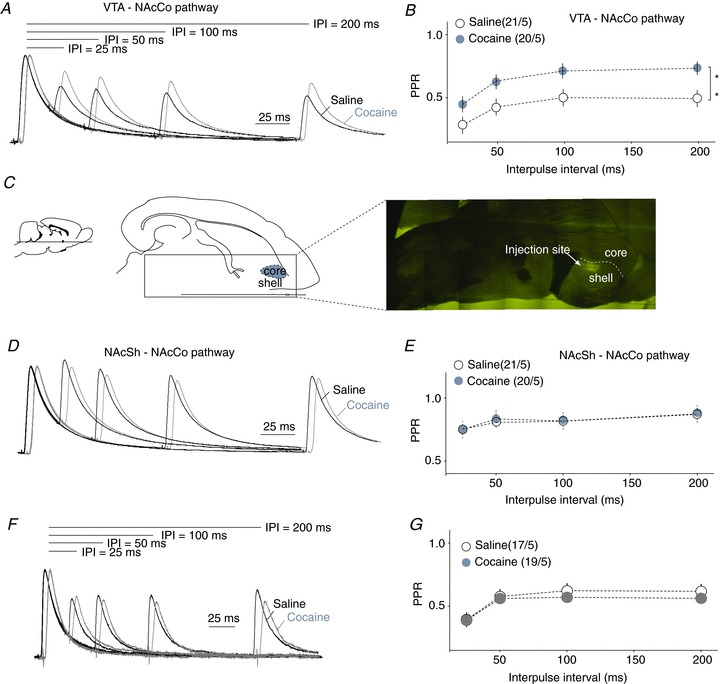
A, Example IPSCs elicited in the rat VTA-to-NAcCo synapses after 1 day of withdrawal by paired-pulse optical stimulations with different interpulse intervals (25, 50, 100 and 200 ms). B, Summarized results showing that the PPRs of VTA-to-NAcCo inhibitory synapses were significantly increased after 1 day of withdrawal from repeated i.p. injections of cocaine. C, Diagrams and image showing that intra-NAcSh injection of AAV2-ChR2Y resulted in the expression of fluorescent signals in the NAcSh. D, Example IPSCs elicited at NAcSh-to-NAcCo synapses by paired-pulse stimulations with different interpulse intervals (25, 50, 100 and 200 ms) in NAcCo MSNs from saline- and cocaine-treated rats. E, Summarized results showing that the PPRs of NAcSh-to-NAcCo inhibitory synapses were not altered significantly after 1 day of withdrawal from repeated i.p. injections of cocaine. F, Example IPSCs elicited at rat VTA-to-NAcCo synapses after 21 days of withdrawal by paired-pulse stimulations with different interpulse intervals (25, 50, 100 and 200 ms). G, Summarized results showing that the PPRs of NAcSh-to-NAcCo inhibitory synapses were not altered significantly after 21 days of withdrawal from repeated i.p. injections of cocaine. **,P < 0.01.
Typically, an increase in the PPR suggests decreased release probability in VTA-to-NAcCo fast inhibitory synaptic transmission. However, alterations in the postsynaptic receptor composition and/or receptor functionality may also contribute to the altered PPR (Heine et al. 2008). We therefore attempted to detect cocaine-induced postsynaptic alterations within this projection. At the postsynaptic membrane of GABAergic synapses, alterations in the subunit composition, typically the a subunits of GABAA receptors, are a common form of cellular adaptation. Following exposure to cocaine, the whole-brain level of a1 subunits is reduced (Koobs, 2001), which can be replaced by a2/3 subunits. The switch from a1 to a2/3 subunits is accompanied by an elongation of the decay of IPSCs. In an attempt to determine potential cocaine-induced subunit switches in postsynaptic GABAA receptors at VTA-to-NAcCo inhibitory synapses, we first measured the time that elapsed from the IPSC peak to one-third of the peak amplitude (T1/3) as a measure of the decay kinetics (see Methods) (Fig. 6A). T1/3 at VTA-to-NAcCo inhibitory synapses was not altered significantly after cocaine exposure (in ms: saline, 16.71 ± 2.08, n= 4/2; cocaine, 17.10 ± 1.21, n= 7/3; P= 0.77, t test). To verify this result, we next took a pharmacological approach. We superfused the slices with L838417 (100 nm), a compound that selectively enhances a2/3-containing GABAA receptors and has a minimal effect on a1-containing GABAA receptors at a concentration of 100 nm (McKernan et al. 2000; Morris et al. 2006; Mathiasen et al. 2007; Gross et al. 2011). Thus, if a2/3 subunits were upregulated by cocaine exposure, application of L838417 should increase the decay kinetics of IPSCs to a higher degree in cocaine-treated animals. However, upon application of L838417, T1/3 was prolonged by very similar degrees in saline- and cocaine-treated animals (F1,18= 0.10, P= 0.75, two-factor ANOVA; Fig. 6A), suggesting a lack of cocaine-induced effect on GABAA receptor subunit composition. We then tested whether there was any cocaine-induced alteration in the subunit composition of GABAA receptors in NAcCo MSNs at the overall level by measuring the decay kinetics of spontaneous IPSCs that were presumably generated from all inhibitory synaptic inputs (evoked by electrical stimulation). Again, no significant alterations were detected (saline, 11.41 ± 0.40, n= 30/9; cocaine, 11.49 ± 0.29, n= 34/7; P= 0.20, t test; Fig. 6B, C).
Figure 6. Cocaine-induced postsynaptic alterations in VTA-to-NAcCo inhibitory synapses were not detected.
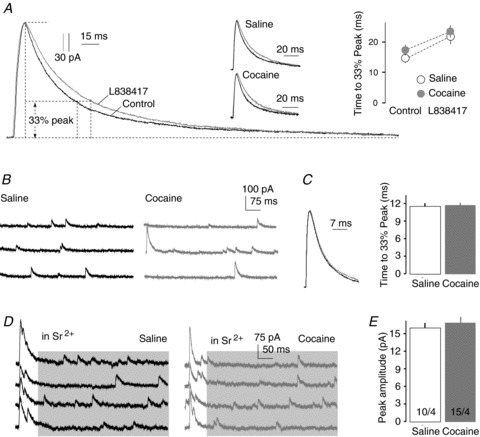
A, Examples from control animal and summarized results showing that the decay kinetics of IPSCs recorded from the VTA-to-NAcCo projection were not altered significantly after repeated cocaine administration, and application of L838417 prolonged the decay of IPSCs to a similar degree between saline- and cocaine-treated rats (on withdrawal day 1). B, Example spontaneous IPSCs in NAcCo MSNs from saline- and cocaine-treated rats. C, Summarized results showing that the decay kinetics of spontaneous IPSCs in NAcCo MSNs were not altered significantly after 1 day of withdrawal from repeated i.p. injections of cocaine. D, Examples showing that, in the presence of Sr2+, asynchronous IPSCs (in shaded area) were recorded following evoked IPSCs from VTA-to-NAcCo inhibitory synapses (via optical stimulation; events before the shaded area). These asynchronous IPSCs are largely attributable to the same set of synapses (i.e. VTA-to-NAcCo synapses) that mediate the evoked IPSCs. E, Summarized results showing a lack of difference in the amplitude of asynchronous IPSCs from VTA-to-NAcCo inhibitory synapses between saline- and cocaine-exposed rats (on withdrawal day 1).
We then made an additional attempt to examine whether the amplitude of postsynaptic responsiveness was altered at VTA-to-NAcCo inhibitory synapses following exposure to cocaine. This can be tested by measuring the amplitude of miniature IPSCs, which reflects the quantal size of the synapse (magnitude of postsynaptic responsiveness to a single released vesicle). To functionally isolate the miniature IPSCs from the VTA-to-NAcCo pathway, we evoked VTA-to-NAcCo IPSCs in the presence of Sr2+ (5 mm) to replace extracellular Ca2+ and Mg2+. This replacement has been shown to desynchronize single evoked IPSCs and to produce a train of quantal events at the tail of individual evoked IPSCs The asynchronous release occurs preferentially at terminals that have recently undergone synchronous release (e.g. evoked release), thus creating a time window to enrich miniature IPSC-like events from the recently activated synapses (Xu-Friedman & Regehr, 2000; Matsui & Jahr, 2003). As shown in Fig. 6D, we were able to evoke asynchronous release by light stimulations at VTA-to-NAcCo synapses. Furthermore, the amplitude of these presumed miniature IPSCs was not altered in cocaine-exposed animals (Fig. 6E), suggesting a lack of cocaine-induced change in postsynaptic responsiveness at VTA-to-NAcCo inhibitory synapses.
Taken together, if potential postsynaptic alterations are excluded, the increased PPR probably reflects a cocaine-induced presynaptic inhibition of VTA-to- NAcCo inhibitory synaptic transmission.
Discussion
Interwoven with mesolimbic DA projection, fast excitatory and inhibitory synaptic inputs from the VTA to NAc may critically contribute to emotional and motivational responses (Steffensen et al. 1998; Birgner et al. 2009). VTA-to-NAc glutamatergic and GABAergic transmissions have been identified, but their cellular properties remain largely elusive. Results from the present study reveal that VTA-to-NAcCo glutamatergic and GABAergic synaptic transmissions undergo differential regulations following exposure to cocaine. These adaptations occur during different exposure/withdrawal phases, and may thus differentially contribute to cocaine-induced behavioural alterations.
Excitatory VTA-to-NAcCo synaptic transmission
The VTA-to-NAc excitatory synaptic inputs may originate from VTA DA neurons that co-release glutamate, from a small portion of glutamatergic neurons or from other neuronal types in the VTA (Sulzer et al. 1998; Joyce & Rayport, 2000; Chuhma et al. 2004; Lavin et al. 2005; Nair-Roberts et al. 2008; Hnasko et al. 2010; Stuber et al. 2010; Tecuapetla et al. 2010). Studies using genetically modified mice have demonstrated that the VTA glutamatergic output regulates drug-induced locomotor and seeking responses, probably via regulation of the DA dynamics within the NAc (Birgner et al. 2009; Hnasko et al. 2010; Alsio et al. 2011). Our present study detected a modest cocaine-induced presynaptic alteration in VTA-to-NAc excitatory synapses. However, we did not detect cocaine-induced alterations in the postsynaptic responses, which were assessed by the relative weight of AMPAR- and NMDAR-mediated EPSCs rather than their absolute values. As such, the results cannot rule out the possibility of postsynaptic changes, such as a parallel up- or downregulation of postsynaptic AMPARs and NMDARs. This parallel scaling may happen during the generation of new mature synapses or the degeneration of pre-existing synapses. Furthermore, although VTA-to-NAc excitatory synapses contribute to the depolarization of NAc MSNs, their contribution is not likely to be predominant compared with other excitatory synaptic inputs. Rather, a potentially more significant role of VTA-to-NAc excitatory synaptic transmission may lie in the regulation of VTA-to-NAc DA terminals, their release of DA or other synaptic inputs to the NAc, as suggested previously (Hnasko et al. 2010; Tecuapetla et al. 2010; Alsio et al. 2011).
Inhibitory VTA-to-NAcCo synaptic transmission
GABAergic neurons constitute ∼30% of VTA neurons; they receive synaptic projections from the same or approximate brain regions that innervate DA neurons, project to the same brain regions as DA neurons and are activated simultaneously with DA neurons upon reward stimulation (Carr & Sesack, 2000; Kiyatkin & Rebec, 2001; Steffensen et al. 2001; Nair-Roberts et al. 2008). However, it remains unclear whether exposure to cocaine affects VTA-to-NAcCo inhibitory synapses. Our results show that VTA-to-NAcCo inhibitory synapses exhibit a higher presynaptic release probability than NAcSh-to-NAcCo input. Given that VTA GABAergic neurons tonically spike at ∼20 Hz in vivo (Steffensen et al. 1998), a frequency much higher than NAcSh MSNs (O’Donnell & Grace, 1995; O’Donnell et al. 1999), VTA-to-NAcCo inhibitory synapses may contribute substantially to the basal inhibition of NAcCo MSNs. As such, decreased presynaptic release probability of these synapses following cocaine exposure may undermine this inhibition, potentially resetting the balance between ‘distant’ and ‘local’ inhibitory controls of NAcCo MSNs.
Although it has been reported that very few VTA GABAergic neurons project to NAc MSNs (Brown et al. 2012), stable VTA-originated IPSCs were detected in our previous (Ishikawa et al. 2013) and current studies. Our recordings were primarily made in the caudal NAcCo, and the discrepant results may reflect the fact that NAcCo MSNs within different subregions possess different synaptic connections.
It is also worth noting that, in addition to GABAergic terminals, the activation of VTA-to-NAcCo dopaminergic/glutamatergic terminals also elicits PTX-sensitive (100 μm) IPSCs (Tritsch et al. 2012). However, IPSCs evoked from VTA-to-NAcCo dopaminergic projection exhibited a significant rundown by ∼90% within 5 min (Ishikawa et al. 2013). Because the baseline of our recordings was typically established after 10 min of recording, it is not likely that GABA release from the dopaminergic projection contributed significantly to the IPSCs analysed.
Effects of cocaine and cocaine withdrawal
Our results suggest that cocaine-induced alterations in VTA-to-NAc excitatory transmission are different from the other excitatory projections to the NAc. After 1 day of withdrawal, we did not detect cocaine-induced alterations in either the PPR or AMPAR/NMDAR ratio (Fig. 2), suggesting the lack of pre- or postsynaptic alterations. However, randomly sampled excitatory synapses on NAcCo neurons do not show presynaptic alterations following similar cocaine regimens (Dobi et al. 2011; Curcio et al. 2013), but exhibit an increase in the AMPAR/NMDAR ratio (Curcio et al. 2013). In comparison, glutamatergic synapses onto NAcSh neurons are more dynamically regulated following similar cocaine regimens. We have shown previously that the presynaptic release probability of medial prefrontal cortex-to-NAcSh excitatory synapses is increased at this withdrawal time point (Suska et al. 2013). Furthermore, randomly sampled excitatory synapses in the NAcSh exhibit a decreased AMPAR/NMDAR ratio (Kourrich et al. 2007; Mameli et al. 2009). This decrease can be mediated by a potential downregulation of synaptic AMPARs (Boudreau & Wolf, 2005; Boudreau et al. 2007) or an upregulation of NMDARs (Huang et al. 2009; Brown et al. 2011). Thus, at this withdrawal time point, pre- or postsynaptic alterations are detected in some other excitatory afferents, but not in the VTA-to-NAc projection.
On withdrawal day 1, when no changes were detected at the glutamatergic transmission at VTA-to-NAcCo synapses, there was a significant suppression of GABAergic transmission within this projection (Fig. 5A, B). By contrast, on withdrawal day 21, as GABAergic transmission returned to control levels (Fig. 5F, G), glutamatergic transmission showed presynaptic enhancement (Fig. 2G, H). Thus, the differential presynaptic alterations may shift the inhibitory–excitatory balance within the VTA-to-NAcCo projection during different stages of cocaine withdrawal, and may thus contribute to the time-dependent withdrawal-associated behavioural alterations. An additional factor that may tip the balance of NAc output is through the differential innervations of subpopulations of NAc MSNs. For example, DA D1 and D2 receptor-expressing MSNs in the NAc have been shown to differ in their cellular responses to addictive drugs and could play opposing roles in cocaine-associated behaviours (Lobo et al. 2010; Smith et al. 2013). Although these two types of neuron may share similar synaptic inputs, the subcellular connectivity patterns may be biased to convey pathway-specific signalling (MacAskill et al. 2012). It remains to be determined whether D1- and D2-expressing MSNs in the NAc also receive differential innervations from the VTA DAergic, glutamatergic and GABAergic transmissions, and whether the innervation patterns are altered by cocaine experience.
In summary, the present study characterized some basic electrophysiological properties of VTA-to-NAcCo fast excitatory and inhibitory synapses, and detected cocaine-induced presynaptic adaptations within these afferents. These results may provide a knowledge and technical basis with which to explore the role of VTA-to-NAc fast synaptic transmission in cocaine-induced emotional and motivational alterations.
Translational perspective
For a long time it has been known that reward-elicited responses as well as pathophysiological reward-related emotional motivational states, such as depression and drug addiction, are critically regulated by the mesolimbic dopamine system, mainly comprising the synaptic projection from the ventral tegmental area (VTA) to the nucleus accumbens (NAc). Recent discoveries have revealed a complex nature of the VTA-to-NAc projection. In addition to the neuromodulator dopamine, activation of the VTA-to-NAc projection also releases a rich repertoire of neurotransmitters, in particular glutamate and GABA, which mediate fast excitatory and inhibitory synaptic transmissions. In contrast to dopamine and other neuromodulators, these fast synaptic transmissions can instantly change the membrane potential of NAc neurons upon activation of the VTA, thus exerting highly timing-contingent regulation of the mesolimbic output. Results from the present study show that, following repeated exposure to cocaine, VTA-to-NAc fast inhibitory synaptic transmission appears to be decreased by reduced presynaptic activity. A perceivable consequence of this drug-induced neuroadaptation is that the NAc neurons receive insufficient inhibition upon contingent activation of the VTA, resulting in compromised contingent responses mediated by the NAc. Thus, this adaptation might contribute to the compromised or impaired NAc-based emotional and motivational responses observed in drug addicts. Thanks to the rapid progress in biomedical research, neurons can now be targeted and manipulated selectively based on the neurotransmitters they make and release. Thus, VTA-to-NAc GABAergic synaptic transmission, which is targeted by drugs of abuse to produce addiction-associated cellular and behavioural consequences, can also be altered clinically to achieve therapeutic benefits.
Glossary
- AAV2
adeno-associated viral vector 2
- aCSF
artificial cerebrospinal fluid
- AMPAR
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- ChR2
channelrhodopsin 2
- DA
dopamine
- EPSC
excitatory postsynaptic current
- IPI
interpulse interval
- IPSC
inhibitory postsynaptic current
- MSN
medium spiny neurons
- NAc
nucleus accumbens
- NAcCo
NAc core
- NAcSh
NAc shell
- NBQX
2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline
- NMDAR
N-methyl-d-aspartate receptor
- PBP
parabrachial pigmented area
- PN
paranigral nucleus
- PPR
paired-pulse ratio
- PTX
picrotoxin
- TEA-Cl
tetraethylammonium chloride
- TH
tyrosine hydroxylase
- VGLUT2
vesicular glutamate transporter 2
- VTA
ventral tegmental area
Additional information
Competing interests
None declared.
Author contributions
MI, MO, JMC, OMS, YHH, and YD contributed to the design of the experiments, design of the analyses, and the writing of the manuscript. MI, MO, PAN, and ZW conducted the experiments and formed the analyses.
Funding
The research reported in this publication was supported by the National Institutes of Health under Award Numbers DA29565, DA36303, DA23206, DA30379, DA34856 and AA16179, and the Pennsylvania Department of Health Commonwealth Universal Research Enhancement. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Alsio J, Nordenankar K, Arvidsson E, Birgner C, Mahmoudi S, Halbout B, Smith C, Fortin GM, Olson L, Descarries L, Trudeau LE, Kullander K, Levesque D, Wallen-Mackenzie A. Enhanced sucrose and cocaine self-administration and cue-induced drug seeking after loss of VGLUT2 in midbrain dopamine neurons in mice. J Neurosci. 2011;31:12593–12603. doi: 10.1523/JNEUROSCI.2397-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Aponte Y, Su HH, Sternson SM. A FLEX switch targets Channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J Neurosci. 2008;28:7025–7030. doi: 10.1523/JNEUROSCI.1954-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birgner C, Nordenankar K, Lundblad M, Mendez JA, Smith C, le Greves M, Galter D, Olson L, Fredriksson A, Trudeau LE, Kullander K, Wallen-Mackenzie A. VGLUT2 in dopamine neurons is required for psychostimulant-induced behavioral activation. Proc Natl Acad Sci. 2009;107:389–394. doi: 10.1073/pnas.0910986107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27:10621–10635. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Wolf ME. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci. 2005;25:9144–9151. doi: 10.1523/JNEUROSCI.2252-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MT, Tan KR, O’Connor EC, Nikonenko I, Muller D, Luscher C. Ventral tegmental area GABA projections pause accumbal cholinergic interneurons to enhance associative learning. Nature. 2012;492:452–456. doi: 10.1038/nature11657. [DOI] [PubMed] [Google Scholar]
- Brown TE, Lee BR, Mu P, Ferguson D, Dietz D, Ohnishi YN, Lin Y, Suska A, Ishikawa M, Huang YH, Shen H, Kalivas PW, Sorg BA, Zukin RS, Nestler EJ, Dong Y, Schluter OM. A silent synapse-based mechanism for cocaine-induced locomotor sensitization. J Neurosci. 2011;31:8163–8174. doi: 10.1523/JNEUROSCI.0016-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DB, Sesack SR. Projections from the rat prefrontal cortex to the ventral tegmental area: target specificity in the synaptic associations with mesoaccumbens and mesocortical neurons. J Neurosci. 2000;20:3864–3873. doi: 10.1523/JNEUROSCI.20-10-03864.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuhma N, Zhang H, Masson J, Zhuang X, Sulzer D, Hen R, Rayport S. Dopamine neurons mediate a fast excitatory signal via their glutamatergic synapses. J Neurosci. 2004;24:972–981. doi: 10.1523/JNEUROSCI.4317-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcio L, Podda MV, Leone L, Piacentini R, Mastrodonato A, Cappelletti P, Sacchi S, Pollegioni L, Grassi C, D’Ascenzo M. Reduced D-serine levels in the nucleus accumbens of cocaine-treated rats hinder the induction of NMDA receptor-dependent synaptic plasticity. Brain. 2013;136:1216–1230. doi: 10.1093/brain/awt036. [DOI] [PubMed] [Google Scholar]
- Dobi A, Seabold GK, Christensen CH, Bock R, Alvarez VA. Cocaine-induced plasticity in the nucleus accumbens is cell specific and develops without prolonged withdrawal. J Neurosci. 2011;31:1895–1904. doi: 10.1523/JNEUROSCI.5375-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Saal D, Thomas M, Faust R, Bonci A, Robinson T, Malenka RC. Cocaine-induced potentiation of synaptic strength in dopamine neurons: behavioral correlates in GluRA(-/-) mice. Proc Natl Acad Sci U S A. 2004;101:14282–14287. doi: 10.1073/pnas.0401553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A, Sims RE, Swinny JD, Sieghart W, Bolam JP, Stanford IM. Differential localization of GABA(A) receptor subunits in relation to rat striatopallidal and pallidopallidal synapses. Eur J Neurosci. 2011;33:868–878. doi: 10.1111/j.1460-9568.2010.07552.x. [DOI] [PubMed] [Google Scholar]
- Heine M, Groc L, Frischknecht R, Beique JC, Lounis B, Rumbaugh G, Huganir RL, Cognet L, Choquet D. Surface mobility of postsynaptic AMPARs tunes synaptic transmission. Science. 2008;320:201–205. doi: 10.1126/science.1152089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnasko TS, Chuhma N, Zhang H, Goh GY, Sulzer D, Palmiter RD, Rayport S, Edwards RH. Vesicular glutamate transport promotes dopamine storage and glutamate corelease in vivo. Neuron. 2010;65:643–656. doi: 10.1016/j.neuron.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Ishikawa M, Lee BR, Nakanishi N, Schluter OM, Dong Y. Searching for presynaptic NMDA receptors in the nucleus accumbens. J Neurosci. 2011;31:18453–18463. doi: 10.1523/JNEUROSCI.3824-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Lin Y, Mu P, Lee BR, Brown TE, Wayman G, Marie H, Liu W, Yan Z, Sorg BA, Schluter OM, Zukin RS, Dong Y. In vivo cocaine experience generates silent synapses. Neuron. 2009;63:40–47. doi: 10.1016/j.neuron.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Ikemoto S. Dopamine reward circuitry: two projection systems from the ventral midbrain to the nucleus accumbens–olfactory tubercle complex. Brain Res Rev. 2007;56:27–78. doi: 10.1016/j.brainresrev.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa M, Mu P, Moyer JT, Wolf JA, Quock RM, Davies NM, Hu XT, Schluter OM, Dong Y. Homeostatic synapse-driven membrane plasticity in nucleus accumbens neurons. J Neurosci. 2009;29:5820–5831. doi: 10.1523/JNEUROSCI.5703-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa M, Otaka M, Huang YH, Neumann PA, Winters BD, Grace AA, Schluter MO, Dong Y. Dopamine triggers heterosynaptic plasticity. J Neurosci. 2013;33:6759–6765. doi: 10.1523/JNEUROSCI.4694-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce MP, Rayport S. Mesoaccumbens dopamine neuron synapses reconstructed in vitro are glutamatergic. Neuroscience. 2000;99:445–456. doi: 10.1016/s0306-4522(00)00219-0. [DOI] [PubMed] [Google Scholar]
- Kawano M, Kawasaki A, Sakata-Haga H, Fukui Y, Kawano H, Nogami H, Hisano S. Particular subpopulations of midbrain and hypothalamic dopamine neurons express vesicular glutamate transporter 2 in the rat brain. J Comp Neurol. 2006;498:581–592. doi: 10.1002/cne.21054. [DOI] [PubMed] [Google Scholar]
- Kelley AE. Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–179. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, Rebec GV. Impulse activity of ventral tegmental area neurons during heroin self-administration in rats. Neuroscience. 2001;102:565–580. doi: 10.1016/s0306-4522(00)00492-9. [DOI] [PubMed] [Google Scholar]
- Koobs DH. The two faces of vitamin C. Science. 2001;293:1993–1995. [PubMed] [Google Scholar]
- Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci. 2007;27:7921–7928. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavin A, Nogueira L, Lapish CC, Wightman RM, Phillips PE, Seamans JK. Mesocortical dopamine neurons operate in distinct temporal domains using multimodal signaling. J Neurosci. 2005;25:5013–5023. doi: 10.1523/JNEUROSCI.0557-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeberg J, Usoskin D, Bengtsson H, Gustafsson A, Kylberg A, Soderstrom S, Ebendal T. Transgenic expression of Cre recombinase from the tyrosine hydroxylase locus. Genesis. 2004;40:67–73. doi: 10.1002/gene.20065. [DOI] [PubMed] [Google Scholar]
- Lobo MK, Covington HE, 3rd, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, Mouzon E, Mogri M, Neve RL, Deisseroth K, Han MH, Nestler EJ. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2010;330:385–390. doi: 10.1126/science.1188472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAskill AF, Little JP, Cassel JM, Carter AG. Subcellular connectivity underlies pathway-specific signaling in the nucleus accumbens. Nat Neurosci. 2012;15:1624–1626. doi: 10.1038/nn.3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mameli M, Halbout B, Creton C, Engblom D, Parkitna JR, Spanagel R, Luscher C. Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci. 2009;12:1036–1041. doi: 10.1038/nn.2367. [DOI] [PubMed] [Google Scholar]
- Mathiasen LS, Rodgers RJ, Mirza NR. Comparative effects of nonselective and subtype-selective gamma-aminobutyric acidA receptor positive modulators in the rat-conditioned emotional response test. Behav Pharmacol. 2007;18:191–203. doi: 10.1097/FBP.0b013e32814fcdd4. [DOI] [PubMed] [Google Scholar]
- Matsui K, Jahr CE. Ectopic release of synaptic vesicles. Neuron. 2003;40:1173–1183. doi: 10.1016/s0896-6273(03)00788-8. [DOI] [PubMed] [Google Scholar]
- McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, Atack JR, Farrar S, Myers J, Cook G, Ferris P, Garrett L, Bristow L, Marshall G, Macaulay A, Brown N, Howell O, Moore KW, Carling RW, Street LJ, Castro JL, Ragan CI, Dawson GR, Whiting PJ. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat Neurosci. 2000;3:587–592. doi: 10.1038/75761. [DOI] [PubMed] [Google Scholar]
- Morgan JL, Soto F, Wong RO, Kerschensteiner D. Development of cell type-specific connectivity patterns of converging excitatory axons in the retina. Neuron. 2011;71:1014–1021. doi: 10.1016/j.neuron.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris HV, Dawson GR, Reynolds DS, Atack JR, Stephens DN. Both alpha2 and alpha3 GABAA receptor subtypes mediate the anxiolytic properties of benzodiazepine site ligands in the conditioned emotional response paradigm. Eur J Neurosci. 2006;23:2495–2504. doi: 10.1111/j.1460-9568.2006.04775.x. [DOI] [PubMed] [Google Scholar]
- Mu P, Moyer JT, Ishikawa M, Zhang Y, Panksepp J, Sorg BA, Schluter OM, Dong Y. Exposure to cocaine dynamically regulates the intrinsic membrane excitability of nucleus accumbens neurons. J Neurosci. 2010;30:3689–3699. doi: 10.1523/JNEUROSCI.4063-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai T, McGeer PL, McGeer EG. Distribution of GABA-T-intensive neurons in the rat forebrain and midbrain. J Comp Neurol. 1983;218:220–238. doi: 10.1002/cne.902180209. [DOI] [PubMed] [Google Scholar]
- Nair-Roberts RG, Chatelain-Badie SD, Benson E, White-Cooper H, Bolam JP, Ungless MA. Stereological estimates of dopaminergic, GABAergic and glutamatergic neurons in the ventral tegmental area, substantia nigra and retrorubral field in the rat. Neuroscience. 2008;152:1024–1031. doi: 10.1016/j.neuroscience.2008.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell P, Grace AA. Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J Neurosci. 1995;15:3622–3639. doi: 10.1523/JNEUROSCI.15-05-03622.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell P, Greene J, Pabello N, Lewis BL, Grace AA. Modulation of cell firing in the nucleus accumbens. Ann N Y Acad Sci. 1999;877:157–175. doi: 10.1111/j.1749-6632.1999.tb09267.x. [DOI] [PubMed] [Google Scholar]
- Petreanu L, Mao T, Sternson SM, Svoboda K. The subcellular organization of neocortical excitatory connections. Nature. 2009;457:1142–1145. doi: 10.1038/nature07709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips AG. Brain reward circuitry: a case for separate systems. Brain Res Bull. 1984;12:195–201. doi: 10.1016/0361-9230(84)90189-8. [DOI] [PubMed] [Google Scholar]
- Phillipson OT, Griffiths AC. The topographic order of inputs to nucleus accumbens in the rat. Neuroscience. 1985;16:275–296. doi: 10.1016/0306-4522(85)90002-8. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Grace AA. Cortico-basal ganglia reward network: microcircuitry. Neuropsychopharmacology. 2010;35:27–47. doi: 10.1038/npp.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RJ, Lobo MK, Spencer S, Kalivas PW. Cocaine-induced adaptations in D1 and D2 accumbens projection neurons (a dichotomy not necessarily synonymous with direct and indirect pathways) Curr Opin Neurobiol. 2013;23:546–552. doi: 10.1016/j.conb.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffensen SC, Lee RS, Stobbs SH, Henriksen SJ. Responses of ventral tegmental area GABA neurons to brain stimulation reward. Brain Res. 2001;906:190–197. doi: 10.1016/s0006-8993(01)02581-1. [DOI] [PubMed] [Google Scholar]
- Steffensen SC, Svingos AL, Pickel VM, Henriksen SJ. Electrophysiological characterization of GABAergic neurons in the ventral tegmental area. J Neurosci. 1998;18:8003–8015. doi: 10.1523/JNEUROSCI.18-19-08003.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuber GD, Hnasko TS, Britt JP, Edwards RH, Bonci A. Dopaminergic terminals in the nucleus accumbens but not the dorsal striatum corelease glutamate. J Neurosci. 2010;30:8229–8233. doi: 10.1523/JNEUROSCI.1754-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Joyce MP, Lin L, Geldwert D, Haber SN, Hattori T, Rayport S. Dopamine neurons make glutamatergic synapses in vitro. J Neurosci. 1998;18:4588–4602. doi: 10.1523/JNEUROSCI.18-12-04588.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suska A, Lee BR, Huang YH, Dong Y, Schluter OM. Selective presynaptic enhancement of the prefrontal cortex to nucleus accumbens pathway by cocaine. Proc Natl Acad Sci U S A. 2013;110:713–718. doi: 10.1073/pnas.1206287110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tecuapetla F, Patel JC, Xenias H, English D, Tadros I, Shah F, Berlin J, Deisseroth K, Rice ME, Tepper JM, Koos T. Glutamatergic signaling by mesolimbic dopamine neurons in the nucleus accumbens. J Neurosci. 2010;30:7105–7110. doi: 10.1523/JNEUROSCI.0265-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Ding JB, Sabatini BL. Dopaminergic neurons inhibit striatal output through non-canonical release of GABA. Nature. 2012;490:262–266. doi: 10.1038/nature11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA. The role of reward pathways in the development of drug dependence. Pharmacol Ther. 1987;35:227–263. doi: 10.1016/0163-7258(87)90108-2. [DOI] [PubMed] [Google Scholar]
- Wise RA. Neurobiology of addiction. Curr Opin Neurobiol. 1996;6:243–251. doi: 10.1016/s0959-4388(96)80079-1. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG. Probing fundamental aspects of synaptic transmission with strontium. J Neurosci. 2000;20:4414–4422. doi: 10.1523/JNEUROSCI.20-12-04414.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]