

Abstract
Background: Epidemiological studies have indicated smoking to be a risk factor for the progression of liver diseases. Nicotine is the chief addictive substance in cigarette smoke and has powerful biological properties throughout the body. Nicotine has been implicated in a number of disease processes, including increased cell proliferation and fibrosis in several organ systems. Aims: The aim of this study was to evaluate the effects of chronic administration of nicotine on biliary proliferation and fibrosis in normal rats. Methods: In vivo, rats were treated with nicotine by osmotic minipumps for two weeks. Proliferation, α7-nicotinic receptor (α7-nAChR) and profibrotic expression were evaluated in liver tissue, cholangiocytes and a polarized cholangiocyte cell line (NRIC). Nicotine-dependent activation of the Ca2+/IP3/ERK 1/2 intracellular signaling pathway was also evaluated in NRIC. Results: Cholangiocytes express α7-nAChR. Chronic administration of nicotine to normal rats stimulated biliary proliferation and profibrotic gene and protein expression such as alpha-smooth muscle actin (α-SMA) and fibronectin 1 (FN-1). Activation of α7-nAChR stimulated Ca2+/ERK1/2-dependent cholangiocyte proliferation. Conclusion: Chronic exposure to nicotine contributes to biliary fibrosis by activation of cholangiocyte proliferation and expression of profibrotic genes. Modulation of α7-nAChR signaling axis may be useful for the management of biliary proliferation and fibrosis during cholangiopathies.
Keywords: Cholangiocytes, fibrosis, nicotinic receptors, biliary epithelia, proliferation
Introduction
Cholangiocytes are epithelial cells that line intra- and extrahepatic bile ducts of the liver. In humans and rodent models, normal cholangiocytes are relatively mitotically dormant (1–3). However, the proliferation of cholangiocytes is critical for the maintenance of biliary mass and secretory function during the pathogenesis of chronic cholestatic liver diseases such as primary biliary cirrhosis (PBC), and primary sclerosing cholangitis (PSC) (3–5). It has been postulated that cholangiocyte proliferation plays a key role in the progression of cholestatic liver diseases including contributing to chronic liver inflammation and fibrosis (6). The IP3/Ca2+ and cAMP/ERK1/2 MAPK signaling pathways play a crucial role in the regulation of cholangiocyte proliferation during cholestasis and liver injury (7).
Smoking has been indicated as a risk factor for the progression of cholestatic liver diseases in several epidemiological studies. Emerging evidence indicates that exposure to cigarette smoke may stimulate the progression of chronic liver disease towards fibrosis (such as PSC, PBC, chronic hepatitis C, and non-alcoholic fatty liver disease) (8–11). A recent study has shown that long-standing nicotine abuse in patients with PSC is a risk factor for the development of hepatobiliary cancers (11).
Cigarette smoke contains a large number of compounds that exert biological effects. Of these, nicotine plays a critical role in addiction and contributes to the pathogenesis of several diseases. Nicotine exerts biological effects throughout the body by binding to nicotinic acetylcholine receptors (nAChRs) located on the cell membrane (12–14).
nAChRs are ligand-gated channels formed by the pentameric assembly of nAChR subunits: αl-αl0, βl-β4, γ, δ, and ɛ (15). These are non-selective ligand-gated ion channels that allow several different positively charged ions such as K+, Na+ and Ca2+ to cross the cell membrane (15). Until recently, it was thought that nAChRs existed only in the nervous system. However, nAChRs are expressed by virtually all mammalian cells and are evolutionary conserved (16).
α7-nAChR has been the focus of numerous studies due to pronounced effects on proliferation and fibrogenesis (17). Binding of nicotine to the α7-nAChR receptor triggers an influx of Ca2+ and downstream signaling mechanisms that contribute to dysregulated growth, angiogenesis, release of growth factors, and modulation of the microenvironment (18). Also, nicotine stimulates activation of several intracellular signaling pathways involved in nAChR-induced cell proliferation, which include Ca2+-dependent activation of ERK1/2 (17). Activation of ERK1/2 is a key signaling mechanism that regulates cholangiocyte proliferation (7).
There is growing information on the role of proliferating cholangiocytes in the pathogenesis of liver fibrosis during chronic cholestatic liver diseases. Nicotine also activates atrial fibrosis (19) and stimulates the expression of factors that contribute to fibrosis such as fibronectin (20). Nicotine-induced fibronectin expression was mediated through ERK1/2 (20). A recent in vitro study demonstrated that nicotine induces hepatic stellate cells (HSC) proliferation and upregulates collagen 1-A2 mRNA expression, which is inhibited by mecamylamine (a non-specific nAChR antagonist) (21).
Similar to muscarinic acetylcholine receptors (mAChR), nAChR is activated by binding to the neurotransmitter acetylcholine (ACh) (22). However, unlike nAChRs, mAChRs are not activated by nicotine (22). ACh plays a crucial role in the regulation of cholangiocyte proliferation and function. Cholangiocytes express mAChR muscarinic 3 (M3) and ACh potentiates secretin-induced cholangiocyte secretion (23). In addition, total vagotomy decreases mAChR M3 expression, impairs cholangiocyte proliferation and enhances apoptosis, leading to decreased ductal mass in response to bile duct ligation (BDL) (24). However, the potential role that nicotine and nAChRs play in the regulation of cholangiocyte proliferation and biliary fibrosis has not been evaluated.
Materials and Methods
Materials
Reagents were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise indicated. Tissue culture reagents were purchased from Invitrogen (Carlsbad, CA). Both AR-R 17779 (α7-nAChR specific agonist) (25) and alpha-bungarotoxin (ABT; α7-nAChR selective antagonist) (26) were purchased from Tocris Bioscience (Minneapolis, MN). The goat polyclonal anti-cytokeratin 19 (CK-19) was purchased from Life Technologies (Grand Island, NY). The mouse monoclonal anti-proliferating cell nuclear antigen (PCNA), mouse monoclonal anti-alpha-smooth muscle actin (α-SMA), rabbit polyclonal anti-ERK1 (which detects p44 and p42) and goat polyclonal anti-pERK (which detects phosphorylated p44 and p42) antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The rabbit polyclonal anti-α7-nAChR and mouse anti-fibronectin 1 (FN-1) antibodies were purchased from Abcam (Cambridge, MA). The cAMP EIA kit was obtained from Cayman Chemical Company (Ann Arbor, MI). The IP-One ELISA was purchased from Cisbio US (Bedford, MA).
Animal models
All animal experiments were performed in accordance with protocols approved by the Scott and White Healthcare and Texas A&M Heath Science Center IACUC. Male Fischer 344 rats (150–175 g) were purchased from Charles River Laboratories International, Inc. (Wilmington, MA) and maintained in a temperature-controlled environment (20–22°C) with 12:12-hr light/dark cycles. Animals were fed ad libitum standard rat chow and had free access to drinking water. The in vivo effect of nicotine administration on biliary proliferation was evaluated in rats treated with nicotine salt (9 mg/kg/d; nicotine-treated rats) or 0.9% NaCl (control) via implanted osmotic minipumps (intraperitoneal, IP) for 2 weeks (27, 28). The surgical procedures were performed under isoflurane anesthesia. Postoperative care included administration of buprenorphine (0.05 mg/kg body weight). This dose of nicotine has been previously used for chronic treatment in rat models (29–32). The serum concentration of nicotine in the rat at this dosage has been reported to be within the range observed in heavy smokers (33, 34). Before terminal procedures, the animals were injected with Euthasol® (50 mg/kg body weight, IP).
Cholangiocyte isolation and culture
Cholangiocytes were isolated as described by immunoaffinity separation by using a rat monoclonal antibody (IgM, kindly provided by Dr. R. Faris, Brown University, Providence, RI) that recognizes an unidentified antigen expressed by all intrahepatic rat cholangiocytes (35). Normal rat intrahepatic cholangiocyte cultures (NRIC), which have morphological, phenotypical and functional features similar to that of freshly isolated cholangiocytes were maintained in culture as described (36).
Evaluation of α7-nAChR expression
The mRNA and protein expression levels for the α7-nAChR were evaluated by: 1) realtime PCR in isolated cholangiocytes and NRIC; 2) immunohistochemistry in liver sections; 3) immunoblots in isolated cholangiocytes; and 4) immunofluorescence in NRIC smears (37, 38). The RT2 Real-Time assay from Qiagen, Inc. (Valencia, CA) was utilized for evaluating the gene expression levels of α7-nAChR RNA was extracted with the RNeasy® Mini Kit (Qiagen) and reverse-transcribed with the Reaction Ready™ First Strand cDNA synthesis kit (Qiagen). SYBR Green PCR master mix (Qiagen) was utilized in the experimental assay with RT2 PCR rat primers designed specifically for α7-nAChR (NM_012832) (39) and the housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase (NM_017008; GAPDH) (40) (Qiagen). Real-time RT-PCR was performed with an ABI Prism 7900HT System using a two-step PCR cycling program at 95°C for 10 minutes followed by 40 cycles of 95°C for 15 sec and 60°C for 1 minute. A AACT analysis was performed using normal rat cholangiocytes as the control. Immunohistochemistry with anti-α7-nAChR (1:100) was performed in liver sections from normal and BDL rats as described (41, 42). Immunofluorescence in NRIC was performed with a rabbit polyclonal antibody for anti-α7-nAChR (1:100) and a Cy2-conjugated anti-rabbit secondary antibody (1:50, Jackson Immunochemicals, West Grove, PA) as described (41). The expression of α7-nAChR was evaluated by immunoblots in whole cell lysates of cholangiocytes isolated from control and nicotine-treated rats and NRIC as described (43).
Evaluation of transaminases, alkaline phosphatase, and bilirubin serum levels, lobular necrosis, inflammation, cholangiocyte proliferation and apoptosis
In in vivo studies, we measured 1) liver weight, body weight, and liver-to-body weight ratio (44) and 2) serum levels of transaminases, alanine aminotransferase (ALT) and aspartate aminotransferase (AST), alkaline phosphatase (ALP) and total bilirubin using a Dimension® RxL Max Integrated Chemistry system (Dade Behring, Deerfield IL). Lobular damage, necrosis and degree of portal inflammation were also evaluated as described (38, 41). Proliferation was evaluated by quantitative evaluation of the number of PCNA- and CK-19-positive cholangiocytes in liver sections and PCNA expression in cholangiocytes isolated from control and nicotine-treated rats (43). Apoptosis was evaluated by terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling (TUNEL) analysis using a commercially available kit (Wako Chemicals, Tokyo, Japan) (45).
Evaluation of profibrotic gene and protein expression and biliary fibrosis
The mRNA expression levels for α-SMA, collagen 1A1 and FN-1 were evaluated in total RNA (0.5 μg) from liver tissue and cholangiocytes from control and nicotine-treated rats as previously described. The primers for FN-1, collagen 1A1 and a-SMA (SABiosciences) were designed according to the NCBI GenBank Accession numbers: NM_019143 (FN-1), NM_053304 (collagen 1A1) (46) and NM_031004 (α-SMA) (47). Collagen deposition in portal areas was evaluated in liver sections from control and nicotine-treated rats by Masson’s trichrome staining according to the manufacturer’s protocol (Sigma-Aldrich). Collagen staining (blue) in light-microscopy images was quantitated in a coded fashion using ImageJ Software (48). Expression of a-SMA and FN-1 were evaluated by immunohistochemistry in liver sections from control and nicotine-treated rats. Immunohistochemistry with anti-α-SMA (1:100) and FN-1 (1:100) was performed as described (41, 42).
Evaluation of the in vitro effects of nicotine on proliferation and intracellular signaling mechanisms
NRIC were treated at 37°C for 48 hours with 0.2% bovine serum albumin (BSA), nicotine (0.01, 0.1, 1.0, 10 and 20 μM) (21, 49) or AR-R 17779 (α7-nAChR specific agonist; 0.01, 0.1, 1.0, 10 and 20 μM) (50) before evaluation of cell proliferation with the CellTiter 96® Cell Proliferation Assay (Promega, Madison, WI) (51). Various dosages of nicotine ranging from 1.0 to 1200 μM have been utilized in previous studies (52–54). In separate experiments, NRIC were treated at 37°C for 48 h with 0.2% BSA (basal) or AR-R 17779 (10 μM) (50) for 48 hours in the absence or presence of preincubation with ABT (α7-nAChR selective antagonist; 1 μM) (55), BAPTA/AM (intracellular Ca2+ chelator, 5 μM) (51), or PD98059 (ERK pathway inhibitor, 10 μM) (56).
For intracellular Ca2+ measurements, NRIC plated on collagen-coated cover slips were preincubated at 37°C with fura-2-AM (1 μM, Life Technologies) for 20 minutes. Labeled cells were washed with serum-free medium before the analysis and stimulated with nicotine (10 μM) or AR-R 17779 (10 μM). A fluorescent microscope equipped with a fura-2 filter was used to image cells emitting excitation wavelengths at 510 nM. For each time point, radiometric analysis using the ratio of 360 nM to 380 nM, was used to approximate intracellular Ca2+ levels using IonWizard v.6 software. The change in Ca2+ was determined by subtracting baseline Ca2+ measurements immediately before, from that immediately following nicotine treatment.
For intracellular cAMP and IP3 levels, NRIC were treated at room temperature for 5 (cAMP) or 10 (IP3) minutes with 0.2% BSA or nicotine (10 μM) before evaluation by ELISA. ERK1/2 activation was evaluated by immunofluorescence for nuclear translocation of phosphorylated-ERK1/2. NRIC cells were seeded on coverslips and stimulated with 0.2% BSA and nicotine (10 μM) in absence/presence of preincubation with ABT. Immunofluorescence was performed as described (51). NRIC were also stimulated with 0.2% BSA or nicotine (10 μM for 0, 2, 5, 10, 20, 30, 60, minutes) before evaluation of ERK1/2 phosphorylation with the phospho-ERK Cell-based ELISA kit (R&D Systems, Minneapolis, MN).
Statistical Analysis
All data are expressed as mean ± SEM. Differences between groups were analyzed by the Student unpaired t test when two groups were analyzed, by Mann-Whitney U test and Kruskal-Wallis H test when more than two groups were analyzed, followed by an appropriate post hoc test. A value of p < 0.05 was considered significant.
Results
α7-nAChR is expressed by bile ducts in liver sections, isolated cholangites and NRIC
Bile ducts in liver sections from control and nicotine-treated rats were positive for α7-nAChR by immunohistochemistry (Figure 1A). Hepatocytes are also positive for α7-nAChR. A previous study has also demonstrated that HSC also express several subtypes of nAChRs (21). However, hepatocytes were not the focus of this study. The expression of α7-nAChR in cholangiocytes isolated from control and nicotine-treated rats was confirmed by immunoblots and real-time PCR (Figure 1B, D). There were no significant differences observed in the expression levels of α7-nAChR in control and nicotine-treated rats. NRIC were positive for α7-nAChR by immunofluorescence (green) and realtime PCR (Figure 1C, D).
Figure 1.
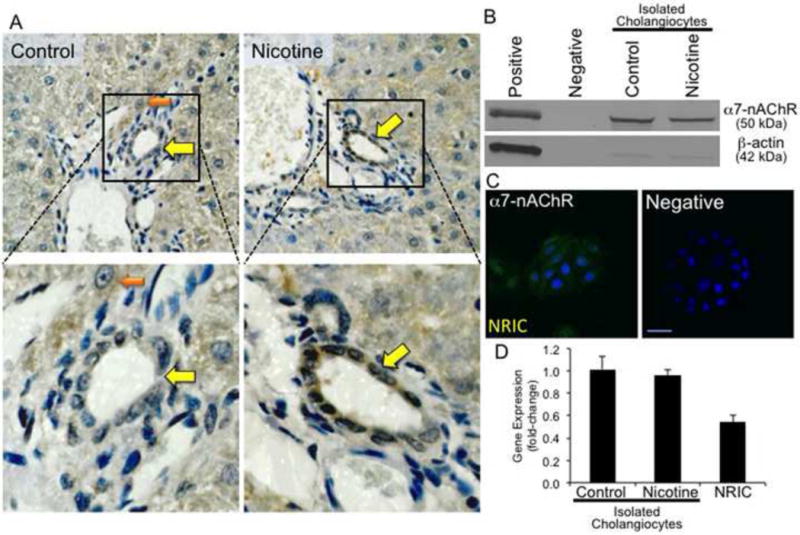
α7-nAChR gene and protein expression in liver sections and isolated cholangiocytes from control and nicotine-treated rats and NRIC. [A] Bile ducts in liver sections from control and nicotine-treated rats express α7-nAChR by immunohistochemistry. Original magnification, x40. Enlargement of areas containing bile ducts positive for α7-nAChR are shown. Positively stained bile ducts are indicated by yellow arrows, whereas hepatocytes are indicated by an orange arrow. [B] Cholangiocytes isolated from control and nicotine-treated rats express α7-nAChR by immunoblotting analysis. ß-actin was utilized as a loading control. [C] NRICs are positive for α7-nAChR (green) by immunofluorescence. Nuclei are stained with DAPI (blue). [D] Cholangiocytes isolated from normal, nicotine-treated rats and NRIC express the message for α7-nAChR by real-time PCR. Data are mean ± SEM of 6 experiments.
Chronic administration of nicotine increases liver enzyme levels and biliary proliferation
The body weight of nicotine-treated rats was significantly lower than that of control animals (Table 1), which is consistent with other studies (57, 58). There were no significant changes in liver weight and liver to body weight ratios. Chronic nicotine administration induced a significant increase in ALT, AST and ALP levels compared to the values of control animals (Table 1). No significant changes were observed for bilirubin levels. Despite elevations in ALT and AST levels, there were no observable effects of nicotine on the levels of apoptosis (Table 1), inflammation and lobular damage in the nicotine-treated rats, which may be due to the short duration of nicotine exposure.
Table 1.
Evaluation of liver and body weight, liver-to-body weight ratio, and serum levels of transaminases, total bilirubin, alkaline phosphatase, CK-19, PCNA and TUNEL positive cholangiocytes per portal area, and Masson’s trichrome staining.
| Treatment Groups
|
||
|---|---|---|
| Control | Nicotine | |
| Liver Weight (g) |
8.4 ± 0.2 | 8.6 ± 0.4 |
|
| ||
| Body Weight (g) |
224.4 ± 4.1 | 209.9 ± 4.0a |
|
| ||
| Liver-to-Body Weight Ratio (%) |
3.8 ± 0.1 | 4.09 ± 0.2 |
|
| ||
| ALT (U/l) |
73.0 ± 5.6 | 120.0 ± 7.7a |
|
| ||
| AST (U/l) |
146.7 ± 1.2 | 184.0 ± 5.5a |
|
| ||
| Total Bilirubin (mg/l) |
< 0.1 | < 0.1 |
|
| ||
| ALP (U/l) |
154.7 ± 2.2 | 268.3 ± 7.52a |
|
| ||
| CK-19 (positive cholangiocytes per portal area) |
17.2 ± 2.3 | 36.0 ± 1.9a |
|
| ||
| PCNA (positive cholangiocytes per portal area) |
0.2 ± 0.2 | 0.8 ± 0.2a |
|
| ||
| TUNEL (positive cholangiocytes per portal area) |
< 1 | < 1 |
|
| ||
| Masson’s Trichrome (Fold-change) |
1.0 ± 0.1 | 31.9 ± 6.9a |
|
| ||
ALT, alanine aminotransferase; AST, aspartate aminotransferase; ALP, alkaline phosphatase; CK-19, cytokeratin-19; PCNA, proliferating cell nuclear antigen.
Values for liver weight, body weight, liver-to-body weight ratio, ALT, AST, total bilirubin and ALP are presented as mean ± SEM for n = 16 animals per treatment group
Values for CK-19, PCNA, TUNEL and Masson’s Trichrome are presented as the mean ± SEM of cumulative values obtained from the evaluation of 10 portal tracts from 5 different slides for each group.
p < 0.05 vs. corresponding values of control.
The number of CK-19 and PCNA-positive cholangiocytes per portal area significantly increased in nicotine-treated rats (Figure 2A, Table 1). A significant increase in PCNA protein expression was observed in isolated cholangiocytes from nicotine-treated rats (Figure 2B).
Figure 2.
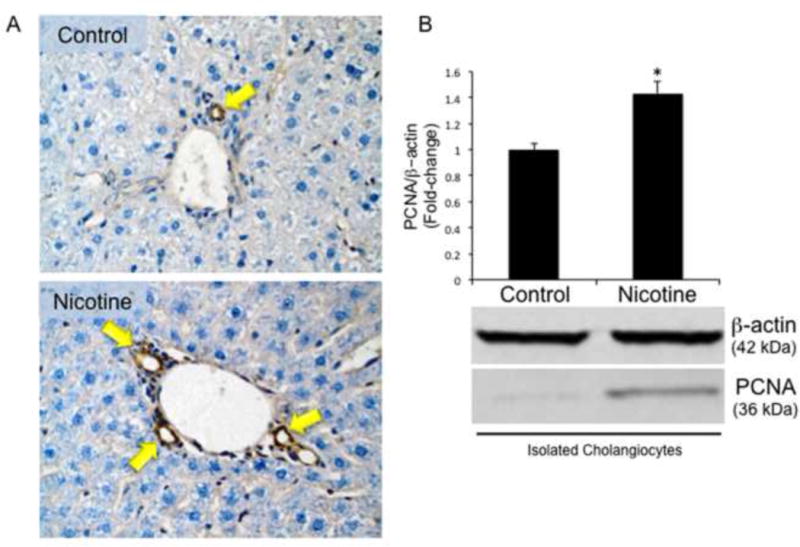
Evaluation of intrahepatic bile duct mass (IBDM) and biliary proliferation in liver sections and cholangiocytes isolated from control and nicotine-treated rats. [A] In liver sections from control and nicotine-treated rats, a significant increase in IBDM was observed in nicotine-treated rats compared with the normal control rats (for semiquantitative data see Table 1). Bile ducts are indicated with yellow arrows. Original magnification, x40. [B] A significant increase in PCNA protein expression is observed in cholangiocytes isolated from nicotine-treated compared to control rats by immunoblots. Data are mean ± SEM of 6 experiments. *p < 0.05 vs. control.
Chronic administration of nicotine stimulates profibrotic gene and protein expression
Nicotine has been shown to stimulate profibrotic gene and protein expression as well as proliferation (21, 59, 60). The effects of nicotine on gene and protein expression of α-SMA, collagen 1A1 and FN-1 were evaluated in total liver samples and cholangiocytes from control and nicotine-treated rats. In total liver, there was a significant increase in the expression levels of α-SMA, collagen 1A1, and FN-1 in nicotine-treated rats (Figure 3 top). In isolated cholangiocytes, there was a significant increase in α-SMA, and collagen 1A1 in nicotine-treated rats (Figure 3 bottom). However, no changes in FN-1 were observed in nicotine-treated rats (Figure 3 bottom). Expression levels of a-SMA and FN-1 were also evaluated in liver sections from control and nicotine-treated rats by immunohistochemistry. The amount of collagen deposition in the portal areas was evaluated by Masson’s trichrome histochemistry. There was a significant increase in the amount of collagen in the portal areas of nicotine-treated rats (Figure 4A, B, Table 1). Chronic administration of nicotine induced an increase in positive staining for FN-1 in the portal areas (Figure 4C, D) and a visible increase in α-SMA expression in the bile ducts of nicotine-treated rats (Figure 4E, F).
Figure 3.
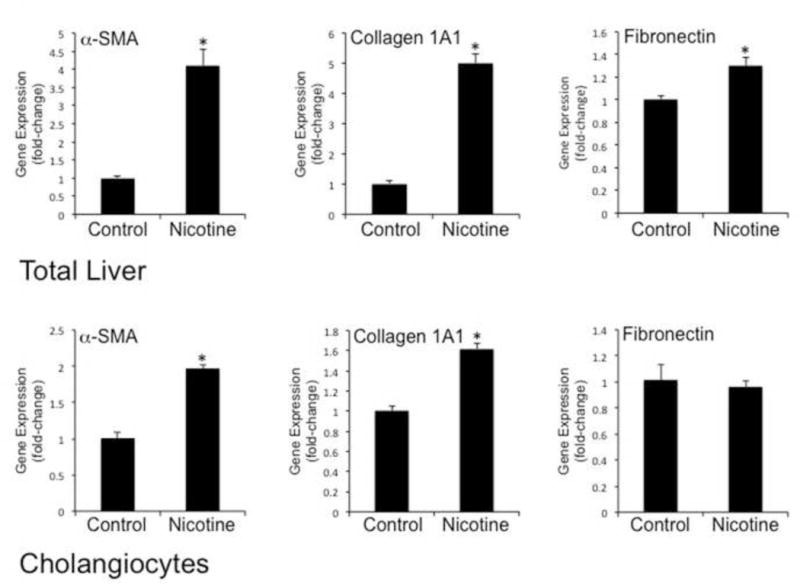
Evaluation of profibrotic gene expression in total liver samples and cholangiocytes isolated from normal and nicotine-treated rats. [upper panel] Chronic administration of nicotine stimulated a significant increase in the gene expression of α-SMA, collagen 1A and fibronectin in total liver samples from nicotine-treated rats compared to control. [lower panel] Chronic administration of nicotine stimulated a significant increase in the gene expression of α-SMA and collagen 1A in cholangiocytes isolated from nicotine-treated rats compared to normal control. No significant changes were observed for fibronectin gene expression. Data are mean ± SEM of 6 experiments. *p < 0.05 vs. normal control.
Figure 4.
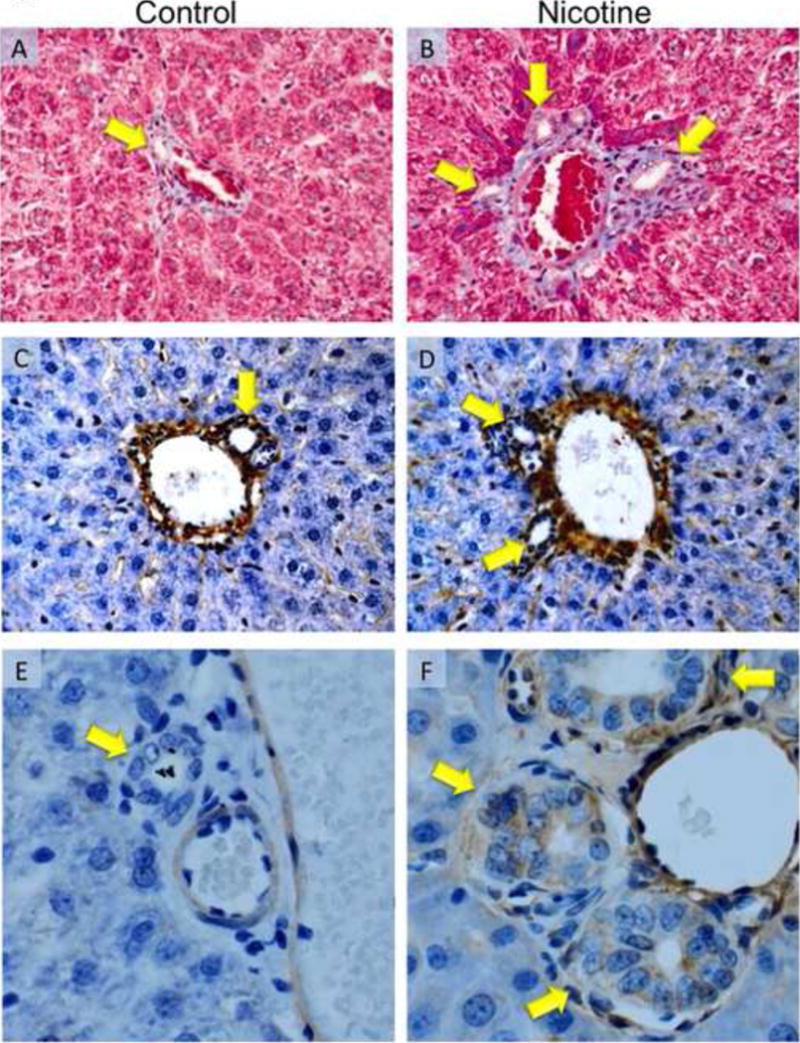
Evaluation of collagen deposition by Masson’s trichrome staining, fibronectin and α-SMA expression by immunohistochemistry in liver sections from control and nicotine-treated rats. Masson’s trichrome staining in control [A] and nicotine-treated rats [B]. Chronic administration of nicotine stimulated an increase in collagen deposition (blue staining) around the portal areas of nicotine-treated [B] compared with normal control rats [A] (for semiquantitative data see Table 1). Immunohistochemistry for fibronectin [C, D] and α-SMA [E, F] expression in control and nicotine-treated rats, respectively. An increase in both fibronectin and α-SMA expression is observed in in nicotine-treated [D, F] compared with normal control rats [C, D], respectively. Bile ducts are indicated with yellow arrows. Original magnification, x40.
Nicotine stimulates NRIC proliferation via activation of α7-nAChR and Ca2+/IP3/ERK1/2 signaling
To confirm the direct effects of nicotine on cholangiocytes, we performed MTS assays in NRIC stimulated in vitro with the selected compound. MTS assays evaluate the number of viable cells (cell proliferation) (61). Nicotine induced a significant increase in NRIC proliferation at 48 hours at all doses evaluated (Figure 5A). AR-R 17779 also stimulated a dose-dependent increase in NRIC proliferation at 48 hours (Figure 5B). AR-R 17779-stimulated proliferation was significantly inhibited by ABT, BAPTA/AM and PD98059 (Figure 5C). Inhibition of AR-R 17779-stimulated proliferation by BAPTA/AM and PD08059 indicates involvement of Ca2+/ERK1/2-dependent signaling mechanism. Activation of α7-nAChR by nicotine has been shown to increase intracellular Ca2+ levels (62). Nicotine (≈16-fold increase) and AR-R 17779 (≈7-fold increase) significantly increased intracellular Ca2+ levels in NRIC. Nicotine induced a significant increase in intracellular IP3 (2.2-fold increase compared to basal) but not cAMP levels. Nicotine stimulated phosphorylation and nuclear translocation of ERK1/2 (Figure 5D), which was blocked by ABT. Nicotine also increased of ERK1/2 phosphorylation by ELISA (Figure 5E)
Figure 5.
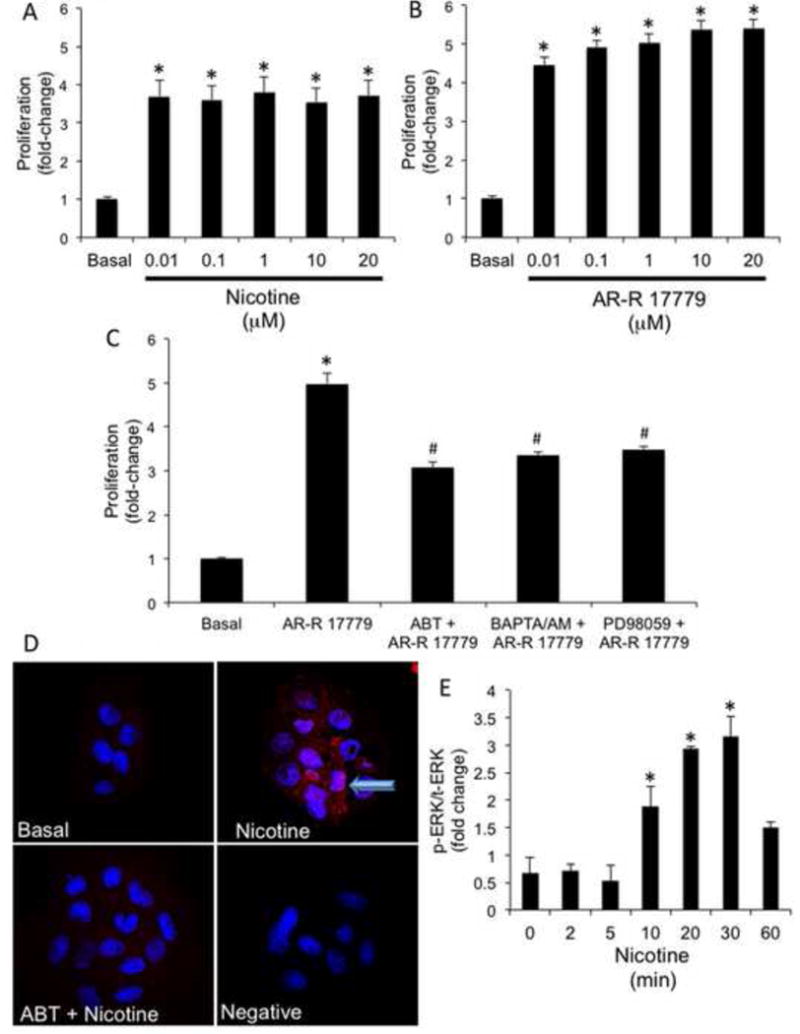
Evaluation of nicotine-induced proliferation and ERK1/2 activation in NRIC. [A] Nicotine (0.01–20 μM) induced a dose-dependent increase in NRIC proliferation at 48 hr. [B] AR-R 17779 (0.01×20 μM; a specific α7-nAChR agonist) stimulated a dose-dependent increase in NRIC proliferation at 48 hr. [C] AR-R 17779 (1 μM)-induced NRIC proliferation is partially blocked by alpha-bungarotoxin (ABT; 0.5 μM; α7-nAChR selective antagonist), BAPTA/AM (5 μM) and PD98059 (10 nM). Data are mean ± SEM of six experiments. *p < 0.05 vs. basal values. [D] Nicotine-stimulated the phosphorylation and nuclear translocation of ERK1/2. p-ERK1/2 (red) and DAPI (blue). Phosphorylation and nuclear translocation of ERK1/2 was inhibited by preincubation with ABT. [E] NRIC were stimulated with 0.2% BSA or nicotine (10 μM for 0, 2, 5, 10, 20, 30, 60, min) and ERK1/2 phosphorylation was evaluated by a phospho-ERK Cell-based ELISA kit. Nicotine stimulated an increase in p-ERK/t-ERK at 10, 20 and 30 minutes of incubation. Data are mean ± SEM of six experiments. *p < 0.05 vs. basal.
Discussion
To date, this is the first study to demonstrate a role for nicotine in the regulation of proliferation and expression of a profibrogenic phenotype in normal cholangiocytes. Several studies have demonstrated that nicotine plays a key role in regulating proliferation and fibrosis in a number of cell types including HSC (19–21). Previous studies have shown that the cholinergic system regulates biliary mass and function during cholestasis through the activation of mAChR M3 (23, 24). However, these studies did not include evaluation of nAChR receptors, which also bind ACh (22). Given that nicotine has been implicated in activation of proliferation and fibrosis in various normal and malignant cell types, we hypothesized that chronic administration of nicotine would stimulate biliary proliferation and fibrosis in normal rats. Therefore, the biological effects of nicotine on biliary proliferation and fibrosis were evaluated in a normal rat in vivo and NRIC in vitro.
The expression of nAChRs is limited to the nervous system and widely distributed in a number of cell types (16). The majority of studies evaluating the biological effects of nicotine (including proliferation, angiogenesis, epithelial to mesenchymal transition and fibrogenesis) have focused on α7-nAChR (17, 63, 64). Soeda et al. have shown that hepatic stellate cells express several subtypes of nAChRs α3, β1 and ɛ in addition to α7-nAChR (21). α7-nAChR is also a mediator of nicotine effects in chronic kidney disease (65). Therefore, we focused on the evaluation of α7-nAChR in cholangiocytes. Bile ducts and isolated cholangiocytes from control (normal) and nicotine-treated rats expressed α7-nAChR by immunohistochemistry, immunoblots and real-time PCR. No differences in α7-nAChR expression levels were observed between the treatment groups. In addition, NRIC were positive for α7-nAChR by immunofluorescence and real-time PCR.
Chronic administration of nicotine for 2 weeks induced biliary proliferation in both liver sections and isolated cholangiocytes compared to controls. These findings are similar to a previous study that demonstrated the chronic administration of nicotine for 1 and 2 weeks stimulated vascular endothelial cell proliferation (66). Elevations of liver enzyme levels (ALT, AST and ALP) indicated mild liver damage and were moderately elevated in the nicotine treated animals. However, overt hepatocellular necrosis or lobular damage was not observed in liver sections from nicotine-treated rats. Total bilirubin levels were also unchanged. These findings are consistent with studies that evaluated biliary proliferation in the cholestatic rat model of BDL (44, 67). ALT, AST and ALP levels are significantly elevated during BDL (44, 67). However, the elevations that were observed in the nicotine-treated rats are lower compared to those shown for cholestatic BDL models (44, 67). The elevation of ALT, AST and ALP levels in the nicotine-treated rats may be due to increased biliary proliferation and fibrosis. These findings were evaluated further in NRIC. Both nicotine and AR-R 17779 (α7-nAChR) induced significant increases in NRIC proliferation. The specific activation of NRIC proliferation by AR-R 17779 indicates that α7-nAChR plays an important role in regulating cholangiocyte proliferation. Taken together, the in vivo and in vitro findings demonstrate that nicotine possesses pro-proliferative effects on cholangiocytes, which appears to occur through α7-nAChR.
It is necessary to emphasize that while the current study demonstrates an increase in liver enzymes in the rat model, studies have shown that nicotine/smoking by itself does not cause a significant increase in liver enzymes in humans (68, 69). However, additional data from a greater number of patients is necessary to define the effects of nicotine on serum liver enzymes and determine the possibility of an intrinsic difference between human and rat response to nicotine. In addition, the upregulation of various nicotinic receptors in humans and rats following nicotine exposure would alter the activation of various signaling pathways, which may contribute to the differences in liver enzymes observed. At the same time, it is possible that the route of administration (inhalation in humans vs. minipumps in this study) and time period of chronic administration may also have contributed to the differences. However, there is strong data demonstrating that nicotine with alcohol causes synergistic increases in liver enzymes (68, 69).
In addition to stimulating proliferation, nicotine has been implicated in the development of fibrogenesis in a number of organs and cell types. For example, nicotine activates atrial fibrosis (19) and stimulates the expression of factors that contribute to fibrosis such as fibronectin (20, 59), which was mediated through upregulation of ERK1/2 (20, 59). Nicotine induces HSC proliferation and upregulated collagen 1-A2 and TGF-β mRNA expression, which was inhibited by mecamylamine (non-specific nAChR antagonist) (21). Nicotine has been shown to increase a-SMA expression in a number of cell types (59, 70). The gene and protein expression levels of FN-1, collagen, and a-SMA (markers of fibrosis) were upregulated in total liver samples from the nicotine-treated groups. In isolated cholangiocytes, the gene and protein expression levels of collagen and α-SMA were upregulated in nicotine-treated rats. However, FN-1 gene expression levels were unchanged in isolated cholangiocytes from the nicotine-treated group compared to control rats. This may be due to earlier activation of FN-1 gene expression in cholangiocytes that was missed by evaluation of FN-1 expression levels from animals treated chronically. Nicotine does stimulate FN-1 gene expression in NRIC at 6 to 24 hours (not shown), which supports this overall concept. Taken together, the findings indicate that nicotine increases both biliary proliferation and fibrosis in normal rats suggesting that chronic nicotine abuse may have deleterious effects on cholangiocytes.
Studies were performed to elucidate the intracellular signaling mechanisms participating in nicotine/α7-nAChR axis-mediated proliferative and profibrotic events. AR-R 17779-(α7-nAChR specific agonist) stimulated NRIC proliferation was partially but significantly inhibited by ABT, indicating that α7-nAChR plays an important role in the regulation cholangiocyte proliferation. In addition, AR-R 17779 induced proliferation was partially but significantly inhibited by BAPTA/AM and PD98059. Supporting this concept, nicotine and AR-R 17779 also increased intracellular Ca2+ and IP3 levels. Nicotine also induced phosphorylation and nuclear translocation of ERK1/2, which was blocked by ABT. Activation of the α7-nAChR has been shown to trigger increased Ca2+ levels and ERK1/2 activation (71). The partial inhibition of AR-R 17779-induced proliferation in NRIC by ABT, BAPTA/AM and PD98059, may be due to the activation of other nicotinic receptor subtypes or other pro-proliferative receptors. Proliferating cholangiocytes have been shown to display a neuroendocrine and secrete and respond to a number of neurotransmitters and neuropeptides that regulate autocrine/paracrine proliferation (7). Future studies are underway to evaluate the roles of other nAChR subtypes and nicotine-induced secretion of pro-proliferative factors. However, the overall findings suggest the involvement of Ca2+/IP3/ERK1/2-dependent signaling mechanism in the regulation of cholangiocyte proliferation by nicotine and α7-nAChR.
Although, this study cannot rule out the effects of other nAChR subtypes and the interplay of pro-proliferative factors secreted by cholangiocytes, it does support the concept that α7-nAChR plays a key role in regulating cholangiocyte proliferation. Future studies to determine the effects of chronic nicotine exposure and the α7-nAChR axis in models of cholestasis, liver injury and fibrosis are warranted. In summary, the findings presented indicate that chronic nicotine exposure plays a significant role in the regulation of biliary proliferation and fibrosis and may participate in the pathogenesis of cholangiopathies.
Acknowledgments
We thank Anna Webb and the Texas A&M Health Science Center Microscopy Imaging Center for assistance with confocal microscopy.
Sources of Support: Portions of this work were supported by a Department of Veteran’s Affairs Career Development Award-2, the Scott & White Hospital, Department of Internal Medicine, a Scott and White Research Advancement award, and NIH RO1 grant (DK081442) awarded to S.G. Undergraduate research program participants, A.F. and M.S., were supported by NSF-STEP Grant “The Central Texas 2-STEP” awarded to Dr. John P. Idoux.
Footnotes
Addresses of Affiliations:
Texas A&M Health Science Center Central Texas Veterans Health Care System 1901 South 1st Street Temple, TX, 76504
Yamagata University Faculty of Medicine Department of Gastroenterology 2-2-2 Iidanishi Yamagata, 9909585, Japan
NSF-STEP Grant “The Central Texas 2-STEP” Texas Bioscience Institute-Temple College 5701 Airport Road Temple, Texas 76502
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.LeSage G, Benedetti A, Glaser S, et al. Acute carbon tetrachloride feeding selectively damages large, but not small, cholangiocytes from normal rat liver. Hepatology. 1999;29:307–319. doi: 10.1002/hep.510290242. [DOI] [PubMed] [Google Scholar]
- 2.LeSage G, Glaser S, Gubba S, et al. Regrowth of the rat biliary tree after 70% partial hepatectomy is coupled to increased secretin-induced ductal secretion. Gastroenterology. 1996;111:1633–1644. doi: 10.1016/s0016-5085(96)70027-6. [DOI] [PubMed] [Google Scholar]
- 3.Alpini G, Prall RT, LaRusso NF. The pathobiology of biliary epithelia. In: Arias IM, Boyer JL, Chisari FV, Fausto N, Jakoby W, Schachter D, Shafritz DA, editors. The Liver; Biology & Pathobiology, 4E. Philadelphia, PA: Lippincott Williams & Wilkins; 2001. pp. 421–435. [Google Scholar]
- 4.Munshi MK, Priester S, Gaudio E, et al. Regulation of biliary proliferation by neuroendocrine factors: implications for the pathogenesis of cholestatic liver diseases. Am J Pathol. 2011;178:472–484. doi: 10.1016/j.ajpath.2010.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alpini G, McGill JM, Larusso NF. The pathobiology of biliary epithelia. Hepatology. 2002;35:1256–1268. doi: 10.1053/jhep.2002.33541. [DOI] [PubMed] [Google Scholar]
- 6.Glaser SS, Gaudio E, Miller T, et al. Cholangiocyte proliferation and liver fibrosis. Expert Reviews Mol Med. 2009;11:e7. doi: 10.1017/S1462399409000994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glaser S, Francis H, DeMorrow S, et al. Heterogeneity of the intrahepatic biliary epithelium. World J Gastroenterol. 2006;12:3523–3536. doi: 10.3748/wjg.v12.i22.3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corpechot C, Gaouar F, Chretien Y, et al. Smoking as an independent risk factor of liver fibrosis in primary biliary cirrhosis. J Hepatol. 2012;56:218–224. doi: 10.1016/j.jhep.2011.03.031. [DOI] [PubMed] [Google Scholar]
- 9.Tsochatzis E, Papatheodoridis GV, Manolakopoulos S, et al. Smoking is associated with steatosis and severe fibrosis in chronic hepatitis C but not B. Scand J Gastroenterol. 2009;44:752–759. doi: 10.1080/00365520902803515. [DOI] [PubMed] [Google Scholar]
- 10.Zein CO, Unalp A, Colvin R, et al. Smoking and severity of hepatic fibrosis in nonalcoholic fatty liver disease. J Hepatol. 2011;54:753–759. doi: 10.1016/j.jhep.2010.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tischendorf JJ, Meier PN, Strassburg CP, et al. Characterization and clinical course of hepatobiliary carcinoma in patients with primary sclerosing cholangitis. Scand J Gastroenterol. 2006;41:1227–1234. doi: 10.1080/00365520600633495. [DOI] [PubMed] [Google Scholar]
- 12.Benowitz NL. Drug therapy. Pharmacologic aspects of cigarette smoking and nicotine addition. N Engl J Med. 1988;319:1318–1330. doi: 10.1056/NEJM198811173192005. [DOI] [PubMed] [Google Scholar]
- 13.Cahill K, Stead LF, Lancaster T. Nicotine receptor partial agonists for smoking cessation. Cochrane Database Syst Rev. 2011 doi: 10.1002/14651858.CD006103.pub2. CD006103. [DOI] [PubMed] [Google Scholar]
- 14.Hukkanen J, Jacob P, 3rd, Benowitz NL. Metabolism and disposition kinetics of nicotine. Pharmacol Rev. 2005;57:79–115. doi: 10.1124/pr.57.1.3. [DOI] [PubMed] [Google Scholar]
- 15.Millar NS, Harkness PC. Assembly and trafficking of nicotinic acetylcholine receptors (Review) Mol Membr Biol. 2008;25:279–292. doi: 10.1080/09687680802035675. [DOI] [PubMed] [Google Scholar]
- 16.Wessler I, Kirkpatrick CJ. Acetylcholine beyond neurons: the non-neuronal cholinergic system in humans. Br J Pharmacol. 2008;154:1558–1571. doi: 10.1038/bjp.2008.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Egleton RD, Brown KC, Dasgupta P. Nicotinic acetylcholine receptors in cancer: multiple roles in proliferation and inhibition of apoptosis. Trends Pharmacol Sci. 2008;29:151–158. doi: 10.1016/j.tips.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 18.Schuller HM. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat Rev Cancer. 2009;9:195–205. doi: 10.1038/nrc2590. [DOI] [PubMed] [Google Scholar]
- 19.Dasgupta P, Rastogi S, Pillai S, et al. Nicotine induces cell proliferation by beta-arrestin-mediated activation of Src and Rb-Raf-1 pathways. J Clin Invest. 2006;116:2208–2217. doi: 10.1172/JCI28164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng Y, Ritzenthaler JD, Roman J, et al. Nicotine stimulates human lung cancer cell growth by inducing fibronectin expression. Am J Respir Cell Mol Biol. 2007;37:681–690. doi: 10.1165/rcmb.2007-0051OC. [DOI] [PubMed] [Google Scholar]
- 21.Soeda J, Morgan M, McKee C, et al. Nicotine induces fibrogenic changes in human liver via nicotinic acetylcholine receptors expressed on hepatic stellate cells. BBRC. 2012;417:17–22. doi: 10.1016/j.bbrc.2011.10.151. [DOI] [PubMed] [Google Scholar]
- 22.Gotti C, Clementi F. Neuronal nicotinic receptors: from structure to pathology. Prog Neurobiol. 2004;74:363–396. doi: 10.1016/j.pneurobio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 23.Alvaro D, Alpini G, Jezequel AM, et al. Role and mechanisms of action of acetylcholine in the regulation of rat cholangiocyte secretory functions. J Clin Invest. 1997;100:1349–1362. doi: 10.1172/JCI119655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lesage G, Alvaro D, Benedetti A, et al. Cholinergic system modulates growth, apoptosis, and secretion of cholangiocytes from bile duct-ligated rats. Gastroenterology. 1999;117:191–199. doi: 10.1016/s0016-5085(99)70567-6. [DOI] [PubMed] [Google Scholar]
- 25.Bodnar AL, Cortes-Burgos LA, Cook KK, et al. Discovery and structure-activity relationship of quinuclidine benzamides as agonists of alpha7 nicotinic acetylcholine receptors. J Med Chem. 2005;48:905–908. doi: 10.1021/jm049363q. [DOI] [PubMed] [Google Scholar]
- 26.Zhang ZW, Vijayaraghavan S, Berg DK. Neuronal acetylcholine receptors that bind alpha-bungarotoxin with high affinity function as ligand-gated ion channels. Neuron. 1994;12:167–177. doi: 10.1016/0896-6273(94)90161-9. [DOI] [PubMed] [Google Scholar]
- 27.Glaser S, Alvaro D, Ueno Y, et al. Gastrin reverses established cholangiocyte proliferation and enhanced secretin-stimulated ductal secretion of BDL rats by activation of apoptosis through increased expression of Ca2+- dependent PKC isoforms. Liver Int. 2003;23:78–88. doi: 10.1034/j.1600-0676.2003.00814.x. [DOI] [PubMed] [Google Scholar]
- 28.Glaser S, Benedetti A, Marucci L, et al. Gastrin inhibits cholangiocyte growth in bile duct-ligated rats by interaction with cholecystokinin-B/Gastrin receptors via D-myoinositol 1,4,5-triphosphate-, Ca(2+)-, and protein kinase C alpha-dependent mechanisms. Hepatology. 2000;32:17–25. doi: 10.1053/jhep.2000.8265. [DOI] [PubMed] [Google Scholar]
- 29.Ohmura Y, Jutkiewicz EM, Zhang A, et al. Dopamine D1/5 and D2/3 agonists differentially attenuate somatic signs of nicotine withdrawal in rats. Pharmacol Biochem Behavior. 2011;99:552–556. doi: 10.1016/j.pbb.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 30.Malin DH, Goyarzu P. Rodent models of nicotine withdrawal syndrome. Handbook Exp Pharmacol. 2009:401–434. doi: 10.1007/978-3-540-69248-5_14. [DOI] [PubMed] [Google Scholar]
- 31.Hamilton KR, Berger SS, Perry ME, et al. Behavioral effects of nicotine withdrawal in adult male and female rats. Pharmacol Biochem Behavior. 2009;92:51–59. doi: 10.1016/j.pbb.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 32.Ohmura Y, Jutkiewicz EM, Domino EF. L-DOPA attenuates nicotine withdrawal-induced behaviors in rats. Pharmacol Biochem Behavior. 2011;98:552–558. doi: 10.1016/j.pbb.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 33.Benowitz NL, Kuyt F, Jacob P., 3rd Circadian blood nicotine concentrations during cigarette smoking. Clinical Pharmacol Ther. 1982;32:758–764. doi: 10.1038/clpt.1982.233. [DOI] [PubMed] [Google Scholar]
- 34.LeSage MG, Keyler DE, Shoeman D, et al. Continuous nicotine infusion reduces nicotine self-administration in rats with 23-h/day access to nicotine. Pharmacol Biochem Behavior. 2002;72:279–289. doi: 10.1016/s0091-3057(01)00775-4. [DOI] [PubMed] [Google Scholar]
- 35.Alpini G, Ulrich C, Roberts S, et al. Molecular and functional heterogeneity of cholangiocytes from rat liver after bile duct ligation. Am J Physiol Gastrointest Liver Physiol. 1997;272:G289–297. doi: 10.1152/ajpgi.1997.272.2.G289. [DOI] [PubMed] [Google Scholar]
- 36.Alpini G, Phinizy JL, Glaser S, et al. Development and characterization of secretin-stimulated secretion of cultured rat cholangiocytes. Am J Physiol Gastrointest Liver Physiol. 2003;284:G1066–1073. doi: 10.1152/ajpgi.00260.2002. [DOI] [PubMed] [Google Scholar]
- 37.Glaser S, Rodgers RE, Phinizy JL, et al. Gastrin inhibits secretin-induced ductal secretion by interaction with specific receptors on rat cholangiocytes. Am J Pathol. 1997;273:G1061–1070. doi: 10.1152/ajpgi.1997.273.5.G1061. [DOI] [PubMed] [Google Scholar]
- 38.Glaser S, DeMorrow S, Francis H, et al. Progesterone stimulates the proliferation of female and male cholangiocytes via autocrine/paracrine mechanisms. Am J Physiol Gastrointest Liver Physiol. 2008;295:G124–G136. doi: 10.1152/ajpgi.00536.2007. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Seguela P, Wadiche J, Dineley-Miller K, et al. Molecular cloning, functional properties, and distribution of rat brain alpha 7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tso JY, Sun XH, Kao TH, et al. Isolation and characterization of rat and human glyceraldehyde-3-phosphate dehydrogenase cDNAs: genomic complexity and molecular evolution of the gene. Nucleic Acids Res. 1985;13:2485–2502. doi: 10.1093/nar/13.7.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Glaser S, Gaudio E, Renzi A, et al. Knockout of the neurokinin-1 receptor reduces cholangiocyte proliferation in bile duct-ligated mice. Am J Physiol Gastrointest Liver Physiol. 2011;301:G297–305. doi: 10.1152/ajpgi.00418.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glaser S, Wang M, Ueno Y, et al. Differential transcriptional characteristics of small and large biliary epithelial cells derived from small and large bile ducts. Am J Physiol Gastrointest Liver Physiol. 2010;299:G769–777. doi: 10.1152/ajpgi.00237.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Glaser S, Alvaro D, Francis H, et al. Adrenergic receptor agonists prevent bile duct injury induced by adrenergic denervation by increased cAMP levels and activation of Akt. Am J Physiol Gastrointest Liver Physiol. 2006;290:G813–826. doi: 10.1152/ajpgi.00306.2005. [DOI] [PubMed] [Google Scholar]
- 44.Alpini G, Lenzi R, Sarkozi L, et al. Biliary physiology in rats with bile ductular cell hyperplasia. Evidence for a secretory function of proliferated bile ductules. J Clin Invest. 1988;81:569–578. doi: 10.1172/JCI113355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mancinelli R, Onori P, Gaudio E, et al. Follicle-stimulating hormone increases cholangiocyte proliferation by an autocrine mechanism via cAMP-dependent phosphorylation of ERK1/2 and Elk-1. Am J Physiol Gastrointest Liver Physiol. 2009;297:G11–26. doi: 10.1152/ajpgi.00025.2009. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.Leheup BP, Federspiel SJ, Guerry-Force ML, et al. Extracellular matrix biosynthesis by cultured fetal rat lung epithelial cells. I. Characterization of the clone and the major genetic types of collagen produced. Lab Invest. 1989;60:791–807. [PubMed] [Google Scholar]
- 47.Hsu CY, Frankel FR. Effect of estrogen on the expression of mRNAs of different actin isoforms in immature rat uterus. Cloning of alpha-smooth muscle actin message. J Biol Chem. 1987;262:9594–9600. [PubMed] [Google Scholar]
- 48.Schneider C, Rasband W, Eliceiri K. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khalil AA, Jameson MJ, Broaddus WC, et al. Nicotine enhances proliferation, migration, and radioresistance of human malignant glioma cells through EGFR activation. Brain Tumor Pathol. 2012 doi: 10.1007/s10014-012-0101-5. [DOI] [PubMed] [Google Scholar]
- 50.Maggi L, Sher E, Cherubini E. Regulation of GABA release by nicotinic acetylcholine receptors in the neonatal rat hippocampus. J Physiol. 2001;536:89–100. doi: 10.1111/j.1469-7793.2001.00089.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alpini G, Franchitto A, DeMorrow S, et al. Activation of alpha(1) -adrenergic receptors stimulate the growth of small mouse cholangiocytes via calcium-dependent activation of nuclear factor of activated T cells 2 and specificity protein 1. Hepatology. 2011;53:628–639. doi: 10.1002/hep.24041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lien YC, Wang W, Kuo LJ, et al. Nicotine promotes cell migration through alpha7 nicotinic acetylcholine receptor in gastric cancer cells. Annals Surg Oncol. 2011;18:2671–2679. doi: 10.1245/s10434-011-1598-2. [DOI] [PubMed] [Google Scholar]
- 53.Wei PL, Kuo LJ, Huang MT, et al. Nicotine enhances colon cancer cell migration by induction of fibronectin. Annals Surgical Oncol. 2011;18:1782–1790. doi: 10.1245/s10434-010-1504-3. [DOI] [PubMed] [Google Scholar]
- 54.Cucina A, Dinicola S, Coluccia P, et al. Nicotine stimulates proliferation and inhibits apoptosis in colon cancer cell lines through activation of survival pathways. J Surg Res. 2012;178:233–241. doi: 10.1016/j.jss.2011.12.029. [DOI] [PubMed] [Google Scholar]
- 55.Momi N, Ponnusamy MP, Kaur S, et al. Nicotine/cigarette smoke promotes metastasis of pancreatic cancer through alpha7nAChR-mediated MUC4 upregulation. Oncogene. 2012 May 21; doi: 10.1038/onc.2012.163. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Francis H, Glaser S, Ueno Y, et al. cAMP stimulates the secretory and proliferative capacity of the rat intrahepatic biliary epithelium through changes in the PKA/Src/MEK/ERK1/2 pathway. J Hepatol. 2004;41:528–537. doi: 10.1016/j.jhep.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 57.Chowdhury P, Hosotani R, Rayford PL. Weight loss and altered circulating GI peptide levels of rats exposed chronically to nicotine. Pharmacol Biochem Behavior. 1989;33:591–594. doi: 10.1016/0091-3057(89)90393-6. [DOI] [PubMed] [Google Scholar]
- 58.Frankham P, Cabanac M. Nicotine lowers the body-weight set-point in male rats. Appetite. 2003;41:1–5. doi: 10.1016/s0195-6663(03)00024-2. [DOI] [PubMed] [Google Scholar]
- 59.Arany I, Reed DK, Grifoni SC, et al. A novel U-STAT3-dependent mechanism mediates the deleterious effects of chronic nicotine exposure on renal injury. Am J Physiol Renal Physiol. 2012;302:F722–729. doi: 10.1152/ajprenal.00338.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frazier K, Williams S, Kothapalli D, et al. Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J Invest Dermatol. 1996;107:404–411. doi: 10.1111/1523-1747.ep12363389. [DOI] [PubMed] [Google Scholar]
- 61.Cory AH, Owen TC, Barltrop JA, et al. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- 62.Dajas-Bailador F, Wonnacott S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci. 2004;25:317–324. doi: 10.1016/j.tips.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 63.Pillai S, Chellappan S. alpha7 nicotinic acetylcholine receptor subunit in angiogenesis and epithelial to mesenchymal transition. Current Drug Targets. 2012;13:671–679. doi: 10.2174/138945012800398847. [DOI] [PubMed] [Google Scholar]
- 64.Schuller HM, Al-Wadei HA. Neurotransmitter receptors as central regulators of pancreatic cancer. Future Oncol. 2010;6:221–228. doi: 10.2217/fon.09.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rezonzew G, Chumley P, Feng W, et al. Nicotine exposure and the progression of chronic kidney disease: role of the alpha7-nicotinic acetylcholine receptor. Am J Physiol Renal Physiol. 2012;303:F304–312. doi: 10.1152/ajprenal.00661.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Strohschneider T, Oberhoff M, Hanke H, et al. Effect of chronic nicotine delivery on the proliferation rate of endothelial and smooth muscle cells in experimentally induced vascular wall plaques. Clin Invest. 1994;72:908–912. doi: 10.1007/BF00190750. [DOI] [PubMed] [Google Scholar]
- 67.Renzi A, Glaser S, DeMorrow S, et al. Melatonin inhibits cholangiocyte hyperplasia in cholestatic rats by interaction with MT1 but not MT2 melatonin receptors. Am J Physiol Gastroint Liver Physiol. 2011;301:G634–643. doi: 10.1152/ajpgi.00206.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kurtul N, Cil MY, Bakan E. The effects of alcohol and smoking on serum, saliva, and urine sialic acid levels. Saudi Med J. 2004;25:1839–1844. [PubMed] [Google Scholar]
- 69.Breitling LP, Arndt V, Drath C, et al. Liver enzymes: interaction analysis of smoking with alcohol consumption or BMI, comparing AST and ALT to gamma-GT. PloS one. 2011;6:e27951. doi: 10.1371/journal.pone.0027951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krebs M, Sakurai R, Torday JS, et al. Evidence for in vivo nicotine-induced alveolar interstitial fibroblast-to-myofibroblast transdifferentiation. Experimental Lung Res. 2010;36:390–398. doi: 10.3109/01902141003714023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chernyavsky AI, Arredondo J, Qian J, et al. Coupling of ionic events to protein kinase signaling cascades upon activation of alpha7 nicotinic receptor: cooperative regulation of alpha2-integrin expression and Rho kinase activity. J Biol Chem. 2009;284:22140–22148. doi: 10.1074/jbc.M109.011395. [DOI] [PMC free article] [PubMed] [Google Scholar]