

Abstract
Reported herein are the synthesis and solid-phase peptide incorporation of N-Fmoc-(2S,3R)-2-amino-3-methyl-4-phosphonobutyric acid bis-pivaloyloxymethyl phopshoryl ester [Fmoc-Pmab(POM)2-OH, 2] as a phosphatase-stable phosphothreonine (pThr) mimetic bearing orthogonal protection suitable for the synthesis of Pmab-containing peptides having bio-reversible protection of the phosphonic acid moiety. This represents the first report of a bio-reversibly protected pThr mimetic in a form suitable for facile solid-phase peptide synthesis.
Keywords: Phosphothreonine mimetic, Prodrug, Pmab, Solid-phase peptide synthesis, Signal transduction
Introduction
Protein phosphorylation is a fundamental form of post-translational modification that affects approximately one third of all proteins. The presence of phosphothreonine (pThr), phosphoserine (pSer) and phosphotyrosine (pTyr)-can introduce unique recognition features, which often facilitate specific protein-protein interactions (PPIs) (Yaffe 2002; Ladbury 2005; Elia and Yaffe 2005). Synthetic phosphopeptides modeled on recognition sequences can serve as useful pharmacological tools for studying these PPIs (Eisele et al. 1999; Lu et al. 2012). However, in cellular systems the bioavailabilty of phosphopeptides may be limited by the enzymatic lability of the phosphoryl ester bond toward phosphatases and poor membrane transport of the phosphoryl di-anionic species. While replacement of the phosphoryl ester oxygen by methylene or fluoromethylenes has addressed issues related to phosphatase hydrolysis for pTyr (Burke and Lee 2003), pSer (Shapiro et al. 1993; Perich 1994; Nair et al. 1995; Panigrahi et al. 2009) and pThr (Berkowitz et al. 1966; Otaka et al. 2000; Liu et al. 2009), cell membrane transit can still be limited by the di-anionic charge of the resulting phosphonic acids.
A general strategy for increasing the cell membrane permeability of phosphates and phosphonic acids is to mask their acidic functionality with neutral “prodrug” groups that can be removed enzymatically once the agent is within the cell. Although a variety of general prodrug strategies have been for phosphonic acids (Schultz 2003; Hecker and Erion 2008), there are few reports detailing incorporation of these functionalities into protected amino acid derivatives that are amenable to solid-phase peptide synthesis. Recent examples of the latter include the application of 5’-nitrofuryl-2’-methyl N-(4”-chlorobutyl)phosphonamido ester prodrug strategy to the pTyr mimetic difluorophosphonomethylphenylalanine (F2Pmp) (Boutselis et al. 2007) and to the pSer mimetic, difluorophosphonoaminobutyric acid (Arrendale et al. 2012). In these two cases, the prodrug-protected reagents were used to prepare dipeptides for examination in whole cell studies. The use of these reagents for the solid-phase synthesis of longer peptides is potentially limited by chemical instability of the phosphonamido group to repetitive cycles of piperidine treatment needed for Fmoc-based solid-phase protocols (Boutselis et al. 2007). In the broader sense, there is a paucity of reagents available for the solid-phase synthesis of polypeptides containing prodrug-protected pThr mimetics.
The pivaloyloxymethyl (POM) group is an esterase-labile moiety that has been widely applied to phosphoryl prodrug protection of nucleotides (Hecker and Erion 2008) and phosphate and phosphonic acid functionality in small molecules and peptide mimetics (Stankovic et al. 1997; Mandal et al. 2009; Mandal et al. 2011; Zhao and Etzkorn 2007). However, in spite of its usefulness in these latter contexts, there are no prior reports of polypeptides containing pThr, pSer or their phosphonic acid-based mimetics, bearing POM protection. Given the importance of pThr in a large number of biological processes (Elia and Yaffe 2005), a reagent that would permit the solid-phase synthesis of polypeptides containing POM-protected pThr mimetics would be highly desirable.
We had previously reported the preparation of N-Fmoc-(2S,3R)-2-amino-3-methyl-4-phosphonobutyric acid bis-tert-butyl phopshoryl ester [Fmoc-Pmab(But2)-OH, 1, Fig. 1] as a hydrolytically-stable pThr mimetic bearing orthogonal protection suitable for the standard Fmoc-based synthesis of peptides containing Pmab. However this reagent yields peptides in which the Pmab residue has its phosphonic acid moiety in the free di-anionic form (Liu et al. 2009). To date, there have been no reports of Pmab suitably derivatized for the solid-phase synthesis of polypeptides that maintained the phosphonic acid in a prodrug-protected form. In the current paper we describe the first preparation of Fmoc-Pmab having the phosphonic acid group masked as its POM bis-esters [Fmoc-Pmab(POM)2-OH, 2, Fig. 1] and demonstrate its use in the solid-phase synthesis of a peptide bearing full POM protection of the Pmab residue.
Fig. 1.
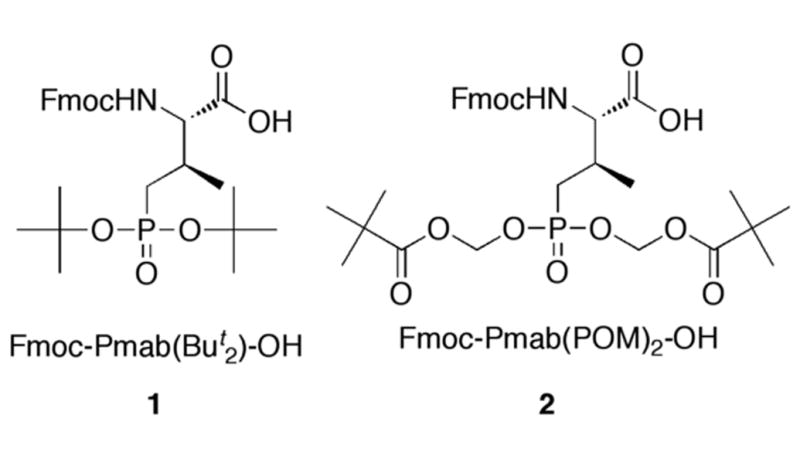
Structures of key reagents
Materials and methods
General methods
All experiments involving moisture-sensitive compounds were conducted under anhydrous conditions. Fmoc-Ser(Trt)-OH, and Fmoc-His(Mtt)-OH were purchased from NovaChioChem. All solvents were purchased in anhydrous form (Aldrich) and used directly. Analytical TLCs were performed using Analtech precoated plates (Uniplate, silica gel GHLF, 250 nm) containing a fluorescence indicator. NMR spectra were recorded using a Varian Inova 400 MHz spectrometer. Coupling constants are reported in hertz, and peak shifts are reported in δ (ppm) relative to TMS. Low-resolution mass spectra (ESI) were measured with an Agilent 260 1200 LC/MSD-SL system. High resolution mass spectra (HRMS) were obtained by positive ion, ESI analysis on a Thermo Scientific LTQ-XL Orbitrap mass spectrometer with HPLC sample introduction using a short narrow-bore C18 reversed-phase column with CH3CN - H2O gradients. Reported m/z values are the average of eight or more scans over the chromatographic peak of interest.
Synthesis of Fmoc-Pmab(POM)2-OH (2)
(2S,3R)-Benzyl 2-(((benzyloxy)carbonyl)amino)-4-(di-tert-butoxyphosphoryl)-3-methylbutanoate (4)
To a solution of 3 (Liu et al. 2009) (0.26 g, 0.57 mmol) in THF–H2O (4 : 1; 5 mL) at 0 °C was added a solution of LiOH•H2O (48 mg, 1.14 mmol) and the mixture was stirred at room temperature until all starting material was consumed as indicated by TLC. The pH was adjusted to 2–3 by the addition of 1N aqueous HCl and THF was removed by evaporation. The residue was extracted (EtOAc) and the combined organic phase was washed with brine, dried (MgSO4), filtered and concentrated. To a solution of the crude residue in DMF (5.0 mL) were added sequentially, NaHCO3 (95 mg, 1.14 mmol) and BnBr (0.10 mL, 0.85 mmol) at room temperature under argon and the mixture was stirred until the starting material was consumed as indicated by TLC. The mixture was diluted with EtOAc and washed with H2O and brine, dried (MgSO4) and filtered and concentrated. Purification by silica gel column chromatography (CH2Cl2 : MeOH from 100:1 to 30:1) afforded 4 as a colorless oil (0.25 g, 80% for two steps, Scheme 1). [α]D21.6 -0.59 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3δ 7.40 – 7.28 (m, 10H), 5.91 (d, J = 8.0 Hz, 1H), 5.20 – 5.06 (m, 4H), 4.38 -4.31 (m, 1H), 2.42 (brs, 1H), 1.84 – 1.69 (m, 1H), 1.57 – 1.48 (m, 1H), 1.47 – 1.45 (m, 18H), 1.10 (d, J = 8.0 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3δ 171.7, 156.4, 136.5, 135.4, 128.8, 128.7, 128.6, 128.3, 82.4 (d, 2JCP = 10 Hz), 82.3 (d, 2JCP = 10 Hz), 67.4, 67.2, 59.8 (d, 3JCP = 10 Hz), 34.0, 32.6, 30.59, 30.56, 17.6 ppm; ESI-HRMS m/z calcd for C28H41NO7P (M+H)+: 534.2621, found: 534.2594.
Scheme 1.
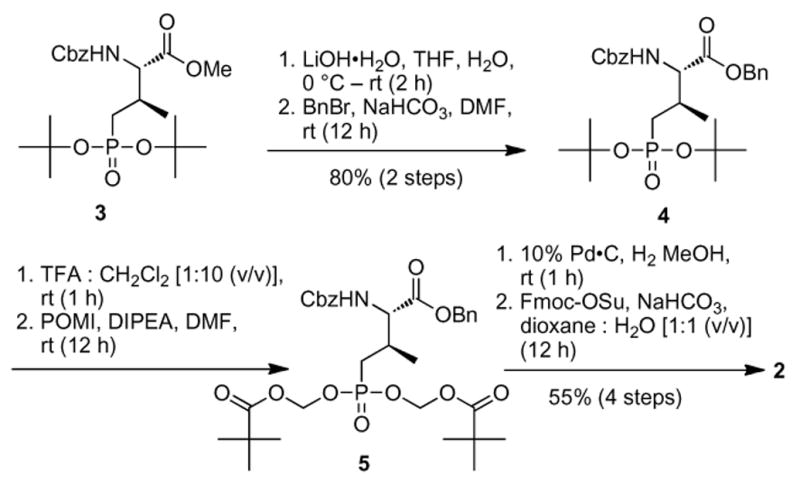
Synthesis of title reagent 2
((((2R,3S)-4-(Benzyloxy)-3-(((benzyloxy)carbonyl)amino)-2-methyl-4-oxobutyl)phosphoryl)bis(oxy))bis(methylene) Bis(2,2-dimethylpropanoate)(5)
A solution of 4 (0.27 g, 0.5 mmol) in 10% TFA (CH2Cl2) was stirred at room temperature (1 h), then volatiles were removed under vacuum and the residue was taken up in DMF with pivaloyloxymethyl iodide (POMI) (Bandgar et al. 2011) (0.32 mL, 2.0 mmol) and DIPEA (0.35 mL, 2.0 mmol) and stirred at room temperature under argon (overnight). The mixture was diluted with H2O, extracted with EtOAc and the combined organic extracts were washed with H2O, and brine, dried (MgSO4), filtered and concentrated. Purification by silica gel chromatography (EtOAc : hexanes from 1:4 to 1:1) afforded 5 as a white semi-solid (0.24 g, 74% yield for two steps, Scheme 1). [α]D21.9 2.88 (c 0.7, CHCl3); 1H NMR (400 MHz, CDCl3δ 7.42 – 7.18 (m, 10H), 5.70 – 5.60 (m, 4H), 5.56 (d, J = 12.0 Hz, 1H), 5.19 (s, 2H), 5.11 (s, 2H), 4.42 – 4.34 (m, 1H), 2.45 (brs, 1H), 2.04 – 1.91 (m, 1H), 1.78 – 1.67 (m, 1H), 1.22 (s, 9H), 1.21 (s, 9H), 1.08 (d, J = 4.0 Hz, 3H) ppm; ; 13C NMR (100 MHz, CDCl3δ 177.1, 171.1, 156.3, 136.3, 135.2, 128.9, 128.8, 128.7, 128.44, 128.35, 81.7 (d, 2JCP = 20 Hz), 67.6, 67.4, 59.2 (d, 3JCP = 10 Hz), 38.9, 31.9, 30.3, 29.9, 29.6 (d, 1JCP = 140 Hz), 27.0, 17.5 ppm; ESI-HRMS m/z calcd for 650.2725, found: 650.2704.
(2S,3R)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-(bis((pivaloyloxy)methoxy)phosphoryl)-3-methylbutanoic Acid [N-Fmoc-Pmab(POM)2-OH] (2)
A solution of 5 (0.18 g, 0.28 mmol) in MeOH was hydrogenated over 10% Pd•C (30 mg) until the reaction was complete as indicated by TLC. The mixture was filtered, evaporated to dryness and reacted with Fmoc-OSu (0.19 g, 0.55 mmol) and NaHCO3 (70 mg, 0.83 mmol) in dioxane : H2O (1:1, 5.5 mL) at room temperature (overnight). The mixture was acidified with 1N HCl, extracted with EtOAc and the combined organic phases were washed with brine, dried (MgSO4), filtered and the filtrate concentrated. Purification by silica gel column chromatography (CH2Cl2 : MeOH from 20:1 to 4:1) provided 2 as a colorless oil (0.14g, 75% yield for two steps, Scheme 1). [α]D21.0 12.8 (c 0.66, CHCl3); 1H NMR (400 MHz, CDCl3δ 7.77 (d, J = 8.0 Hz, 2H), 7.60 (d, J = 8.0 Hz, 2H), 7.40 (t, J = 8.0 Hz, 2H), 7.32 (td, J1 = 8.0 Hz, J2 = 4.0 Hz, 2H), 5.78 – 5.64 (m, 5H), 4.51 – 4.44 (m, 1H), 4.40 (d, J = 8.0 Hz, 2H), 4.22 (t, J = 8.0 Hz, 1H), 2.54 (brs 1H), 2.18 – 2.03 (m, 1H), 1.97 -1.83 (m, 1H), 1.24 (s, 9H), 1.23 (s, 9H), 1.13 (d, J = 8.0 Hz, 3H) ppm; 13C NMR (100 MHz, CDCl3δ 177.2, 172.8, 156.4, 144.0, 143.8, 141.5, 128.0, 127.3, 125.3, 120.2, 82.0, 67.5, 58.3 (d, 3JCP = 10 Hz), 47.3, 39.0, 31.4, 29.4 (d, 1JCP = 140 Hz), 27.0, 17.6 (d, 3JCP = 7 Hz) ppm; ESI-HRMS m/z calcd for C32H42NO11PNa (M+Na)+: 670.2393, found: 670.2376.
Use of reagent 2 for the solid-phase synthesis of peptide 6
Standard Fmoc-protected amino acids were purchased from Novabiochem. Peptides were synthesized on NovaSyn® TG Sieber resin (Novabiochem, cat. no. 01-64-0092) using Fmoc-based solid-phase protocols in N-methyl-2-pyrrolidone (NMP). 1-O-Benzotriazole-N,N,N’,N’-tetramethyl-uronium-hexafluoro-phosphate (HBTU) (5.0 eq.), hydroxybenzotriazole (HOBT) (5.0 eq.) and N,N-diisopropylethylamine (DIPEA) (10.0 eq.) were used as coupling reagents. Reagent 2 was employed for introduction of the first residue. Following completion of peptide elongation and Fmoc-deprotection, amino terminal acetylation was achieved using 1-acetylimidazole. The finished resin (7, Scheme 2) was washed with DMF, MeOH, CH2Cl2 and diethyl ether and then dried under vacuum (overnight). Peptide 6 was cleaved from the resin using 1% TFA in CH2Cl2. The resin was removed by filtration and the filtrate was concentrated under vacuum, then precipitated with cold ether and the precipitate washed with cold ether. The resulting solid was dissolved in 50% aqueous acetonitrile (5 mL) and purified by reverse phase preparative HPLC using a Phenomenex C18 column (21 mm dia x 250 mm, cat. no: 00G-4436-P0) with a linear gradient from 0% aqueous acetonitrile (0.1% TFA) to 100% acetonitrile (0.1% trifluoroacetic acid) over 30 minutes at a flow rate of 10.0 mL/minute. Lyophilization gave peptide 6 as a white powder (>99% pure by HPLC, Scheme 2). ESI-MS m/z calcd for C39H66N8O14P (M+H)+: 901.4, found: 901.4.
Scheme 2.
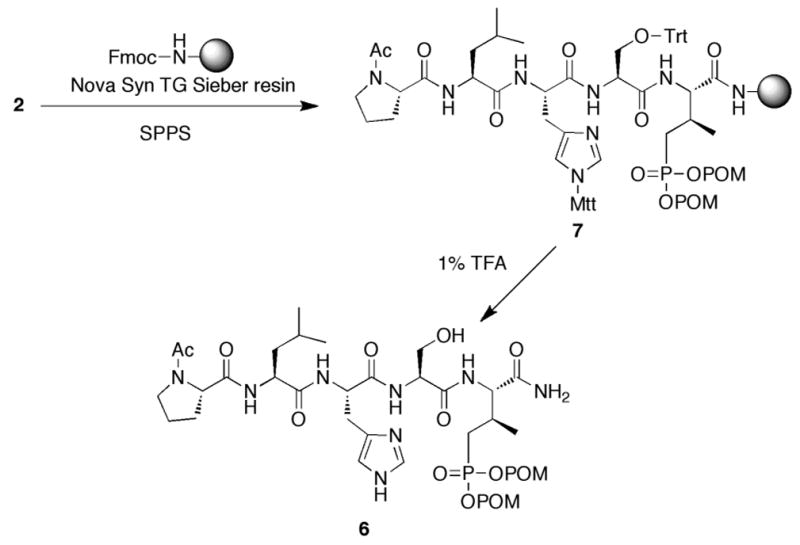
Synthesis of bis-POM-protected peptide 6 using reagent 2
Pig liver esterase hydrolysis studies on peptide 6
Following literature procedures (Srivastva and Farquhar 1984), 0.05M potassium phosphate buffer (pH 7.4) was placed in a centrifuge tube and a solution of peptide 6 in MeOH was added to achieve a concentration of 200 μM of 6 with MeOH being less than 1%. To a 1 mL aliquot of the above solution was added pig liver esterase (57.6 units) and the reaction mixture was incubated at 37 °C with gentle agitation. At various time points, aliquots of the reaction mixture (50 μL) were transferred to an Eppendorf tube containing MeCN (50 μL). Following filtration, the hydrolysis product was monitored by LC-MS. Data are shown in Fig. 2.
Fig. 2.
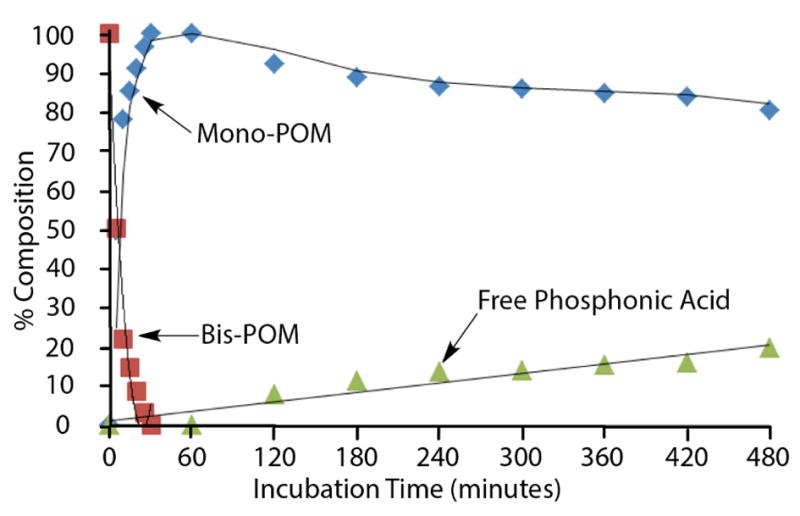
Results of pig liver esterase treatment of peptide 6
Results and discussion
Preparation of reagent 2 began with Cbz-Pmab(But2)-OMe (3), which is an intermediate in our previously reported synthesis of 1 (Liu et al. 2009). Benzyl trans-esterification of 3 to 4 was accomplished by initial LiOH-mediated hydrolysis of the methyl ester followed by reaction of the free acid with benzyl bromide (NaHCO3 in DMF, 80% yield for two steps, Scheme 1). Subsequent treatment of 4 with pivaloyloxymethyl iodide (POMI) (Bandgar et al. 2011) and diisopropylethyl amine (DIPEA) in DMF provided the corresponding bis-POM-protected intermediate (5). Hydrogenolytic removal of amino and carboxyl protecting groups and introduction of N-Fmoc-protection by treatment with 9-fluorenylmethyl succinimidyl carbonate (Fmoc-OSu) in aqueous THF with NaHCO3, gave the desired reagent 2 (55% yield from 4) (Scheme 1).
In order to demonstrate the usefulness of 2 for the incorporation of Pmab(POM)2 into peptides by solid-phase techniques, we chose the sequence, Ac-Pro-Leu-His-Ser-Pmab-amide, which has previously shown to bind with good affinity to the polo-like kinase 1 (Plk1) polo box domain (PBD) (Yun et al. 2009; Liu et al. 2011; Liu et al. 2012b, a). PBD-binding ligands may potentially serve as anticancer agents by blocking the spatial organization required for Plk1 to function in oncogenic processes (van de Weerdt et al. 2008). The syntheses of Ac-Pro-Leu-His-Ser-Pmab(POM)2-amide (6) was accomplished on NovaSyn TG Sieber resin using standard Fmoc protocols. Histidine and serine were employed in their 4-methoxytrityl (Mtt) and trityl (Trt) side chain-protected forms, respectively, to allow their cleavage under mildly acidic conditions (Scheme 2). Following synthesis completion, the resin-bound Ac-Pro-Leu-His(Mtt)-Ser(Trt)-Pmab(POM)2-amide (7) was subjected to treatment with 1% TFA, which resulted in removal of histidine and serine protecting groups and cleavage of the peptide from the resin with retention of Pmab-bis-POM functionality. Purification by HPLC provided Ac-Pro-Leu-His-Ser-Pmab(POM)2-amide (6) as a white solid (Scheme 2).
In order to examine the bio-reversibility of the Pmab POM protection of 6, we performed in vitro assays using porcine liver estase (PLE) in phosphate buffer at pH 7.4 (Srivastva and Farquhar 1984). We observed that the parent Pmab bis-POM-containing 6 was rapidly converted to the mono-POM-containing product (t1/2 ≈ 5 minutes) with further deprotection to the free Pmab-containing peptide occurring at a slower rate (20% conversion over 8 h) (Fig. 2). Reduction in the rate of enzymatic cleavage of a second POM group is known (Srivastva and Farquhar 1984).
Conclusions
Our current paper presents the synthesis of a reagent [Fmoc-Pmab(POM)2-OH (2)] that allows the facile synthesis of peptides containing the phosphatase-stable pThr mimetic, Pmab, bearing bio-reversible POM protection. This represents a rare example of a reagent that allows the solid-phase synthesis of polypeptides having a pThr mimetic in bio-reversible prodrug form. Reagent 2 should find utility in a variety of pharmacological applications.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, Center for Cancer Research, NCI-Frederick and the National Cancer Institute, National Institutes of Health.
Abbreviations
- Ac
Acetyl
- [α]D
Specific rotation
- BnBr
Benzyl bromide
- But
tert-Butyl
- Cbz
Benzyloxycarbonyl
- CH2Cl2
Dimethyl chloride
- CH3CN
Acetonitrile
- DIPEA
Diisopropylethylamine
- DMF
Dimethylformamide
- ESI
Electrospray ionization
- EtOAc
Ethyl acetate
- F2Pmp
Difluorophosphonomethylphenylalanine
- Fmoc
9-Fluorenylmethoxycarbonyl
- Fmoc-OSu
9-Fluorenylmethyl succinimidyl carbonate
- HBTU
1-O-Benzotriazole-N,N,N’,N’-tetramethyl-uromium-hexafluoro-phosphate
- HCl
Hydrochloric acid
- His
Histidine
- HOBt
1-Hydroxybenzotriazole
- HPLC
High-performance liquid chromatography
- HRMS
High resolution mass spectra
- Leu
Leucine
- LiOH
Lithium hydroxide
- MeOH
Methanol
- MgSO4
Magnesium sulfate
- Mtt
Methyltrityl
- m/z
Mass-to-charge ratio
- NaHCO3
Sodium bicarbonate
- NMP
N-Methyl-2-pyrrolidone
- NMR
Nuclear magnetic resonance
- PBD
polo box domain
- Pd•C
Palladium on carbon
- PLE
Porcine liver esterase
- Plk1
Polo-like kinase 1
- Pmab
(2S,3R)-2-Amino-3-methyl-4-phosphonobutyric Acid
- POM
Pivaloyloxymethyl
- POMI
Pivaloyloxymethyl iodide
- PPIs
Protein-protein interactions
- pSer
Phosphoserine
- pThr
Phosphothreonine
- pTyr
Phosphotyrosine
- Ser
Serine
- SPPS
Solid-phase peptide synthesis
- t ½
Half life
- TFA
Trifluoroacetic acid
- THF
Tetrahydrofuran
- TLC
Thin layer chromatography
- TMS
Trimethylsilane
- Trt
Trityl
Footnotes
Conflict of interest
The authors have declared no conflict of interest.
Electronic supplementary material
The online version of the article (doi:) contains the 1H and 13C NMR spectra of compounds 2 – 5 and the analytical HPLC of peptide 6, which is available to authorized users.
References
- Arrendale A, Kim K, Choi J, Li W, Geahlen RL, Borch RF. Synthesis of a phosphoserine mimetic prodrug with potent 14-3-3 protein inhibitory activity. Chem Biol. 2012;19:764–771. doi: 10.1016/j.chembiol.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandgar BP, Sarangdhar RJ, Viswakarma S, Ahamed FA. Synthesis and biological evaluation of orally active prodrugs of indomethacin. J Med Chem. 2011;54:1191–1201. doi: 10.1021/jm101085j. [DOI] [PubMed] [Google Scholar]
- Berkowitz DB, Eggen M, Shen Q, Shoemaker RK. Ready access to fluorinated phosphonate mimics of secondary phosphates Synthesis of the (α,α–difluoroalkyl)phosphonate analogs of L-phosphoserine, L-phosphoallothreonine, and L-phosphothreonine. J Org Chem. 1996;61:4666–4675. doi: 10.1021/jo9604752. [DOI] [PubMed] [Google Scholar]
- Boutselis IG, Yu X, Zhang Z-Y, Borch RF. Synthesis and cell-based activity of a potent and selective protein tyrosine phosphatase 1B inhibitor prodrug. J Med Chem. 2007;50:856–864. doi: 10.1021/jm061146x. [DOI] [PubMed] [Google Scholar]
- Burke TR, Jr, Lee K. Phosphotyrosyl mimetics in the development of signal transduction inhibitors. Acc Chem Res. 2003;36:426–433. doi: 10.1021/ar020127o. [DOI] [PubMed] [Google Scholar]
- Eisele F, Owen DJ, Waldmann H. Peptide conjugates as tools for the study of biological signal transduction. Bioorg Med Chem. 1999;7:193–224. doi: 10.1016/s0968-0896(98)00204-1. [DOI] [PubMed] [Google Scholar]
- Elia AEH, Yaffe MB. Phosphoserine/threonine binding domains. In: Cesare G, editor. Modulr Protein Domain. Wiley-VCH Verlag GmbH & Co KGaA; Weinheim Germany: 2005. pp. 163–179. [DOI] [Google Scholar]
- Hecker SJ, Erion MD. Prodrugs of phosphates and phosphonates. J Med Chem. 2008;51:2328–2345. doi: 10.1021/jm701260b. [DOI] [PubMed] [Google Scholar]
- Ladbury JE. Protein-protein recognition in phosphotyrosine-mediated intracellular signaling. Protein Rev. 2005;3:165–184. [Google Scholar]
- Liu F, Park J-E, Lee KS, Burke TR., Jr Preparation of orthogonally protected (2S,3R)-2-amino-3-methyl-4-phosphonobutyric acid (Pmab) as a phosphatase-stable phosphothreonine mimetic and its use in the synthesis of polo-box domain-binding peptides. Tetrahedron. 2009;65:9673–9679. doi: 10.1016/j.tet.2009.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Park J-E, Qian W-J, Lim D, Graber M, Berg T, Yaffe MB, Lee KS, Burke TR., Jr Serendipitous alkylation of a Plk1 ligand uncovers a new binding channel. Nat Chem Biol. 2011;7:595–601. doi: 10.1038/nchembio.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Park J-E, Qian W-J, Lim D, Scharow A, Berg T, Yaffe MB, Lee KS, Burke TR. Identification of high affinity polo-like kinase 1 (Plk1) polo-box domain binding peptides using oxime-based diversification. ACS Chem Biol. 2012a;7:805–810. doi: 10.1021/cb200469a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Park J-E, Qian W-J, Lim D, Scharow A, Berg T, Yaffe MB, Lee KS, Burke TR. Peptoid-peptide hybrid ligands targeting the polo box domain of polo-like kinase 1. ChemBioChem. 2012b;13:1291–1296. doi: 10.1002/cbic.201200206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CHS, Liu K, Tan LP, Yao SQ. Current chemical biology tools for studying protein phosphorylation and dephosphorylation. Chem Eur J. 2012;18:28–39. doi: 10.1002/chem.201103206. [DOI] [PubMed] [Google Scholar]
- Mandal PK, Gao F, Lu Z, Ren Z, Ramesh R, Birtwistle JS, Kaluarachchi KK, Chen X, Bast RC, Liao WS, McMurray JS. Potent and selective phosphopeptide mimetic prodrugs targeted to the Src homology 2 (SH2) domain of signal transducer and activator of transcription 3. J Med Chem. 2011;54:3549–3563. doi: 10.1021/jm2000882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal PK, Liao WSL, McMurray JS. Synthesis of phosphatase-stable, cell-permeable peptidomimetic prodrugs that target the SH2 domain of Stat3. Org Lett. 2009;11:3394–3397. doi: 10.1021/ol9012662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair SA, Lee B, Hangauer DG. Synthesis of orthogonally protected L-homocysteine and L-2-amino-4-phosphonobutanoic acid from L-homoserine. Synthesis. 1995:810–814. [Google Scholar]
- Otaka A, Mitsuyama E, Kinoshita T, Tamamura H, Fujii N. Stereoselective synthesis of CF2-substituted phosphothreonine mimetics and their incorporation into peptides using newly developed deprotection procedures. J Org Chem. 2000;65:4888–4899. doi: 10.1021/jo000169v. [DOI] [PubMed] [Google Scholar]
- Panigrahi K, Eggen M, Maeng J-H, Shen Q, Berkowitz DB. The alpha, alpha-difluorinated phosphonate L-pSer-analogue: An accessible chemical tool for studying kinase- dependent signal transduction. Chem Biol. 2009;16:928–936. doi: 10.1016/j.chembiol.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perich JW. Efficient Fmoc/solid-phase synthesis of Abu(P)-containing peptides using Fmoc-Abu(PO3Me2)-OH. Int J Pept Protein Res. 1994;44:288–294. doi: 10.1111/j.1399-3011.1994.tb00172.x. [DOI] [PubMed] [Google Scholar]
- Schultz C. Prodrugs of biologically active phosphate esters. Bioorg Med Chem. 2003;11:885–898. doi: 10.1016/s0968-0896(02)00552-7. [DOI] [PubMed] [Google Scholar]
- Shapiro G, Buechler D, Ojea V, Pombo-Villar E, Ruiz M, Weber HP. Synthesis of both D- and L-Fmoc-Abu[PO(OCH2CH:CH2)2]-OH for solid phase phosphonopeptide synthesis. Tetrahedron Lett. 1993;34:6255–6258. [Google Scholar]
- Srivastva DN, Farquhar D. Bioreversible phosphate protective groups: synthesis and stability of model acyloxymethyl phosphates. Bioorg Chem. 1984;12:118–129. [Google Scholar]
- Stankovic CJ, Surendran N, Lunney EA, Plummer MS, Para KS, Shahripour A, Fergus JH, Marks JS, Herrera R, Hubbell SE, Humblet C, Saltiel AR, Stewart BH, Sawyer TK. The role of 4-phosphonodifluoromethyl- and 4-phosphono-phenylalanine in the selectivity and cellular uptake of SH2 domain ligands. Bioorg Med Chem Lett. 1997;7:1909–1914. [Google Scholar]
- van de Weerdt BCM, Littler DR, Klompmaker R, Huseinovic A, Fish A, Perrakis A, Medema RH. Polo-box domains confer target specificity to the Polo-like kinase family. Biochim Biophys Acta, Mol Cell Res. 2008;1783:1015–1022. doi: 10.1016/j.bbamcr.2008.02.019. [DOI] [PubMed] [Google Scholar]
- Yaffe MB. Phosphotyrosine-binding domains in signal transduction. Nat Rev Mol Cell Biol. 2002;3:177–186. doi: 10.1038/nrm759. [DOI] [PubMed] [Google Scholar]
- Yun S-M, Moulaei T, Lim D, Bang JK, Park J-E, Shenoy SR, Liu F, Kang YH, Liao C, Soung N-K, Lee S, Yoon D-Y, Lim Y, Lee D-H, Otaka A, Appella E, McMahon JB, Nicklaus MC, Burke TR, Jr, Yaffe MB, Wlodawer A, Lee KS. Structural and functional analyses of minimal phosphopeptides targeting the polo-box domain of polo-like kinase 1. Nat Struct Mol Biol. 2009;16:876–882. doi: 10.1038/nsmb.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Etzkorn FA. A phosphorylated prodrug for the inhibition of Pin1. Bioorg Med Chem Lett. 2007;17:6615–6618. doi: 10.1016/j.bmcl.2007.09.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.