

Abstract
The DDR1 receptor tyrosine kinase is activated by matrix collagens and has been implicated in numerous cellular functions such as proliferation, differentiation, adhesion, migration, and invasion. Here we report the discovery of a potent and selective DDR1 inhibitor, DDR1-IN-1, and present the 2.2 Å DDR1 co-crystal structure. DDR1-IN-1 binds to DDR1 in the ‘DFG-out’ conformation and inhibits DDR1 autophosphorylation in cells at submicromolar concentrations with good selectivity as assessed against a panel of 451 kinases measured using the KinomeScan technology. We identified a mutation in the hinge region of DDR1, G707A, that confers >20-fold resistance to the ability of DDR1-IN-1 to inhibit DDR1 autophosphorylation and can be used to establish what pharmacology is DDR1-dependent. A combinatorial screen of DDR1-IN-1 with a library of annotated kinase inhibitors revealed that inhibitors of PI3K and mTOR such as GSK2126458 potentiate the antiproliferative activity of DDR1-IN-1 in colorectal cancer cell lines. DDR1-IN-1 provides a useful pharmacological probe for DDR1-dependent signal transduction.
The discoidin domain receptor (DDR) kinases bind to several collagens and have been shown to be key regulators of cellular morphogenesis, differentiation, proliferation, adhesion, migration, and invasion.1 There are two types of DDR kinases, DDR1 and DDR2, which are characterized by an approximately 155-aa discoidin homology domain in the extracellular region of the protein. DDR1 is primarily expressed in epithelial cells of a variety of tissues, whereas DDR2 is expressed in interstitial cells. DDR1 was originally identified in a screen for tyrosine kinase proteins expressed in human malignancies.2 Recent studies have reported altered expression of DDR1 in human tumors, including lung, esophagous, breast, ovary, and pediatric brain cancers, suggesting a potential role for DDR1 in tumor progression.3−6 Moreover, the elevated expression of DDR1 in a number of fast-growing invasive tumors has suggested that this matrix-activated RTK may be involved in the proliferation of and stromal invasion by tumor cells.7 The precise mechanisms by which this receptor may contribute to oncogenesis is unknown; however, given its important role in transmitting signals from the extracellular matrix (ECM), it has been postulated that it may act as a critical regulator of cell proliferation, adhesion, migration, and subsequent tumor metastasis.8 Recently, DDR1 was identified as one of several major activated tyrosine kinases carrying somatic mutations in non-small cell lung tumors as well as in acute myeloid leukemia.9,10 Moreover, through a chemical proteomics approach, DDR1 was identified as a previously unanticipated target of imatinib, a clinically approved multitargeted inhibitor of Bcr-Abl, c-Kit, and PDGFR,11 which raises the possibility that inhibition of DDR1 could contribute to a subset of pharmacological effects of the drug. Overexpression of DDR1 in several human cancer cell lines enhanced anchorage-independent growth and tumorigenic potential in nude mice.12 In addition, knock-down of DDR1 may suppress the tumorigensis in vitro and in vivo.12 The identity of direct DDR1 substrates and downstream effectors is currently unknown. DDR1 is a direct p53 transcriptional target and is essential for survival of wild-type p53 cells when challenged with genotoxic stress, suggesting that inhibition of DDR1 function may provide a potential approach to selectively enhance treatment of such tumors.13 In order to determine the pharmacological consequences of acute inhibition of DDR1 kinase activity in a variety of cancer cell lines, we sought to develop potent and selective inhibitors.
It has been reported that the clinically approved BCR-ABL kinase inhibitors imatinib, nilotinib, and dasatinib are also potent inhibitors of DDR1 and DDR2.14,15 However, these drugs potently target a number of other important kinases, making them difficult to use as pharmacological probes of DDR1-dependent cellular phenomena.16 Recently Ding et al. reported the development of pyrazolopyrimidine derivatives that inhibit DDR1 kinase activity with an IC50 of 6.8 nM and exhibit good selectivity using the KinomeScan approach (S(10) = 0.008 at 0.1 μM).17 Noting imatinib and nilotinib as typical type II kinase inhibitors, we used this structural information in conjunction with a general pharmacophore model for type II kinase inhibitors to develop a library of potential kinase inhibitors (Figure 1A).18 This pharmacophore model divides the inhibitors into three sections: a ‘head’ hinge interacting motif that occupies the adenine portion of the ATP-pocket, a ‘linker’ motif that traverses the area proximal to the ‘gatekeeper’ position, and a ‘tail’ motif that occupies the region created by the flip of the ‘DFG’ sequence of the activation loop. A collection of close to 100 type II inhibitors created by this approach was screened across a panel of 451 kinases using the KinomeScan approach, which resulted in the identification of DDR1-IN-1 and DDR1-IN-2 as two chemotypes that possessed potent and selective binding to DDR1 (Figure 1B).
Figure 1.

Developing selective type II kinase inhibitors. (A) Docking imatinib into the X-ray co-crystal structure of DDR1. (B) Chemical structure of DDR1-IN-1/2 and representative developing rationale. (C) DDR1-IN-1 efficacy on blocking DDR1 autophosphorylation. (D) DDR1-IN-2 efficacy on blocking DDR1 autophosphorylation. (E) KinomeScanTreeSpot description of the DDR1-IN-1/2 selectivity profile.
We confirmed the observed binding to DDR1 using an enzymatic kinase assay employing the Lanthascreen technology. In this assay DDR1-IN-1 exhibits an IC50 of 105 nM against DDR1 and 413 nM against DDR2. DDR1-IN-2 exhibits an IC50 of 47 nM against DDR1 and 145 nM against DDR2 (Table 1). In order to determine whether these inhibitors could inhibit DDR1 kinase activity in cells, we measured their ability to block collagen induced DDR1 autophosphorylation in U2OS cells. DDR1-IN-1 and DDR1-IN-2 inhibited basal DDR1 autophosphorylation with an EC50 of 86 nM and 9 nM, respectively (Figure 1C,D). To determine whether inhibitor efficacy was maintained against activated DDR1, we compared the ability of the two compounds to inhibit DDR1 autophosphorylation in cells in the presence and absence of collagen stimulation. Both DDR1-IN-1 and DDR1-IN-2 demonstrated weaker inhibition of DDR1 autophosphorylation in the absence of collagen stimulation (Supplemental Figure 1).
Table 1. Biochemical Characterization of DDR1-IN-1/2.
| IC50 (nM) |
||||
|---|---|---|---|---|
| nilotinib | imatinib | DDR1-IN-1 | DDR1-IN-2 | |
| DDR1 | 43 | 337 | 105 | 47.6 |
| DDR2 | 0.5 | 675 | 413 | 145 |
DDR1-IN-1 exhibits excellent selectivity for DDR1 with a selectivity score (S(1) at 1 μM) of 0.01 as assessed using the KinomeScan approach with binding observed for ABL, KIT, and PDGFRβ, none of which were confirmed using enzymatic assays (Figure 1E and Supplemental Tables 1 and 2). In contrast, DDR1-IN-2 is considerably less selective with a selectivity score (S(1) at 1 μM) of 0.07 and potential additional targets including Abl, BLK, CSK, EGFR, LCK, and PDGFRβ (Supplemental Tables 3 and 4).
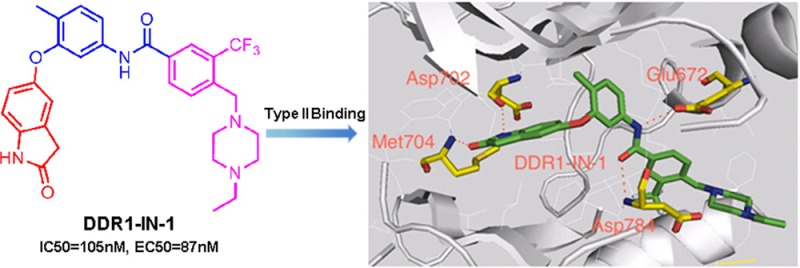
Despite the excellent kinase selectivity observed for DDR1-IN-1, there always remains the possibility that other unanticipated and functionally important intracellular targets exist. We therefore sought to develop an ‘inhibitor-resistant’ mutant of DDR1 that could be used as a ‘chemical-genetic’ means to evaluate inhibitor selectivity. To confirm the inhibitor binding mode and select the key drug-resistant amino acid residue, we determined the 2.2 Å co-crystal structure of DDR1 in complex with DDR1-IN-1, which confirmed the presumed type II binding mode (Figure 2A and Supplemental Table 5). As expected, two hydrogen bonds formed in the hinge binding area with Met704 and Asp702. Glu672 in the c-Helix and Asp784 in DFG motif formed two hydrogen bonds with the linker amide moiety. One potential drug-resistant mutation site is the ‘gatekeeper’ residue Thr701; indeed the corresponding gatekeeper mutation in BCR-ABL, PDGF, and c-KIT has been demonstrated to confer resistance to type II inhibitors such as imatinib and nilotinib. However, this mutation in DDR1 leads to a loss of enzymatic activity (Supplemental Figure 2). We therefore chose to test an alternative mutation in the hinge region, G707A, where modeling and previous efforts have demonstrated that inhibitor resistance can be conferred.19 Introduction of G707A into DDR1 resulted in a 20- and 100-fold increase in the EC50 with which DDR1-IN-1 and DDR1-IN-2 could inhibit DDR1 autophosphorylation in cells (Figure 2B,C).19 Docking DDR1-IN-2 into the DDR1 crystal structure with a modeled G707A mutation suggested that the side-chain methyl group of alanine would potentially clash with the azaindole ring of DDR1-IN-2 (Figure 2D,E). In addition, the model suggests that gatekeeper residue Thr701 may form a hydrogen bond with nitrogen between the “ Head” and “Linker” region of DDR1-IN-2, which may explain its ability to more potently inhibit DDR1 relative to DDR1-IN-1 (Figure 2D,A).
Figure 2.

Binding information of DDR1-IN-1/2 against DDR1. (A) X-ray co-crystal structure of DDR1-IN-1 with DDR1 kinase. (B) G707A mutation rescued the DDR1-IN-1 inhibitory effect against DDR1 in U2OS cell line. (C) G707A mutation rescued the DDR1-IN-2 inhibitory effect against DDR1 in U2OS cell line. (D) DDR1-IN-2 docked into the DDR1 kinase. (E) DDR1-IN-2 docked into the DDR1 G707A mutation.
We next used DDR1-IN-1 and DDR1-IN-2 to assess the consequences of inhibiting DDR1 activity in a panel of different cancer cell lines that have been reported to possess DDR1 gain-of-function mutations and/or overexpression.1 To our surprise, the more selective DDR1 inhibitor, DDR1-IN-1, did not inhibit proliferation below a concentration of 10 μM, while the multitargeted inhibitor, DDR1-IN-2, inhibited the proliferation of a variety of cell lines at single digit micromolar and lower concentrations, although both inhibitors did affect the autophosphorylation of the DDR1 during the drug treatment (Table 2 and Supplemental Figure 3). In order to assess whether the antiproliferative potency of DDR1-IN-2 was a consequence of inhibition of DDR1, we transiently expressed the G707A DDR1 mutation to U2OS cells. Introduction of this mutation did not change the antiproliferative potency of DDR1-IN-2 (Supplemental Figure 4 and Supplemental Table 6). These results suggest that acute inhibition of DDR1 kinase activity is insufficient to inhibit the proliferation of these cancer cell lines under the conditions investigated.
Table 2. DDR1-IN-1/2 Anti-proliferation Effect on Cancer Cell Lines.
| cell linesa | origin | DDR1-IN-1 (μM) | DDR1-IN-2 (μM) |
|---|---|---|---|
| HCT-116(OWT) | colon | 8.7 | 0.81 |
| T47D(OWT) | breast | >10 | 1.11 |
| A549(WT) | lung | >10 | 2.29 |
| H1975(WT) | lung | >10 | 0.89 |
| SaoS2(WT) | sarcoma | 8.14 | 0.39 |
| SkBr3(WT) | breast | >10 | 3.07 |
| SW480(OWT) | colon | >10 | 2.76 |
| SNU-1040(OMT) | colorectal | >10 | 3.52 |
| EJ(WT) | uterus | >10 | 4.60 |
| SNU-449(OMT) | HCC | >10 | 1.10 |
OWT = overexpression of wide type, WT = wide type, OMT = overexpression of mutant type.
We next sought to identify which kinase targets might potentiate the effects of DDR1 inhibition. This would guide potential combinatorial therapy and suggest what pathways might dynamically compensate for acute loss of DDR1 signaling. To accomplish this, we performed a 3-day combinatorial kinase inhibitor screening where we combined DDR1-IN-1 in a pairwise fashion with 200 different well characterized kinase inhibitors (LINCS library, see: https://lincs.hms.harvard.edu) targeting a variety of pathways. We selected the SNU-1040 cell line for the combinatorial screen since it is one of the most resistant to both DDR1-IN-1 and 2 (GI50: >10 μM and 3.52 μM, respectively, and Supplemental Figure 5). DDR1-IN-1 was fixed at a concentration of 1 μM, which is sufficient to inhibit DDR1 autophosphorylation. The combinatorial screen revealed a number of inhibitors targeting kinases such as EGFR, Src, Cdk1, mTOR, and PI3K that appeared to positively combine with DDR1-IN-1 to inhibit cell growth. A similar screen was performed with DDR1-IN-2, which revealed inhibitors of 12 targets able to positively combine with DDR1-IN-2. PI3K and mTOR inhibitors were revealed by screening of a kinase inhibitor chemical library to positively combine with both DDR1-IN-1 and DDR1-IN-2 (Figure 3A,B and Supplemental Tables 7 and 8). Full dose–response curves were generated for DDR1-IN-1 and DDR1-IN-2, both alone and in combination with GSK2126458 and AZD8055, respectively. These studies revealed positive combination effects between DDR1-IN-1 and GSK2126458 as well as between DDR1-IN-2 and AZD8055. Calcusyn analysis of the DDR1-IN-2+AZD8055 revealed synergy (Figure 3C,D,E).
Figure 3.

Combinatorial Screening of DDR1-IN-1/2 with the LINCS library against the SNU-1040 cell line. (A) Positive hits with DDR1-IN-1. CSF1R/DDR1/EGFR/TIE1/PDGFR2 = WZ-4-145; mTOR = WYE-125132; Cdk1/CyclinB = CGP60474; Chk = PF477736; PI3K = PI103; PI3K/mTOR = GSK2126458; Src/BCR-ABL = AP24534; EGFR = EKB-569; mTOR = Torin2. (B) Positive hits with DDR1-IN-2. CSF1R/DDR1/EGFR/TIE1/PDGFR2 = WZ-4-145; mTOR = WYE-125132; CLK2/CNSK1E/FLT3/ULK1 = WZ3105; Akt1 = A443644; LRRK2 = XMD11-50; mTOR = AZD8055; c-MET = ARQ197; CDK = BMS-387032; PI3K = GSK1059615; PLK1 = GW843682; MEK = AZD6244; mTOR = Torin2. (C) Combinatorial effect of DDR1-IN-1 with GSK2126458. (D) Synergistic effect of DDR1-IN-2 with AZD8055. (E) Combinatorial effect on the blockade of signaling pathway factors.
Discussion
Receptor tyrosine kinases (RTKs) are critical transducers of extracellular cues to intracellular signaling networks that integrate and orchestrate key cellular decisions such as migration, proliferation, differentiation, and survival.20 Deregulation of the RTKs is often observed in pathological associations such as cancers. Numerous mutations have been reported in DDR1, but whether it may represent an attractive anticancer target is currently unclear. In this paper we implemented a focused medicinal chemistry library approach to discover two new potent DDR1 kinase inhibitors. DDR1-IN-1 is a selective DDR1 kinase inhibitor that through kinome-wide selectivity profiling and biochemical assay confirmation was not observed to target other protein kinases when used at concentrations of less than 10 μM. DDR1-IN-2 is a more potent DDR1 kinase inhibitor but also potently inhibits a number of additional kinase targets. In addition, there were apparent effects on the downstream signaling of DDR1. However, the most selective DDR1 kinase inhibitor DDR1-IN-1 does not seem to have significant antiproliferation effects on DDR1 deregulated cancer cell lines, whereas the multitarget inhibitor DDR1-IN-2 shows a more profound antigrowth effect. This might indicate that DDR1-IN-1 may be an incomplete inhibitor of DDR1-mediated signaling perhaps because other kinases such as the Src-family of kinases, which are known to associate with DDR1 also contribute to DDR1-dependent signaling.21 The stronger antiproliferative effect of DDR1-IN-2 is likely a consequence of its ability to potently inhibit several kinases other than DDR1. Other multitargeted inhibitors such as imatinib, which inhibits Abl, c-Kit, PDGFR, and DDR1, do not possess strongly antiproliferative effects, so it remains to be determined which additional kinase targets of DDR1-IN-2 contribute to its antiproliferative potency (Supplemental Figure 6). A combinatorial kinase inhibitor screening revealed that inhibition of either the PI3K or mTOR can potentiate the effect of DDR1 inhibition. These findings suggested that the DDR1 kinase activity alone may be insufficient to inhibit cell proliferation and that DDR1 directed therapy will need to be deployed in a combinatorial format. Further investigation is required to determine whether inhibition of DDR1 kinase activity can impact other phenomena such as cell adhesion, migration, or invasion. Given the fact that the pathological role of DDR1 is not yet understood, we believe the new potent and selective inhibitors DDR1-IN-1/2 will serve as useful pharmacological tools to help the dissect its functions and lay the foundation for the development of DDR1 targeted anticancer therapies.
Acknowledgments
We thank National Institutes of Health (U54 HG006097 and CA154303) for support of L.T., H.G.C., J.W., Q.L., and N.S.G. and the Chinese Academy of Sciences for training grant in support of F.L. and W.H.; H.-G.K., S.A.E., and S.W.L. were supported by NIH grants and MGH Interim funds. The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Boehringer Ingelheim, the Canada Foundation for Innovation, the Canadian Institutes for Health Research, Genome Canada, GlaxoSmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and the Wellcome Trust [092809/Z/10/Z]
Supporting Information Available
Supplemental experimental procedures, tables, figures, and references. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
# These authors contribute equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Valiathan R. R.; Marco M.; Leitinger B.; Kleer C. G.; Fridman R. (2012) Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev. 31, 295–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foehr E. D.; Tatavos A.; Tanabe E.; Raffioni S.; Goetz S.; Dimarco E.; De Luca M.; Bradshaw R. A. (2000) Discoidin domain receptor 1 (DDR1) signaling in PC12 cells: activation of juxtamembrane domains in PDGFR/DDR/TrkA chimeric receptors. FASEB J. 14, 973–981. [DOI] [PubMed] [Google Scholar]
- Yamanaka R.; Arao T.; Yajima N.; Tsuchiya N.; Homma J.; Tanaka R.; Sano M.; Oide A.; Sekijima M.; Nishio K. (2006) Identification of expressed genes characterizing long-term survival in malignant glioma patients. Oncogene 25, 5994–6002. [DOI] [PubMed] [Google Scholar]
- Johansson F. K.; Goransson H.; Westermark B. (2005) Expression analysis of genes involved in brain tumor progression driven by retroviral insertional mutagenesis in mice. Oncogene 24, 3896–3905. [DOI] [PubMed] [Google Scholar]
- Heinzelmann-Schwarz V. A.; Gardiner-Garden M.; Henshall S. M.; Scurry J.; Scolyer R. A.; Davies M. J.; Heinzelmann M.; Kalish L. H.; Bali A.; Kench J. G.; Edwards L. S.; Vanden Bergh P. M.; Hacker N. F.; Sutherland R. L.; O’Brien P. M. (2004) Overexpression of the cell adhesion molecules DDR1, Claudin 3, and Ep-CAM in metaplastic ovarian epithelium and ovarian cancer. Clin. Cancer Res. 10, 4427–4436. [DOI] [PubMed] [Google Scholar]
- Weiner H. L.; Huang H.; Zagzag D.; Boyce H.; Lichtenbaum R.; Ziff E. B. (2000) Consistent and selective expression of the discoidin domain receptor-1 tyrosine kinase in human brain tumors. Neurosurgery 47, 1400–1409. [PubMed] [Google Scholar]
- Park H. S.; Kim K. R.; Lee H. J.; Choi H. N.; Kim D. K.; Kim B. T.; Moon W. S. (2007) Overexpression of discoidin domain receptor 1 increases the migration and invasion of hepatocellular carcinoma cells in association with matrix metalloproteinase. Oncol. Rep. 18, 1435–1441. [PubMed] [Google Scholar]
- Ford C. E.; Lau S. K.; Zhu C. Q.; Andersson T.; Tsao M. S.; Vogel W. F. (2007) Expression and mutation analysis of the discoidin domain receptors 1 and 2 in non-small cell lung carcinoma. Br. J. Cancer 96, 808–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasson M. H.; Xiang Z.; Walgren R.; Zhao Y.; Kasai Y.; Miner T.; Ries R. E.; Lubman O.; Fremont D. H.; McLellan M. D.; Payton J. E.; Westervelt P.; DiPersio J. F.; Link D. C.; Walter M. J.; Graubert T. A.; Watson M.; Baty J.; Heath S.; Shannon W. D.; Nagarajan R.; Bloomfield C. D.; Mardis E. R.; Wilson R. K.; Ley T. J. (2008) Somatic mutations and germline sequence variants in the expressed tyrosine kinase genes of patients with de novo acute myeloid leukemia. Blood 111, 4797–4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H.; Hunter C.; Smith R.; Stephens P.; Greenman C.; Bignell G.; Teague J.; Butler A.; Edkins S.; Stevens C.; Parker A.; O’Meara S.; Avis T.; Barthorpe S.; Brackenbury L.; Buck G.; Clements J.; Cole J.; Dicks E.; Edwards K.; Forbes S.; Gorton M.; Gray K.; Halliday K.; Harrison R.; Hills K.; Hinton J.; Jones D.; Kosmidou V.; Laman R.; Lugg R.; Menzies A.; Perry J.; Petty R.; Raine K.; Shepherd R.; Small A.; Solomon H.; Stephens Y.; Tofts C.; Varian J.; Webb A.; West S.; Widaa S.; Yates A.; Brasseur F.; Cooper C. S.; Flanagan A. M.; Green A.; Knowles M.; Leung S. Y.; Looijenga L. H.; Malkowicz B.; Pierotti M. A.; Teh B. T.; Yuen S. T.; Lakhani S. R.; Easton D. F.; Weber B. L.; Goldstraw P.; Nicholson A. G.; Wooster R.; Stratton M. R.; Futreal P. A. (2005) Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res. 65, 7591–7595. [DOI] [PubMed] [Google Scholar]
- Bantscheff M.; Eberhard D.; Abraham Y.; Bastuck S.; Boesche M.; Hobson S.; Mathieson T.; Perrin J.; Raida M.; Rau C.; Reader V.; Sweetman G.; Bauer A.; Bouwmeester T.; Hopf C.; Kruse U.; Neubauer G.; Ramsden N.; Rick J.; Kuster B.; Drewes G. (2007) Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat. Biotechnol. 25, 1035–1044. [DOI] [PubMed] [Google Scholar]
- Kim H. G.; Hwang S. Y.; Aaronson S. A.; Mandinova A.; Lee S. W. (2011) DDR1 receptor tyrosine kinase promotes prosurvival pathway through Notch1 activation. J. Biol. Chem. 286, 17672–17681. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ongusaha P. P.; Kim J. I.; Fang L.; Wong T. W.; Yancopoulos G. D.; Aaronson S. A.; Lee S. W. (2003) p53 induction and activation of DDR1 kinase counteract p53-mediated apoptosis and influence p53 regulation through a positive feedback loop. EMBO J. 22, 1289–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rix U.; Hantschel O.; Durnberger G.; Remsing Rix L. L.; Planyavsky M.; Fernbach N. V.; Kaupe I.; Bennett K. L.; Valent P.; Colinge J.; Kocher T.; Superti-Furga G. (2007) Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood 110, 4055–4063. [DOI] [PubMed] [Google Scholar]
- Day E.; Waters B.; Spiegel K.; Alnadaf T.; Manley P. W.; Buchdunger E.; Walker C.; Jarai G. (2008) Inhibition of collagen-induced discoidin domain receptor 1 and 2 activation by imatinib, nilotinib and dasatinib. Eur. J. Pharmacol. 599, 44–53. [DOI] [PubMed] [Google Scholar]
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- Gao M.; Duan L.; Luo J.; Zhang L.; Lu X.; Zhang Y.; Zhang Z.; Tu Z.; Xu Y.; Ren X.; Ding K. (2013) Discovery and optimization of 3-(2-(pyrazolo[1,5-a]pyrimidin-6-yl)ethynyl)benzamides as novel selective and orally bioavailable discoidin domain receptor 1 (DDR1) inhibitors. J. Med. Chem. 56, 3281–3295. [DOI] [PubMed] [Google Scholar]
- Okram B.; Nagle A.; Adrian F. J.; Lee C.; Ren P.; Wang X.; Sim T.; Xie Y.; Wang X.; Xia G.; Spraggon G.; Warmuth M.; Liu Y.; Gray N. S. (2006) A general strategy for creating ″inactive-conformation″ abl inhibitors. Chem. Biol. 13, 779–786. [DOI] [PubMed] [Google Scholar]
- Balzano D.; Santaguida S.; Musacchio A.; Villa F. (2011) A general framework for inhibitor resistance in protein kinases. Chem. Biol. 18, 966–975. [DOI] [PubMed] [Google Scholar]
- Lemmon M. A.; Schlessinger J. (2010) Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemeer S.; Bluwstein A.; Wu Z.; Leberfinger J.; Muller K.; Kramer K.; Kuster B. (2012) Phosphotyrosine mediated protein interactions of the discoidin domain receptor 1. J. Proteomics 75, 3465–3477. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.