

Abstract
In all eukaryotic organisms, pre-mRNA splicing and alternative splicing processes play an essential role in regulating the flow of information required to drive complex developmental and metabolic pathways. As a result, eukaryotic cells have developed a very efficient macromolecular machinery, called the spliceosome, to correctly recognize the pre-mRNA sequences that need to be inserted in a mature mRNA (exons) from those that should be removed (introns). In healthy individuals, alternative and constitutive splicing processes function with a high degree of precision and fidelity in order to ensure the correct working of this machinery. In recent years, however, medical research has shown that alterations at the splicing level play an increasingly important role in many human hereditary diseases, neurodegenerative processes, and especially in cancer origin and progression. In this minireview, we will focus on several genes whose association with cancer has been well established in previous studies, such as ATM, BRCA1/A2, and NF1. In particular, our objective will be to provide an overview of the known mechanisms underlying activation/repression of pseudoexons and pseudointrons; the possible utilization of these events as biomarkers of tumor staging/grading; and finally, the treatment options for reversing pathologic splicing events.
1. Introduction
Starting from the first description of alternative splicing and constitutive splicing processes in 1977 [1–3], the importance of this process that guarantees the correct flow of information from transcription to translation in eukaryotic cells has continued to grow exponentially.
In particular, one major branch of research in this area has the aim to investigate and characterize the cellular macromolecular machine (i.e., the spliceosome) that is physically responsible for the cutting and joining of intronsexons by catalyzing two transesterification reactions [4, 5] and the mechanisms that ensure its fidelity [6]. As a result, research in spliceosome composition and functioning has been complemented by studies aiming to understand the sequences and molecules that determine under which conditions a particular exon or intron is selectively recognized and included in the mature transcript. Therefore, after the basic elements that define introns and exons and are composed by donor and acceptor splice sites plus the branch point sequence, there was the discovery of enhancer and silencer elements that can affect either positively or negatively the way these basic elements are recognized by the spliceosome [7–9]. Usually, enhancer and silencer elements are bound by members of the SR and hnRNP protein families, respectively.
The recruitment of these proteins to specific sites is crucial for their activity as depending on their position with respect to the basic elements their roles can be antagonistic or, in some case, agonistic [10, 11]. Global analyses of the ways these factors cooperate and influence each other in shaping the splicing process has only recently begun to shed light on how they promote or hinder exon recognition in a co-ordinated manner [12–14].
Rather unexpectedly, these studies have shown that SR proteins interact not only with alternatively spliced exons but also with constitutively spliced exons, and that their major role consists in recruiting the splicing machinery to splice sites [15, 16].
On the other hand, hnRNP proteins bind to nascent pre-mRNA and influence splicing decision through a complex and finely tuned network of protein/protein interactions. The mechanisms of hnRNPs actions on splicing is generally less defined than those of SR proteins [17].
In addition to these elements, we now also know that splicing choices are affected by a myriad of other factors, ranging from chromatin modifications [18, 19], transcriptional factors [20], RNA secondary structure [21], short noncoding RNAs [22, 23], and various cellular stresses [24].
In parallel to these mechanistic studies, another branch of splicing research has also addressed the functional importance of alternative splicing processes in biological pathways and in particular the way that alternative splicing isoforms of proteins can acquire different or even antagonistic biological properties [25, 26]. Because of this ability to expand the proteome of cells, alternative splicing has represented a very useful and powerful tool that allows cells to execute the various expression programs which underlie many fundamental needs of higher organisms: from general needs such as controlling normal development and tissue-specific expression of proteins, to highly specialized processes such as DNA damage response or microRNA biogenesis [27–33]. Moreover, the rearrangements that a pre-mRNA undergoes during the splicing process are also advantageous in terms of providing a longer half-life and better translational capacity, something that has recently begun to be exploited by the biotechnology industry [34].
Considering all these necessities and advantages, it is therefore not surprising that the number of genes that are subject to alternative splicing in eukaryotic organisms has been steadily growing. Indeed, it was recently estimated that more than 90% of mammalian protein coding genes can produce at least one splicing-derived isoform expressed at potentially significant biological levels [35].
The complexity of this system, however, also puts spliceosome functioning at risk of being impaired by the occurrence of single-point mutations in splicing regulatory elements, deletions/insertions, genomic rearrangements, and alterations at the splicing factor expression level [36]. Any of these alterations can result in a variety of aberrant splicing outcomes that usually include aberrant exon skipping, cryptic splice site selection, intron retention, and pseudoexon activation [37, 38]. As expected, many of these changes can lead directly to the occurrence of disease in humans, and it has now been estimated that a sizable proportion of all gene mutations leading to disease can be directly connected with the presence of a splicing defect [7, 39].
Importantly, the introduction of novel technologies that allow fast profiling of the transcriptome seems promising for simplifying investigation of which genomic variability and plasticity events allow cancer cells to tailor specific functional units from the available exons of a gene. For example, recent RNA sequencing of the breast cancer transcriptome has revealed many new splicing alterations that were not previously described. In particular, for example, it was reported that 423 primary transcripts (derived from 377 genes) were differentially spliced in triple-negative breast cancer samples (generating 496 novel isoforms) [40]. By analyzing non-triple-negative breast cancer samples, the same study found that 270 and 460 primary transcripts (derived from 242 and 387 differentially spliced genes, resp.) generated 331 and 550 novel isoforms that were not present in normal breast tissue [40].
2. Aberrant Splicing Events in Cancer Genes
It has become clear that in several human pathologies, including cancer, the alternative splicing profile is aberrantly modified in a specific manner and in ways that can favor the growth and survival of cancer cells [41–44]. The generation of these aberrant splicing profiles can occur in many ways, such as through the re-expression of developmentally regulated isoforms that had previously been shut off following early developmental stages [45, 46], by affecting the splicing profiles of genes that are implicated in tumor progression [47, 48], or through other mechanisms that generally have anti-apoptotic/metastatic consequences and that can affect response to therapies [49, 50].
In addition, several studies have defined that cancer-associated splicing alterations arise not only from mutations in the cancer-related genes but derive also from variations in the expression and/or activity of splicing regulatory factors [51]. For example, it has been well established that the levels of SR- and hnRNP-proteins undergo changes associated with transformation and progression of cancers [44, 52]. Strikingly, it has been recently reported that some members of the SR protein family of splicing factors and other components of the spliceosomal cellular machinery can actually act as oncoproteins and play a direct role in promoting tumor origin and progression [53–55] and that external stimuli such as hypoxia can cause the aberrant redistribution of important splicing factors such as Tra2 and promote the expression of tumor-promoting splicing isoforms [56].
The case of Bcl-X (BCL2L1) gene, belonging to the BclII family and implicated in the control of mitochondrial breakdown during apoptosis, is emblematic of this concept. For example, two splicing Bcl-X isoforms can arise from the use of two alternative 5′ splice sites within exon 2 and lead to the synthesis of a short apoptosis-promoting protein (Bcl-XS) and to a long antiapoptotic form (Bcl-XL) [57]. Different splicing factors, including Sam68, hnRNPA1, SF2/ASF, hnRNP F/H, hnRNP K, SAP155, and SRp30c have been found to be involved in the selection of the two competing alternative 5′ splice sites that give rise to these two isoforms [58–61]. In addition, recent studies have shown that the elongation and splicing-related factor TCERG1 can bind to the Bcl- X pre-mRNA and promote the proapoptotic Bcl-XS 5′ splice site in a promoter-dependent manner [62].
Finally, the production of the proapoptotic Bcl-XS splice variant seems to be improved by the core (Y14 and eIF4A3) and auxiliary (RNPS1, Acinus, and SAP18) components of the exon junction complex (EJC) [63], suggesting that EJC-associated components can regulate apoptosis at the alternative splicing level and represent a further level of vulnerability of cancer.
Therefore, one of the most interesting research areas in this field consists in the identification of cancer-specific splice variants or the aberrant expression of splicing-affecting proteins that could lead to their generation. Examples of both these events, in fact, have already been shown to occur in some individual types of cancer, such as breast and ovarian cancer [64, 65]. Another interesting research area in aberrant splicing events connected with tumors is the occurrence of particular types of splicing defects that involve the inclusion of “new” sequences (known as pseudoexons) in the mature mRNA of cancer-related genes. Rather more rarely, the opposite has also been shown to occur: the aberrant recognition of intronic sequences (pseudointrons) within normal exons. In this review, we have also decided to provide particular attention to these events as they are probably more common than previously considered and have not yet been the subject of particular attention.
One of the reasons why these two events are particularly interesting is that these types of defects are ideally suited for novel therapeutic effector molecules that are based in RNA biology. In the case of pseudoexons and pseudointrons, in fact, the major advantage of targeting this type of inclusion events is that the antisense oligonucleotides would be targeted against normal intronic sequences and thus would not remain bound to the mature mRNA (possibly to interfere with later stages of RNA processing such as export/translation).
3. Pseudoexon Activation in Cancer
In the pre-mRNA splicing field, the term “pseudoexon” has been introduced to describe exonic-like sequences that are present within intronic regions but are ignored by the spliceosomal machinery. A closer look at these sequences has often provided a reason for their inability to be recognized as normal exons: the presence of intrinsic defects in their apparently viable donor and acceptor sites [66] or of silencer elements [67–69] and the formation of inhibiting RNA secondary structures [70–72].
From a functional point of view, in most cases of pseudoexon insertion, the presence of an extraneous exon within the mature mRNA causes either the disruption of the translational reading frame or the insertion of novel amino acid sequences following translation. As a result, the normal biological properties of the resulting protein are very likely disrupted, and this can be associated with the development of disease.
Unfortunately, it is still quite hard to identify reliable pseudoexon insertion events in human genes implicated in cancers by just performing a general interrogation of databases (matching 22719 ENSEMBL protein coding genes versus 31057 entries of CanGEM Gene list) [73]. In fact, at present, it is only possible to retrieve strong candidates for alternative splicing events in protein coding genes (Table 1). As expected, a similar situation was seen when pseudogenes were investigated (defined as genomic DNA sequences similar to normal genes but nonfunctional, although some can still be transcribed). In this case, inspection of 14775 pseudogenes returned a list of alternative splicing hits from which it is not easy to distinguish real events in expressed pseudogenes (Table 1).
Table 1.
Alternative splicing events detected in protein coding genes and pseudogenes implicated in cancer.
| Alternative splicing event | Protein coding genes | CanGEM gene list (31057 entries) | % cancer protein coding genes |
|---|---|---|---|
| Cassette exon (CE) | 13506 | 11771 | 87 |
| Intron retention (IR) | 8190 | 6775 | 83 |
| Alternative 3′ss (A3SS) | 6701 | 5685 | 85 |
| Alternative 5′ss (A5SS) | 6624 | 5640 | 85 |
| Alternative first exons (AFE) | 8050 | 7136 | 89 |
| Alternative last exons (ALE) | 3631 | 3233 | 89 |
| Mutually exclusive exons (MXE) | 3376 | 3006 | 89 |
|
| |||
| Alternative splicing event | Pseudogenes | CanGEM gene LIST (31057 entries) | % cancer pseudogenes |
|
| |||
| Cassette exon (CE) | 434 | 139 | 32 |
| Intron retention (IR) | 465 | 187 | 40 |
| Alternative 3′ss (A3SS) | 241 | 91 | 38 |
| Alternative 5′ss (A5SS) | 218 | 82 | 38 |
| Alternative first exons (AFE) | 91 | 27 | 30 |
| Alternative last exons (ALE) | 104 | 32 | 31 |
| Mutually exclusive exons (MXE) | 50 | 16 | 32 |
Homo sapiens genes (GRCh37.p10) dataset (62252 total genes) was used to filter alternative splicing events in protein coding genes (22719 entries) and pseudogenes (14775 entries) and verified their presence in the Cancer GEnome Mine database (31057 entries—CanGEM, http://www.cangem.org/).
More recently, some bioinformatic studies have tried to extrapolate the presence of pseudoexons in cancer tissues by developing new analysis methods of GeneChip gene expression array data [74, 75]. In this way, by comparing normal cerebellum and medulloblastoma tumors, it was possible to predict 811 significantly different expressed pseudoexons (derived from 577 genes). In addition, when nonmetastatic and metastatic medulloblastomas were compared, 13 pseudoexonic sequences were significantly expressed in a differential manner (derived from 8319 strong candidates). However, it should be noted that no experimental validation was carried out to support these predictions. Therefore, presently, manual annotations remain the most reliable system for identifying real pseudoexons.
Fortunately, the scientific community has recently identified a certain number of cases where pseudoexon inclusion has been validated in detail. For this reason, Table 2 reports all the cryptic exons described in the literature that are localized within genes whose expression was altered in cancer.
Table 2.
Pseudoexon insertion events directly involved in cancer pathology.
| Gene name | Description | Entrez gene | Size (bp) pseudoexon | Activating mutation | Reference |
|---|---|---|---|---|---|
| APC | Adenomatosis polyposis coli | 324 | 167 | 5′ss creation | [78] |
| ATM | Ataxia telangiectasia mutated | 470 | 58 | 3′ss creation | [79] |
| ATM | 65 | SRE deletion | [80] | ||
| ATM | 137 | 5′ss creation | [81] | ||
| ATM | 212 | 5′ss creation | [82] | ||
| BCR-ABL | Breakpoint cluster region V-abl Abelson murine leukemia viral oncogene homolog 1 fusion |
613 25 |
42 | Genomic rearrangement | [83] |
| BCR-ABL | 35 | Unknown | [84, 85] | ||
| BRCA1 | Breast cancer 1, early onset | 672 | 66 | 3′ss creation | [86] |
| BRCA2 | Breast cancer 2, early onset | 675 | 93 | Downstream 3′ss deletion | [87] |
| BRCA2 | 95 | 5′ss creation | [88] | ||
| ESR1 | Estrogen receptor 1 | 3467 | 69 | 5′ss creation | [89] |
| MSH2 | MutS homolog 2, colon cancer, nonpolyposis type 1 (E. coli) | 4436 | 75 | 5'ss creation | [90] |
| NF1 | Neurofibromin 1 (neurofibromatosis, von Recklinghausen disease, Watson disease) | 4763 | 67/99 | 3′ss creation | [91] |
| NF1 | 70 | 5′ss creation | [92] | ||
| NF1 | 107 | 5′ss creation | [92] | ||
| NF1 | 172 | 3′ss creation | [93] | ||
| NF1 | 58 | 3′ss creation | [94] | ||
| NF1 | 76 | 5′ss creation | [94] | ||
| NF1 | 54 | 5′ss creation | [95] | ||
| NF1 | 177 | 5′ss creation | [92, 96, 97] | ||
| NF2 | Neurofibromin 2 (bilateral acoustic neuroma) | 4771 | 106 | BP creation | [98] |
| RB1 | Retinoblastoma 1 (including osteosarcoma) | 5925 | 103 | 3′ss creation | [99] |
| TSC2 | Tuberous sclerosis 2 | 7249 | 89 | 5′ss creation | [100] |
| WRN | Werner syndrome | 7486 | 106 | 5′ss creation | [101] |
| WRN | 69 | 3′ss creation | [102] |
First of all, regarding the mechanisms underlying pseudoexon activation, it is interesting to note that these phenomena are caused by inherited mutations resulting in the insertion of intronic sequences in the mature mRNA [38]. In this respect, it is interesting to note that among the genes presenting pseudoexon “awakening” directly associated with cancer origin (Table 2), the creation of new splicing donor (12 events) and acceptor (7 events) sites represents the more frequent occurrence (48% and 28% of the listed events, resp.). Some of the most paradigmatic examples of cancer genes such as BRCA1, BRCA2, NF1, and ATM are included in these two sets.
On the other hand, among pseudoexons described in cancer-related genes (Table 3), the creation of 5′ splice sites and of 3′ splice sites comprise 55% and 15% of the listed records, respectively. Moreover, pseudoexon insertion can also be triggered by the deletion of nearby donor or acceptors splice sites (4 events in both lists), highlighting the importance of the genetic milieu in splicing decisions.
Table 3.
Pseudoexons described in cancer-related genes.
| Gene name | Description | Entrez gene | Size (bp) pseudoexon | Activating mutation | Reference |
|---|---|---|---|---|---|
| ABCC8 | ATP-binding cassette, sub-family C (CFTR MRP), member 8 | 6833 | 76 | 5′ss creation | [103] |
| ALDH7A1 | Aldehyde dehydrogenase 7 family, member A1 | 501 | 36 | 5′ss mutation | [104] |
| CD40LG | CD40 ligand (TNF superfamily, member 5, hyper-IgM syndrome) | 959 | 59 | 5′ss creation | [105] |
| CEP290 | Centrosomal protein 290 kDa | 80184 | 128 | 5′ss creation | [106, 107] |
| CHM | Choroideremia (Rab escort protein 1) | 1121 | 98 | 3′ss creation | [108] |
| COL4A3 | Collagen, type IV, alpha 3 (Goodpasture antigen) | 1285 | 74 | 3′ss creation | [109] |
| COL11A1 | Collagen, type XI, alpha 1 | 1301 | 50 | 5′ss creation | [110] |
| CTDP1 | CTD (carboxy-terminal domain, RNA polymerase II, polypeptide A) phosphatase, subunit 1 | 9150 | 95 | 5′ss creation | [111] |
| CYBB | Cytochrome b-245, beta polypeptide (chronic granulomatous disease) | 1536 | 56 | 5′ss creation | [112] |
| CYBB | 61 | 5′ss creation | [113] | ||
| CYP17A1 | Cytochrome P450, family 17, subfamily A, polypeptide 1 | 1586 | 94 | Upstream 5′ss mutation | [114] |
| QDPR | Quinoid dihydropteridine reductase | 5860 | 152 | 5′ss creation | [115] |
| DPYD | Dihydropyrimidine dehydrogenase | 1806 | 44 | 5′ss creation | [116] |
| FBN1 | Fibrillin 1 | 2200 | 93 | 5′ss creation | [117] |
| GHR | Growth hormone receptor | 2690 | 102 | SRE deletion | [118, 119] |
| GUSB | Glucuronidase, beta | 2990 | 68 | 5′ss creation | [120] |
| HADH | Hydroxyacyl-coenzyme A dehydrogenase | 3033 | 141 | 5′ss creation | [103] |
| HADHB | Hydroxyacyl-coenzyme A dehydrogenase 3-ketoacyl-coenzyme A thiolase enoyl-coenzyme A hydratase (trifunctional protein), beta subunit |
3032 | 56 106 |
5′ss creation | [121] |
| HSPG2 | Heparan sulfate proteoglycan 2 | 3339 | 141 | 5′ss creation | [103] |
| SMARCB1 | SWI SNF related, matrix associated, actin dependent regulator of chromatin, subfamily b, member 1 |
6598 | 72 | 5′ss creation | [122] |
| ISCU | Iron-sulfur cluster scaffold homolog (E. coli) | 23479 | 86 100 |
3′ss creation | [123–125] |
| SLC14A1 (JK) | Solute carrier family 14 (urea transporter), member 1 (Kidd blood group) | 136 | Internal 7 kb deletion | [126] | |
| MCCC2 | Methylcrotonyl-coenzyme A carboxylase 2 (beta) | 64087 | 64 | SRE deletion | [127] |
| MFGE8 | Milk fat globule-EGF factor 8 protein | 4240 | 102 | SRE creation | [128] |
| MLC1 | Megalencephalic leukoencephalopathy with subcortical cysts 1 | 23209 | 246 | 5′ss creation | [129] |
| MTRR | 5-methyltetrahydrofolate-homocysteine methyltransferase reductase | 4552 | 140 | SRE creation | [130] |
| GPR143 | G protein-coupled receptor 143 | 4935 | 165 | 3′ss creation | [131] |
| OAT | Ornithine aminotransferase (gyrate atrophy) | 4942 | 142 | 5′ss creation | [132] |
| OFD1 | Oral-facial-digital syndrome 1 | 8481 | 62 | 5′ss creation | [133] |
| OTC | Ornithine carbamoyltransferase | 5009 | 135 | 3′ss creation | [134] |
| PCCA | Propionyl-coenzyme A carboxylase, alpha polypeptide | 5095 | 84 | SRE creation | [135] |
| PCCB | Propionyl-coenzyme A carboxylase, beta polypeptide | 5096 | 72 | 5′ss creation | [135] |
| PHEX | Phosphate regulating endopeptidase homolog, X-linked (hypophosphatemia, vitamin D resistant rickets) | 5251 | 50 100 170 |
5′ss creation | [136] |
| PKHD1 | Polycystic kidney and hepatic disease 1 (autosomal recessive) | 5314 | 116 | 5′ss creation | [137] |
| PMM2 | Phosphomannomutase 2 | 5373 | 66 | 3′ss creation | [138] |
| PMM2 | 123 | 5′ss creation | [138, 139] | ||
| PRPF31 | PRP31 pre-mRNA processing factor 31 homolog (S. cerevisiae) | 26121 | 175 | 5′ss creation | [140] |
| RHD | Rh blood group, D antigen | 6007 | 170 | Upstream 3′ss deletion and SNP in int7 | [141] |
| RYR1 | Ryanodine receptor 1 (skeletal) | 6261 | 119 | 5′ss creation | [142] |
| SLC12A3 | Solute carrier family 12 (sodium chloride transporters), member 3 | 6559 | 238 | 5′ss creation | [143] |
| USH2A | Usher syndrome 2A (autosomal recessive, mild) | 7399 | 152 | 5′ss creation | [144] |
A deeper look into the mechanisms underlying pseudoexon activation has revealed the involvement of hnRNPs in regulation of these events. In particular, the partial inclusion of a pseudoexon of NF1 gene has been found to arise from a novel intronic mutation c.31−279A>G intron 30, creating a new acceptor splice site and activation of a cryptic 5′ splice site [76]. It has been found that both PTB and nPTB play an active role in the pseudoexon splicing by repressing its inclusion [76]. This finding is particularly interesting since it further supports the hypothesis that PTB/nPTB might have a general role in repressing weak exons and in particular most of pseudoexons [77].
Another example of mutation causing pseudoexon activation consists in the creation of novel branch site (Table 2, one event). This latter case is associated with a peculiar mutation (IVS5 ds +232 G−A) that leads to pseudoexon inclusion in NF2 gene by creating a consensus branch point. In particular, it has been found that the G>A transition, occurring at position −18 from the acceptor site of NF2 IVS5, works in combination with existing consensus splice acceptor and donor sites and causes the formation of an extra exon 5a in the NF2 gene that introduces a premature stop codon [98].
Finally, in spite of being lowly represented, gross genomic rearrangements can bring together or expose splice site sequences that would normally be very distant from each other or absent form the original sequence (one event).
Pseudoexon activation can also be caused by mutations that cause the creation/deletion of splicing regulatory elements (SREs, 6 events). Among these events, one of the most representative examples of the complex regulatory networks that can underlie pseudoexon insertion is represented by the identification of an unusual splicing defect directly related with neoplasia (Figure 1).
Figure 1.
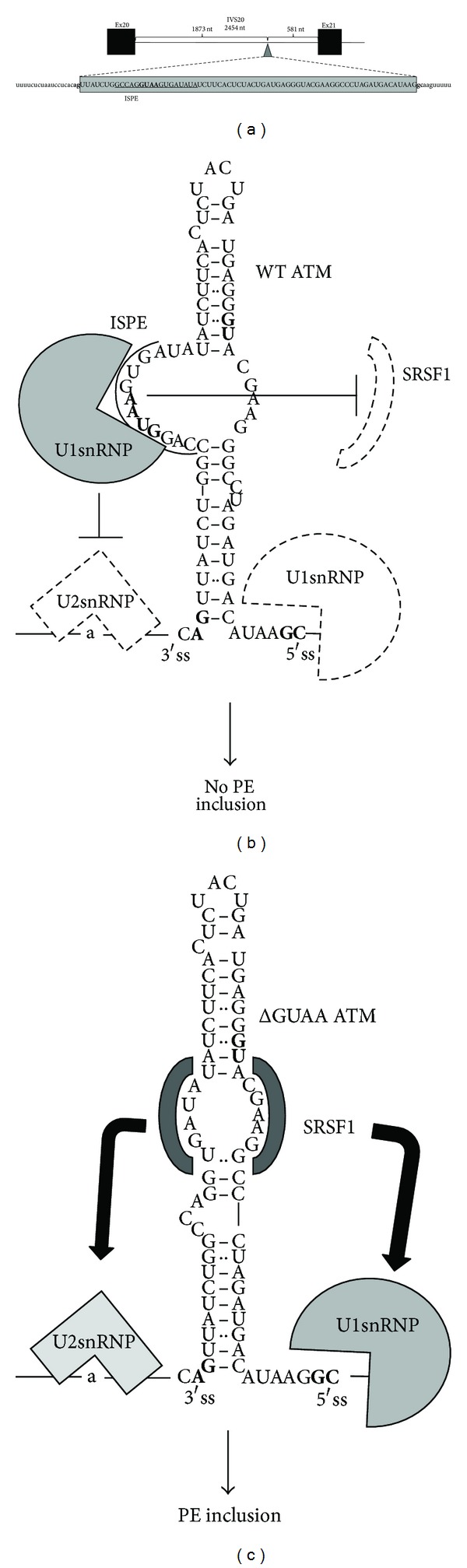
ATM pseudoexon activated in cancer. (a) Scheme of the ATM pseudoexon insertion caused by a 4 nt deletion (GUAA) occurring 1873 nt downstream from the donor splice site of ATM exon 20. The solid lines indicate introns. Dotted lines include the 65 nt-ATM pseudoexon (gray box). The intron-splicing processing element (ISPE) complementary to U1 snRNA, the RNA component of the U1 small nuclear ribonucleoprotein (snRNP), is underlined. (b) Schematic model of U1snRNP mediated inhibition of pseudoexon insertion in normal conditions. In this model, U1snRNP binding to the ISPE blocks pseudoexon inclusion by inhibiting recruitment of U2snRNP to the 3′splice site region and SRSF1 binding to the stem loop of the pseudoexon (shown as dotted lines). (c) In the case of the disease associated ATM ΔGUAA deletion, the internal U1snRNP binding site is no longer present due to the deletion of the ISPE. This leads to a more efficient binding of SRSF1 to the enhancer site and stabilization of the U2snRNP interaction with the branch site and U1snRNP with the 5′gc donor site.
In the case of this particular event, a patient affected by ataxia-telangiectasia was found to carry a 4 nt-deletion (GTAA) within intron 20 of ATM gene [80]. This deletion, termed intron-splicing processing element (ISPE), caused the inclusion of a 65 nucleotide “cryptic exon” in the ATM mRNA (Figure 1(a)). The mechanism through which this occurs has been well characterized in recent studies.
In normal conditions, the repression of the 3′ss in the wild-type pseudoexon sequence (ATM WT) was shown to be a direct consequence of U1snRNP binding in correspondence to the ISPE sequence. The importance of this U1snRNP binding was twofold: first of all, to sterically hinder 3′ss recognition by U2snRNP and secondly, to also inhibit binding of the SRSF1 splice factor to the stem-loop sequence formed by this pseudoexon (Figure 1(b)) [71, 72]. As a result, this internal U1snRNP binding event to the ISPE sequence causes an unproductive U2snRNP association with the 3′ss that results in the complete inhibition of pseudoexon inclusion in normal conditions (Figure 1(b)). Following the deletion of the GUAA motif observed in the patient, U1snRNP binding to the ISPE is relieved, and SRSF1 is also free to bind to the internal enhancer site that becomes available (Figure 1(c)). Taken together, these two events result in efficient recruitment of U2snRNP to the branchsite and a better recruitment of U1snRNP to the rather inefficient 5′gc splice site that lead to efficient inclusion of the pseudoexon in the mature ATM mRNA [72].
This example clearly shows the complexity of pseudoexon activation events that relies on a variety of factors well beyond the simple initial mutation (i.e., 5′ss or 3′ss creation), and that can include the eventual presence of enhancer and silencer elements within the cryptic exon or intro, RNA secondary structures, and probably several other mechanisms that have not yet been described in detail.
4. Pseudointron Activation in Cancer
Pseudointrons (PSIs) represent an intriguing set of intron-like sequences localized within exons that can undergo alternative splicing and that therefore can be included in or excluded from the ORF within the final mRNA species.
Although the examples of pseudointrons reported in the review are not associated with activating mutations, we have included them because of the peculiarity of events insofar that they do not represent simple intron retention events, but rather alternatively spliced intraexonic sequences whose behaviour can resemble that of introns under particular circumstances (e.g., occurrence of previous splicing events in the processed transcript). It is this regulated use of the intraexonic splice sites that fits well with the idea that these intraexonic splice site represent “false” introns, even in the absence of genetic alterations.
In general, however, pseudointrons are less well described compared withpseudoexons probably because of the difficulty in detecting them using available technology (a situation that may well soon change for the better because of the introduction of more sensitive and comprehensive technical approaches such as RNA sequencing). At the moment, only three examples of PSI have been characterized, and for at least two of these pseudointrons there are data supporting their relevance in the pathogenesis of cancer.
4.1. Fibronectin: IIICS-C5
Fibronectin (FN) is an extracellular matrix protein whose functions range from cell adhesion and migration to wound healing and oncogenic transformation [145]. The functional complexity of FN is related to the structural diversity arising from cell type-specific alternative splicing [146]. The fibronectin pre-mRNA undergoes alternative splicing primarily at three sites: two extra domain exons encoding extra structural repeats and a region of nonhomologous sequence called the type-III connecting segment (IIICS).
The IIICS domain is divided into three subdomains of 75 bp, 192 bp, and 93 bp, respectively, that can be alternatively spliced, thus generating up to fivevariant mRNAs in humans [147, 148]. The subdomains of 75 bp and 93 bp encode for two cell-specific binding sites, CS1 (residues 1–25 of the IIICS), and CS5 (residues 90–109 of the IIICS), respectively, that interact with the integrin α4β1 with different affinity [149, 150]. Indeed, the CS1 site has approximately 20-fold higher affinity for integrins than the CS5 site [151].
Because of its intraexonic localization and splicing behavior, the 93 bp segment can be considered a pseudointron (Figure 2(a)). Different tissue-specific and disease-associated changes in the variants of the IIICS region have been reported [152–154], and the CS5 pseudointron is upregulated in foetal tissue and in adult liver [155].
Figure 2.
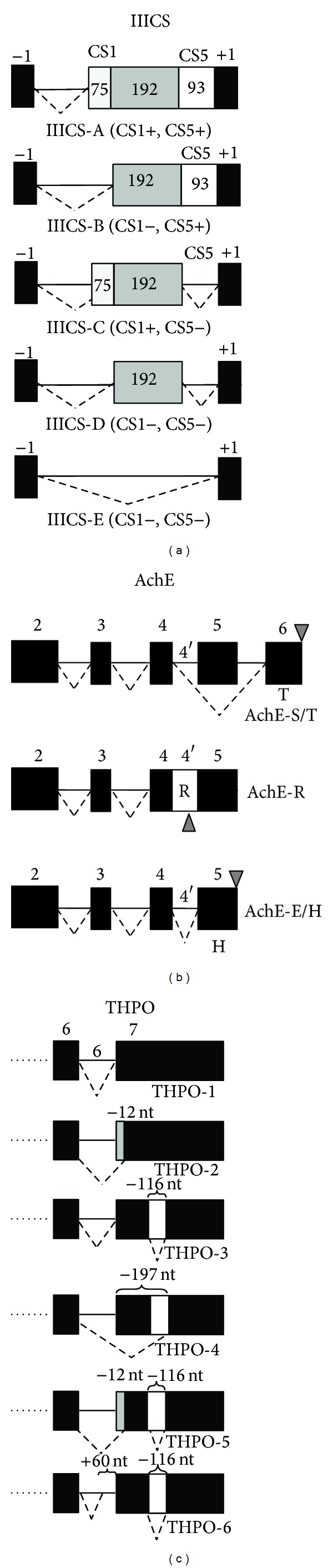
Pseudointrons whose alternative splicing can be altered in tumors. Schemes of pseudointron alternative splicing events shown to occur within the Fibronectin (IIICS, (a)), Acetylcholinesterase (AchE, (b)), and Thrombopoietin (THPO, (c)) genes. Pseudointrons are shown as white boxes in IIICS (CS5-93 nt), AchE (4′-R), and THPO (116 nt). Solid lines indicate introns and dotted lines distinguish the different splicing events. The C-terminal sequence of each splicing variant for each gene is also shown. For IIICS and THPO, additional exonic elements undergoing alternative splicing are shown as gray boxes. In the AchE gene, gray triangles indicate stop codon within different ORFs. See text for definition of each splice variant.
For the purpose of this review, it is important to note that alternative splicing of the IIICS-CS5 subdomain can influence the onset and progression of cancers by affecting Fibronectin activities related to tissue organization and cell-ECM interactions. In keeping with this hypothesis, in vitro studies have shown that CS5 can modulate the spreading of melanoma cells [151].
4.2. Acetylcholinesterase: AChE-R
Acetylcholinesterase is a serine specific hydrolase that hydrolyzes acetylcholine, and its main function is related to the clearance of the neurotransmitter from the synaptic cleft [156]. Nonetheless, the observation that the expression of AChE is not limited to cholinergic tissues has suggested the presence of additional functions [157].
From the splicing process point of view, mammal AChE has been shown to undergo alternative splicing resulting in expression of 3 isoforms, called R (“readthrough”), H (“hydrophobic”), and T/S (“tailed” or “synaptic”), that differ in their subcellular localization, tissue distribution, and developmental pattern of expression [158]. Whereas, AChE-H or AChE-T is the most abundant and common isoforms, the AChE-R expression is low and restricted in specific cell lines, cell differentiation stage, or muscle development [159, 160].
Interestingly, the AChE-R mRNA arises by retention of pseudointron4, that leads to generation of the E1-E2-E3-E4-I4-E5 mRNA transcript (Figure 2(b)). Since pseudointron 4 includes a stop codon when it is included in mature RNA, the resulting protein will be prematurely terminated by a C-terminus translated from the 5′ region of I4. This variant is expected to remain monomeric [161].
In general, the AChE-R transcript can be coexpressed with the AChE-T or AChE-H transcripts, and variations in their relative levels seem to be important for modulation of AChE functions, as well as for the pathogenesis of neurological and autoimmune disorders [161, 162]. Intriguingly, recent studies have found that AChE-R mRNA accumulates in primary human astrocytomas, and that its presence is correlated with their grade of aggressiveness [163]. In human U87MG glioblastoma cells, it was found that AChE-R protein variant can support proliferation of glioblastoma tumors by forming a complex with the scaffold protein RACK1 and protein kinase Ce [164].
In addition, increased levels of an N-terminally extended N-AChE-R isoform were detected in human testicular tumors indicating that the generation of the AChE-R variant can be associated with the utilization of an alternative promoter in the transformed cells.
Finally, related functional studies have suggested that both AChE-R variants (AChE-R and N-AChE-R) might be crucial for increasing the cellular ATP levels and might support selective metabolic advantages as well as genotoxic resistance by altering p73 gene expression [165]. Although it is not yet clear whether the AChE-R (as well as the other AChE splicing variants) can be related directly to tumorigenesis, the observed downregulation of all these splicing isoforms in colorectal carcinoma suggests a crucial role for tumor development [166].
4.3. Thrombopoietin: THPO-3,THPO-5, and THPO-6
A third example of pseudointron possibly implicated in cancer pathogenesis is found within the Thrombopoietin (THPO) gene. THPO is the most important cytokine for regulation of platelet production, and its expression has also been reported in skeletal muscle, ovary, testis, and central nervous system (CNS) [167–169].
The THPO gene shows a complex pattern of alternative splicing [170]; among the six splice variants that can be generated, three of them (THPO-3, THPO-5, and THPO-6) show the peculiar alternative splicing of 116 nucleotides within exon 6 (that can therefore be classified as a pseudointron) (Figure 2(c)) [171].
Contrary to the Fibronectin IIICS-C5 and AchE-R pseudointrons, the involvement in cancerogenesis of the variants deriving from the alternative splicing of the 116 nt pseudointron is less characterized. Nonetheless, functional studies have demonstrated that the protein translated from mouse THPO-3 is not secreted efficiently [172–174]. These results have suggested that THPO-3 might be implicated in the fine regulation of THPO full-length levels and in the modulation of several biological functions beyond thrombopoiesis. Interestingly, parallel studies have also shown an altered expression pattern of the THPO-3, THPO-5, and THPO-6 splicing variants in human carcinomas [175]. As a result, these observations have led researchers to hypothesize that alterations in alternative splicing of the 116 nt-THPO pseudointron might be employed as biomarker of tumor formation or progression [170].
In conclusion, although currently there are few examples of pseudointron removal, at least three of these events have been associated with different levels of cancerogenesis, ranging from transformation to cancer progression. Therefore, it is advisable that future high throughput screening analysis should be able to better associate the alternative splicing process of these pseudointrons with tumoral events. In particular, with regard to their use as possible biomarkers of transformation or of cancer staging/grading.
5. Therapeutic Outlooks
Regarding therapies, it is likely that all this information on the importance of alternative splicing for tumor development and progression may soon become extremely important for the development of novel therapeutic effectors in the fight against cancer [176, 177]. Indeed, one of the reasons why these defects are particularly interesting is the opportunity they provide for RNA therapies and the chance to actually make the jump from the bench to the bedside.
Currently, the field of RNA therapy is rapidly growing especially in the inhibition of undesirable splice site choices by the use of antisense oligos [178–180]. This technique, pioneered by Dominski and Kole [181], is based on the use of suitably modified antisense oligonucleotides that target specific sequences within introns and exons and block the recognition by their cellular binding factors. Therefore, they are ideally suited to block unwanted splice sites that may become activated following their creation or activation. It is interesting that the role of using antisense strategies to regulate exon inclusion also occurs in physiological conditions. An example of this is snoRNA HBII-52 in the regulation of exon Vb inclusion in the serotonin receptor 2C [182].
To this moment, a few pilot studies aimed at inhibiting the inclusion of a pseudoexon in patient cell lines have already been reported. In particular, antisense oligonucleotides have been used to target newly created 5′ or 3′ splice sites deep within intronic regions of the NF1 gene to restore the normal splicing profile [91, 92]. Similar approaches have also been successfully used to target deep intronic mutations causing pseudoexon activation within BRCA2 intron 12 (c.6937+594T>G) [88], ATM intron 11 (c.1236−405C>T) [82], and ATM IVS19 (c.2639−384A>G) [79].
Recent advances in antisense-mediated exon skipping for DMD suggest that not only the splice sites but also exonic splicing enhancer sites or branch points might represent potential targets for inducing pseudoexon skipping [183, 184], and that the action of ASOs might be potentiated by coadministration of drugs, as shown for Duchenne muscular dystrophy [185].
An intriguing variation on the theme of the antisense oligonucleotides, successfully tested for inhibiting the 5′ splice site of Bcl-XL, might consist in the use of tailed oligonucleotides containing a portion annealing to sequences immediately upstream of the target donor splice site joined to a nonhybridizing 5′ tail that includes binding sites for the hnRNP A1/A2 proteins [186].
In spite of being prominent, antisense technologies are not the only possibility available for targeting these aberrant splicing events. An alternative to antisense usage is the development of siRNA molecules capable of targeting the newly created system. In this manner, only the mRNAs containing the pseudoexon might be selectively degraded leaving any residual normal mRNA to be properly processed [187]. The disadvantage of simply degrading the pseudoexon-bearing transcript (rather than acting on the splicing event itself) is that everything depends on the level of pseudoexon inclusion (less than 100%) and on the status of the other allele (i.e., whether it is normally expressed or not).
Finally, small molecule compounds might be also considered as possible therapeutic options in order to prevent the recognition of pseudoexons or modulate the recognition of pseudointrons. For example, it has been shown that sodium butyrate, an histone deacetylase inhibitor known to upregulate the expression of Htra2-beta1 and SC35, promotes skipping of the pseudoexon activated by the CFTR 3849+10kbC>T mutation and can restore functional CFTR channels [188]. In addition, the splicing of different NF1 skipped exons as a result of mutations in cis-acting sequences has been restored with administration of the small molecule kinetin [189].
More recently, a growing body of research indicates that TALE nucleases (TALENs) have been used with great success in a number of organisms to generate site-specific DNA variations [190]. As a result, this approach is another good candidate for an innovative therapeutic anticancer strategy for correction of deep intronic mutations that create novel splice sites.
Of course, crossing the difficult gap between the bench to the bedside will not be immediate even in the presence of highly efficient therapeutic molecules, whether antisense oligos, siRNAs, or others. Indeed, before any of these can be used in humans to treat cancer, several additional factors need to be considered. First of all, there is the development of an efficient and safe carrier system to deliver these compounds into the human body. Secondly, these systems will need to be optimized in order to achieve the best balance between successful delivery, intrinsic toxicity (if any), and avoidance of undesired immune responses (in the case of antisense oligos). Finally, even when all these achievements are met, there will still be the need to optimize recurrent-administration protocols to determine the uptake levels, clearance, and accumulation in various tissues (this is an often overlooked, since none of these methods will cause permanent correction of mRNA splicing defects).
Nonetheless, the first results of this exciting news strategy are available, and there is the distinct possibility that RNA-based treatment of cancer may soon enter the application stage.
Acknowledgments
This work was supported by the University of Trieste-Finanziamento per Ricercatori di Ateneo to Maurizio Romano and the Higher Education Founding Council for England (HEFCE) and Cancer research UK (CRUK) to Diana Baralle.
References
- 1.Chow LT, Gelinas RE, Broker TR, Roberts RJ. An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell. 1977;12(1):1–8. doi: 10.1016/0092-8674(77)90180-5. [DOI] [PubMed] [Google Scholar]
- 2.Berget SM, Moore C, Sharp PA. Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proceedings of the National Academy of Sciences of the United States of America. 1977;74(8):3171–3175. doi: 10.1073/pnas.74.8.3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Darnell JE., Jr. Reflections on the history of pre-mRNA processing and highlights of current knowledge: a unified picture. RNA. 2013;19(4):443–460. doi: 10.1261/rna.038596.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nilsen TW. The spliceosome: the most complex macromolecular machine in the cell? Bioessays. 2003;25(12):1147–1149. doi: 10.1002/bies.10394. [DOI] [PubMed] [Google Scholar]
- 5.Will CL, Lührmann R. Spliceosome structure and function. Cold Spring Harbor Perspectives in Biology. 2011;3(7) doi: 10.1101/cshperspect.a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Semlow DR, Staley JP. Staying on message: ensuring fidelity in pre-mRNA splicing. Trends in Biochemical Sciences. 2012;37(7):263–273. doi: 10.1016/j.tibs.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nature Reviews Genetics. 2002;3(4):285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 8.Hertel KJ. Combinatorial control of exon recognition. The Journal of Biological Chemistry. 2008;283(3):1211–1215. doi: 10.1074/jbc.R700035200. [DOI] [PubMed] [Google Scholar]
- 9.Kornblihtt AR, Schor IE, Allo M, Dujardin G, Petrillo E, Munoz MJ. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nature Reviews Molecular Cell Biology. 2013;14:153–165. doi: 10.1038/nrm3525. [DOI] [PubMed] [Google Scholar]
- 10.Chabot B. Directing alternative splicing: cast and scenarios. Trends in Genetics. 1996;12(11):472–478. doi: 10.1016/0168-9525(96)10037-8. [DOI] [PubMed] [Google Scholar]
- 11.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annual Review of Biochemistry. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Xiao X, Zhang J, et al. A complex network of factors with overlapping affinities represses splicing through intronic elements. Nature Structural & Molecular Biology. 2013;20:36–45. doi: 10.1038/nsmb.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huelga SC, Vu AQ, Arnold JD, et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Reports. 2012;1(2):167–178. doi: 10.1016/j.celrep.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pandit S, Zhou Y, Shiue L, et al. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Molecular Cell. 2013;50:223–235. doi: 10.1016/j.molcel.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schaal TD, Maniatis T. Multiple distinct splicing enhancers in the protein-coding sequences of a constitutively spliced pre-mRNA. Molecular and Cellular Biology. 1999;19(1):261–273. doi: 10.1128/mcb.19.1.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graveley BR. Sorting out the complexity of SR protein functions. RNA. 2000;6(9):1197–1211. doi: 10.1017/s1355838200000960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinez-Contreras R, Cloutier P, Shkreta L, Fisette JF, Revil T, Chabot B. hnRNP proteins and splicing control. Advances in Experimental Medicine and Biology. 2007;623:123–147. doi: 10.1007/978-0-387-77374-2_8. [DOI] [PubMed] [Google Scholar]
- 18.Hnilicová J, Staněk D. Where splicing joins chromatin. Nucleus. 2011;2(3):182–188. doi: 10.4161/nucl.2.3.15876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Acuña LIG, Fiszbein A, Alló M, Schor IE, Kornblihtt AR. Connections between chromatin signatures and splicing. Wiley Interdisciplinary Reviews. 2013;4(1):77–91. doi: 10.1002/wrna.1142. [DOI] [PubMed] [Google Scholar]
- 20.Dujardin G, Lafaille C, Petrillo E, et al. Transcriptional elongation and alternative splicing. Biochimica et Biophysica Acta. 2013;1829(1):134–140. doi: 10.1016/j.bbagrm.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 21.Buratti E, Baralle FE. Influence of RNA secondary structure on the pre-mRNA splicing process. Molecular and Cellular Biology. 2004;24(24):10505–10514. doi: 10.1128/MCB.24.24.10505-10514.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khanna A, Stamm S. Regulation of alternative splicing by short non-coding nuclear RNAs. RNA Biology. 2010;7(4):480–485. doi: 10.4161/rna.7.4.12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luco RF, Misteli T. More than a splicing code: integrating the role of RNA, chromatin and non-coding RNA in alternative splicing regulation. Current Opinion in Genetics and Development. 2011;21(4):366–372. doi: 10.1016/j.gde.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biamonti G, Caceres JF. Cellular stress and RNA splicing. Trends in Biochemical Sciences. 2009;34(3):146–153. doi: 10.1016/j.tibs.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 25.Kelemen O, Convertini P, Zhang Z, et al. Function of alternative splicing. Gene. 2013;514:1–30. doi: 10.1016/j.gene.2012.07.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson TL, Vilardell J. Regulated pre-mRNA splicing: the ghostwriter of the eukaryotic genome. Biochimica et Biophysica Acta. 2012;1819(6):538–545. doi: 10.1016/j.bbagrm.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng S, Black DL. Alternative pre-mRNA splicing in neurons: growing up and extending its reach. Trends in Genetics. 2013;29(8):442–448. doi: 10.1016/j.tig.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Norris AD, Calarco JA. Emerging roles of alternative pre-mRNA splicing regulation in neuronal development and function. Frontiers in Neuroscience. 2012;6(122) doi: 10.3389/fnins.2012.00122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mattick JS. The central role of RNA in human development and cognition. FEBS Letters. 2011;585(11):1600–1616. doi: 10.1016/j.febslet.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 30.Grabowski P. Alternative splicing takes shape during neuronal development. Current Opinion in Genetics and Development. 2011;21(4):388–394. doi: 10.1016/j.gde.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 31.Yap K, Makeyev EV. Regulation of gene expression in mammalian nervous system through alternative pre-mRNA splicing coupled with RNA quality control mechanisms. Molecular and Cellular Neuroscience. 2013 doi: 10.1016/j.mcn.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Montecucco A, Biamonti G. Pre-mRNA processing factors meet the DNA damage response. Frontiers in Genetics. 2013;4(102) doi: 10.3389/fgene.2013.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melamed Z, Levy A, Ashwal-Fluss R, et al. Alternative splicing regulates biogenesis of miRNAs located across exon-intron junctions. Molecular Cell. 2013;50(6):869–881. doi: 10.1016/j.molcel.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 34.Skoko N, Baralle M, Tisminetzky S, Buratti E. InTRONs in biotech. Molecular Biotechnology. 2011;48(3):290–297. doi: 10.1007/s12033-011-9390-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nature Genetics. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 36.Buratti E, Baralle M, Baralle FE. Defective splicing, disease and therapy: searching for master checkpoints in exon definition. Nucleic Acids Research. 2006;34(12):3494–3510. doi: 10.1093/nar/gkl498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krawczak M, Thomas NST, Hundrieser B, et al. Single base-pair substitutions in exon-intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Human Mutation. 2007;28(2):150–158. doi: 10.1002/humu.20400. [DOI] [PubMed] [Google Scholar]
- 38.Dhir A, Buratti E. Alternative splicing: role of pseudoexons in human disease and potential therapeutic strategies: minireview. FEBS Journal. 2010;277(4):841–855. doi: 10.1111/j.1742-4658.2009.07520.x. [DOI] [PubMed] [Google Scholar]
- 39.Baralle D, Lucassen A, Buratti E. Missed threads: the impact of pre-mRNA splicing defects on clinical practice. EMBO Reports. 2009;10(8):810–816. doi: 10.1038/embor.2009.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eswaran J, Horvath A, Godbole S, et al. RNA sequencing of cancer reveals novel splicing alterations. Scientific Reports. 2013;3, article 1689 doi: 10.1038/srep01689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Venables JP. Unbalanced alternative splicing and its significance in cancer. Bioessays. 2006;28(4):378–386. doi: 10.1002/bies.20390. [DOI] [PubMed] [Google Scholar]
- 42.Relógio A, Ben-Dov C, Baum M, et al. Alternative splicing microarrays reveal functional expression of neuron-specific regulators in Hodgkin lymphoma cells. The Journal of Biological Chemistry. 2005;280(6):4779–4784. doi: 10.1074/jbc.M411976200. [DOI] [PubMed] [Google Scholar]
- 43.Klinck R, Bramard A, Inkel L, et al. Multiple alternative splicing markers for ovarian cancer. Cancer Research. 2008;68(3):657–663. doi: 10.1158/0008-5472.CAN-07-2580. [DOI] [PubMed] [Google Scholar]
- 44.Ghigna C, Valacca C, Biamonti G. Alternative splicing and tumor progression. Current Genomics. 2008;9(8):556–570. doi: 10.2174/138920208786847971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes and Development. 2010;24(21):2343–2364. doi: 10.1101/gad.1973010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Venables JP. Aberrant and alternative splicing in cancer. Cancer Research. 2004;64(21):7647–7654. doi: 10.1158/0008-5472.CAN-04-1910. [DOI] [PubMed] [Google Scholar]
- 47.Kalniņa Z, Zayakin P, Siliņa K, Line A. Alterations of pre-mRNA splicing in cancer. Genes Chromosomes and Cancer. 2005;42(4):342–357. doi: 10.1002/gcc.20156. [DOI] [PubMed] [Google Scholar]
- 48.Srebrow A, Kornblihtt AR. The connection between splicing and cancer. Journal of Cell Science. 2006;119(13):2635–2641. doi: 10.1242/jcs.03053. [DOI] [PubMed] [Google Scholar]
- 49.Pettigrew CA, Brown MA. Pre-mRNA splicing aberrations and cancer. Frontiers in Bioscience. 2008;13(3):1090–1105. doi: 10.2741/2747. [DOI] [PubMed] [Google Scholar]
- 50.Grosso AR, Martins S, Carmo-Fonseca M. The emerging role of splicing factors in cancer. EMBO Reports. 2008;9(11):1087–1093. doi: 10.1038/embor.2008.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghigna C, Moroni M, Porta C, Riva S, Biamonti G. Altered expression of heterogeneous nuclear ribonucleoproteins and SR factors in human colon adenocarcinomas. Cancer Research. 1998;58(24):5818–5824. [PubMed] [Google Scholar]
- 52.Zerbe LK, Pino I, Pio R, et al. Relative amounts of antagonistic splicing factors, hnRNP A1 and ASF/SF2, change during neoplastic lung growth: implications for pre-mRNA processing. Molecular Carcinogenesis. 2004;41(4):187–196. doi: 10.1002/mc.20053. [DOI] [PubMed] [Google Scholar]
- 53.Karni R, De Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nature Structural and Molecular Biology. 2007;14(3):185–193. doi: 10.1038/nsmb1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Padgett RA. New connections between splicing and human disease. Trends in Genetics. 2012;28(4):147–154. doi: 10.1016/j.tig.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fu Y, Huang B, Shi Z, et al. SRSF1 and SRSF9 RNA binding proteins promote Wnt signalling-mediated tumorigenesis by enhancing beta-catenin biosynthesis. EMBO Molecular Medicine. 2013;5:737–750. doi: 10.1002/emmm.201202218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hirschfeld M, Jaeger M, Buratti E, et al. Expression of tumor-promoting Cyr61 is regulated by hTRA2-β1 and acidosis. Human Molecular Genetics. 2011;20(12):2356–2365. doi: 10.1093/hmg/ddr128. [DOI] [PubMed] [Google Scholar]
- 57.Boise LH, Gonzalez-Garcia M, Postema CE, et al. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74(4):597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 58.Garneau D, Revil T, Fisette JF, Chabot B. Heterogeneous nuclear ribonucleoprotein F/H proteins modulate the alternative splicing of the apoptotic mediator Bcl-x. The Journal of Biological Chemistry. 2005;280(24):22641–22650. doi: 10.1074/jbc.M501070200. [DOI] [PubMed] [Google Scholar]
- 59.Massiello A, Roesser JR, Chalfant CE. SAP155 binds to ceramide-responsive RNA cis-element 1 and regulates the alternative 5′ splice site selection of Bcl-x pre-mRNA. The FASEB Journal. 2006;20(10):1680–1682. doi: 10.1096/fj.05-5021fje. [DOI] [PubMed] [Google Scholar]
- 60.Cloutier P, Toutant J, Shkreta L, Goekjian S, Revil T, Chabot B. Antagonistic effects of the SRp30c protein and cryptic 5′ splice sites on the alternative splicing of the apoptotic regulator Bcl-x. The Journal of Biological Chemistry. 2008;283(31):21315–21324. doi: 10.1074/jbc.M800353200. [DOI] [PubMed] [Google Scholar]
- 61.Revil T, Toutant J, Shkreta L, Garneau D, Cloutier P, Chabot B. Protein kinase C-dependent control of Bcl-x alternative splicing. Molecular and Cellular Biology. 2007;27(24):8431–8441. doi: 10.1128/MCB.00565-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Montes M, Cloutier A, Sánchez-Hernández N, et al. TCERG1 regulates alternative splicing of the Bcl-x gene by modulating the rate of RNA polymerase II transcription. Molecular and Cellular Biology. 2012;32(4):751–762. doi: 10.1128/MCB.06255-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Michelle L, Cloutier A, Toutant J, et al. Proteins associated with the exon junction complex also control the alternative splicing of apoptotic regulators. Molecular and Cellular Biology. 2012;32(5):954–967. doi: 10.1128/MCB.06130-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stickeler E, Kittrell F, Medina D, Berget SM. Stage-specific changes in SR splicing factors and alternative splicing in mammary tumorigenesis. Oncogene. 1999;18(24):3574–3582. doi: 10.1038/sj.onc.1202671. [DOI] [PubMed] [Google Scholar]
- 65.Scorilas A, Kyriakopoulou L, Katsaros D, Diamandis EP. Cloning of a gene (SR-A1), encoding for a new member of the human Ser/Arg-rich family of pre-mRNA splicing factors: overexpression in aggressive ovarian cancer. British Journal of Cancer. 2001;85(2):190–198. doi: 10.1054/bjoc.2001.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sun H, Chasin LA. Multiple splicing defects in an intronic false exon. Molecular and Cellular Biology. 2000;20(17):6414–6425. doi: 10.1128/mcb.20.17.6414-6425.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sironi M, Menozzi G, Riva L, et al. Silencer elements as possible inhibitors of pseudoexon splicing. Nucleic Acids Research. 2004;32(5):1783–1791. doi: 10.1093/nar/gkh341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang XHF, Chasin LA. Computational definition of sequence motifs governing constitutive exon splicing. Genes and Development. 2004;18(11):1241–1250. doi: 10.1101/gad.1195304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fairbrother WG, Chasin LA. Human genomic sequences that inhibit splicing. Molecular and Cellular Biology. 2000;20(18):6816–6825. doi: 10.1128/mcb.20.18.6816-6825.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang XHF, Leslie CS, Chasin LA. Dichotomous splicing signals in exon flanks. Genome Research. 2005;15(6):768–779. doi: 10.1101/gr.3217705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buratti E, Dhir A, Lewandowska MA, Baralle FE. RNA structure is a key regulatory element in pathology ATM and CFTR pseudoexon inclusion events. Nucleic Acids Research. 2007;35(13):4369–4383. doi: 10.1093/nar/gkm447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dhir A, Buratti E, Van Santen MA, Lührmann R, Baralle FE. The intronic splicing code: multiple factors involved in ATM pseudoexon definition. The EMBO Journal. 2010;29(4):749–760. doi: 10.1038/emboj.2009.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Scheinin I, Myllykangas S, Borze I, Böhling T, Knuutila S, Saharinen J. CanGEM: mining gene copy number changes in cancer. Nucleic Acids Research. 2008;36(1):D830–D835. doi: 10.1093/nar/gkm802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hu GK, Madore SJ, Moldover B, et al. Predicting splice variant from DNA chip expression data. Genome Research. 2001;11(7):1237–1345. doi: 10.1101/gr.165501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fan W, Khalid N, Hallahan AR, Olson JM, Lue PZ. A statistical method for predicting splice variants between two groups of samples using GeneChip expression array data. Theoretical Biology and Medical Modelling. 2006;3, article 19 doi: 10.1186/1742-4682-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Raponi M, Buratti E, Llorian M, Stuani C, Smith CWJ, Baralle D. Polypyrimidine tract binding protein regulates alternative splicing of an aberrant pseudoexon in NF1. FEBS Journal. 2008;275(24):6101–6108. doi: 10.1111/j.1742-4658.2008.06734.x. [DOI] [PubMed] [Google Scholar]
- 77.Wagner EJ, Garcia-Blanco MA. Minireview: polypyrimidine tract binding protein antagonizes exon definition. Molecular and Cellular Biology. 2001;21(10):3281–3288. doi: 10.1128/MCB.21.10.3281-3288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Spier I, Horpaopan S, Vogt S, et al. Deep intronic APC mutations explain a substantial proportion of patients with familial or early-onset adenomatous polyposis. Human Mutation. 2012;33(7):1045–1050. doi: 10.1002/humu.22082. [DOI] [PubMed] [Google Scholar]
- 79.Nakamura K, Du L, Tunuguntla R, et al. Functional characterization and targeted correction of atm mutations identified in japanese patients with ataxia-telangiectasia. Human Mutation. 2012;33(1):198–208. doi: 10.1002/humu.21632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pagani F, Buratti E, Stuani C, Bendix R, Dörk T, Baralle FE. A new type of mutation causes a splicing defect in ATM. Nature Genetics. 2002;30(4):426–429. doi: 10.1038/ng858. [DOI] [PubMed] [Google Scholar]
- 81.McConville CM, Stankovic T, Byrd PJ, et al. Mutations associated with variant phenotypes in ataxia-telangiectasia. American Journal of Human Genetics. 1996;59(2):320–330. [PMC free article] [PubMed] [Google Scholar]
- 82.Cavalieri S, Pozzi E, Gatti RA, Brusco A. Deep-intronic ATM mutation detected by genomic resequencing and corrected in vitro by antisense morpholino oligonucleotide (AMO) European Journal of Human Genetics. 2013;21(7):774–778. doi: 10.1038/ejhg.2012.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sorel N, Mayeur-Rousse C, Deverrière S, et al. Comprehensive characterization of a novel intronic pseudo-exon inserted within an e14/a2 BCR-ABL rearrangement in a patient with chronic myeloid leukemia. Journal of Molecular Diagnostics. 2010;12(4):520–524. doi: 10.2353/jmoldx.2010.090218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Laudadio J, Deininger MWN, Mauro MJ, Druker BJ, Press RD. An intron-derived insertion/truncation mutation in the BCR-ABL kinase domain in chronic myeloid leukemia patients undergoing kinase inhibitor therapy. Journal of Molecular Diagnostics. 2008;10(2):177–180. doi: 10.2353/jmoldx.2008.070128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Quigley NB, Henley DC, Hubbard RA, Laudadio J, Press RD. ABL kinase domain pseudoexon insertion is not uncommon in BCR-ABL transcripts. Journal of Molecular Diagnostics. 2008;10(5):475–476. doi: 10.2353/jmoldx.2008.080055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Balz V, Prisack HB, Bier H, Bojar H. Analysis of BRCA1, TP53, and TSG101 germline mutations in German breast and/or ovarian cancer families. Cancer Genetics and Cytogenetics. 2002;138(2):120–127. doi: 10.1016/s0165-4608(02)00601-5. [DOI] [PubMed] [Google Scholar]
- 87.Chen X, Truong TTN, Weaver J, et al. Intronic alterations in BRCA1 and BRCA2: effect on mRNA splicing fidelity and expression. Human Mutation. 2006;27(5):427–435. doi: 10.1002/humu.20319. [DOI] [PubMed] [Google Scholar]
- 88.Anczukow O, Buisson M, Leone M, et al. BRCA2 deep intronic mutation causing activation of a cryptic exon: opening toward a new preventive therapeutic strategy. Clinical Cancer Research. 2012;18:4903–4909. doi: 10.1158/1078-0432.CCR-12-1100. [DOI] [PubMed] [Google Scholar]
- 89.Wang M, Dotzlaw H, Fuqua SAW, Murphy LC. A point mutation in the human estrogen receptor gene is associated with the expression of an abnormal estrogen receptor mRNA containing a 69 novel nucleotide insertion. Breast Cancer Research and Treatment. 1997;44(2):145–151. doi: 10.1023/a:1005753117205. [DOI] [PubMed] [Google Scholar]
- 90.Clendenning M, Buchanan DD, Walsh MD, et al. Mutation deep within an intron of MSH2 causes Lynch syndrome. Familial Cancer. 2011;10(2):297–301. doi: 10.1007/s10689-011-9427-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fernández-Rodríguez J, Castellsagué J, Benito L, et al. A mild neurofibromatosis type 1 phenotype produced by the combination of the benign nature of a leaky NF1-splice mutation and the presence of a complex mosaicism. Human Mutation. 2011;32(7):705–709. doi: 10.1002/humu.21500. [DOI] [PubMed] [Google Scholar]
- 92.Pros E, Fernández-Rodríguez J, Canet B, et al. Antisense therapeutics for neurofibromatosis type 1 caused by deep intronic mutations. Human Mutation. 2009;30(3):454–462. doi: 10.1002/humu.20933. [DOI] [PubMed] [Google Scholar]
- 93.Raponi M, Upadhyaya M, Baralle D. Functional splicing assay shows a pathogenic intronic mutation in neurofibromatosis type 1 (NF1) due to intronic sequence exonization. Human mutation. 2006;27(3):294–295. doi: 10.1002/humu.9412. [DOI] [PubMed] [Google Scholar]
- 94.Pros E, Gómez C, Martín T, Fábregas P, Serra E, Lázaro C. Nature and mRNA effect of 282 different NF1 point mutations: focus on splicing alterations. Human mutation. 2008;29(9):E173–E193. doi: 10.1002/humu.20826. [DOI] [PubMed] [Google Scholar]
- 95.Wimmer K, Roca X, Beiglböck H, et al. Extensive in silico analysis of NF1 splicing defects uncovers determinants for splicing outcome upon 5′ splice-site disruption. Human Mutation. 2007;28(6):599–612. doi: 10.1002/humu.20493. [DOI] [PubMed] [Google Scholar]
- 96.Perrin G, Morris MA, Antonarakis SE, Boltshauser E, Hutter P. Two novel mutations affecting mRNA splicing of the neurofibromatosis type 1 (NF1) gene. Human Mutation. 1996;7:172–175. doi: 10.1002/(SICI)1098-1004(1996)7:2<172::AID-HUMU15>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 97.Ars E, Serra E, Garcia J, et al. Mutations affecting mRNA splicing are the most common molecular defects in patients with neurofibromatosis type 1. Human Molecular Genetics. 2000;9(2):237–247. doi: 10.1093/hmg/9.2.237. [DOI] [PubMed] [Google Scholar]
- 98.de Klein A, Riegman PHJ, Bijlsma EK, et al. A G→A transition creates a branch point sequence and activation of a cryptic exon, resulting in the hereditary disorder neurofibromatosis 2. Human Molecular Genetics. 1998;7(3):393–398. doi: 10.1093/hmg/7.3.393. [DOI] [PubMed] [Google Scholar]
- 99.Dehainault C, Michaux D, Pagès-Berhouet S, et al. A deep intronic mutation in the RB1 gene leads to intronic sequence exonisation. European Journal of Human Genetics. 2007;15(4):473–477. doi: 10.1038/sj.ejhg.5201787. [DOI] [PubMed] [Google Scholar]
- 100.Mayer K, Ballhausen W, Leistner W, Rott HD. Three novel types of splicing aberrations in the tuberous sclerosis TSC2 gene caused by mutations apart from splice consensus sequences. Biochimica et Biophysica Acta. 2000;1502(3):495–507. doi: 10.1016/s0925-4439(00)00072-7. [DOI] [PubMed] [Google Scholar]
- 101.Masala MV, Scapaticci S, Olivieri C, et al. Epidemiology and clinical aspects of Werner's syndrome in North Sardinia: description of a cluster. European Journal of Dermatology. 2007;17(3):213–216. doi: 10.1684/ejd.2007.0155. [DOI] [PubMed] [Google Scholar]
- 102.Friedrich K, Lee L, Leistritz DF, et al. WRN mutations in Werner syndrome patients: genomic rearrangements, unusual intronic mutations and ethnic-specific alterations. Human Genetics. 2010;128(1):103–111. doi: 10.1007/s00439-010-0832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Flanagan SE, Xie W, Caswell R, et al. Next-generation sequencing reveals deep intronic cryptic ABCC8 and HADH splicing founder mutations causing hyperinsulinism by pseudoexon activation. The American Journal of Human Genetics. 2013;92:131–136. doi: 10.1016/j.ajhg.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Milh M, Pop A, Kanhai W, et al. Atypical pyridoxine-dependent epilepsy due to a pseudoexon in ALDH7A1. Molecular Genetics and Metabolism. 2012;105(4):684–686. doi: 10.1016/j.ymgme.2012.01.011. [DOI] [PubMed] [Google Scholar]
- 105.Lee WI, Torgerson TR, Schumacher MJ, Yel L, Zhu Q, Ochs HD. Molecular analysis of a large cohort of patients with the hyper immunoglobulin M (IgM) syndrome. Blood. 2005;105(5):1881–1890. doi: 10.1182/blood-2003-12-4420. [DOI] [PubMed] [Google Scholar]
- 106.den Hollander AI, Koenekoop RK, Yzer S, et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. American Journal of Human Genetics. 2006;79:556–561. doi: 10.1086/507318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Collin RW, den Hollander AI, van der Velde-Visser SD, Bennicelli J, Bennett J, Cremers FP. Antisense Oligonucleotide (AON)-based Therapy for Leber Congenital Amaurosis caused by a Frequent Mutation in CEP290. Molecular Therapy. 2012;1, article e14 doi: 10.1038/mtna.2012.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.van den Hurk JAJM, van de Pol DJR, Wissinger B, et al. Novel types of mutation in the choroideremia (CHM) gene: a full-length L1 insertion and an intronic mutation activating a cryptic exon. Human Genetics. 2003;113(3):268–275. doi: 10.1007/s00439-003-0970-0. [DOI] [PubMed] [Google Scholar]
- 109.Knebelmann B, Forestier L, Drouot L, et al. Splice-mediated insertion of an Alu sequence in the COL4A3 mRNA causing autosomal recessive Alport syndrome. Human Molecular Genetics. 1995;4(4):675–679. doi: 10.1093/hmg/4.4.675. [DOI] [PubMed] [Google Scholar]
- 110.Akawi N, Al-Gazali L, Ali BR. Clinical and molecular analysis of UAE fibrochondrogenesis patients expands the phenotype and reveals two COL11A1 homozygous null mutations. Clinical Genetics. 2012;82:147–156. doi: 10.1111/j.1399-0004.2011.01734.x. [DOI] [PubMed] [Google Scholar]
- 111.Varon R, Gooding R, Steglich C, et al. Partial deficiency of the C-terminal-domain phosphatase of RNA polymerase II is associated with congenital cataracts facial dysmorphism neuropathy syndrome. Nature Genetics. 2003;35(2):185–189. doi: 10.1038/ng1243. [DOI] [PubMed] [Google Scholar]
- 112.Rump A, Rösen-Wolff A, Gahr M, et al. A splice-supporting intronic mutation in the last bp position of a cryptic exon within intron 6 of the CYBB gene induces its incorporation into the mRNA causing chronic granulomatous disease (CGD) Gene. 2006;371(2):174–181. doi: 10.1016/j.gene.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 113.Noack D, Heyworth PG, Newburger PE, Cross AR. An unusual intronic mutation in the CYBB gene giving rise to chronic granulomatous disease. Biochimica et Biophysica Acta. 2001;1537(2):125–131. doi: 10.1016/s0925-4439(01)00065-5. [DOI] [PubMed] [Google Scholar]
- 114.Hwang DY, Hung CC, Riepe FG, et al. CYP17A1 intron mutation causing cryptic splicing in 17α-hydroxylase deficiency. PloS ONE. 2011;6, article e25492(9) doi: 10.1371/journal.pone.0025492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ikeda H, Matsubara Y, Mikami H, et al. Molecular analysis of dihydropteridine reductase deficiency: identification of two novel mutations in Japanese patients. Human Genetics. 1997;100(5-6):637–642. doi: 10.1007/s004390050566. [DOI] [PubMed] [Google Scholar]
- 116.van Kuilenburg ABP, Meijer J, Mul ANPM, et al. Intragenic deletions and a deep intronic mutation affecting pre-mRNA splicing in the dihydropyrimidine dehydrogenase gene as novel mechanisms causing 5-fluorouracil toxicity. Human Genetics. 2010;128(5):529–538. doi: 10.1007/s00439-010-0879-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Guo DC, Gupta P, Tran-Fadulu V, et al. An FBN1 pseudoexon mutation in a patient with Marfan syndrome: confirmation of cryptic mutations leading to disease. Journal of Human Genetics. 2008;53(11-12):1007–1011. doi: 10.1007/s10038-008-0334-7. [DOI] [PubMed] [Google Scholar]
- 118.Metherell LA, Akker SA, Munroe PB, et al. Pseudoexon activation as a novel mechanism for disease resulting in atypical growth-hormone insensitivity. American Journal of Human Genetics. 2001;69(3):641–646. doi: 10.1086/323266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Akker SA, Misra S, Aslam S, et al. Pre-spliceosomal binding of U1 small nuclear ribonucleoprotein (RNP) and heterogenous nuclear RNP E1 is associated with suppression of a growth hormone receptor pseudoexon. Molecular Endocrinology. 2007;21(10):2529–2540. doi: 10.1210/me.2007-0038. [DOI] [PubMed] [Google Scholar]
- 120.Vervoort R, Gitzelmann R, Lissens W, Liebaers I. A mutation (IVS8+0.6kbdelTC) creating a new donor splice site activates a cryptic exon in an Alu-element in intron 8 of the human β-glucuronidase gene. Human Genetics. 1998;103(6):686–693. doi: 10.1007/pl00008709. [DOI] [PubMed] [Google Scholar]
- 121.Purevsuren J, Fukao T, Hasegawa Y, Fukuda S, Kobayashi H, Yamaguchi S. Study of deep intronic sequence exonization in a Japanese neonate with a mitochondrial trifunctional protein deficiency. Molecular Genetics and Metabolism. 2008;95(1-2):46–51. doi: 10.1016/j.ymgme.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 122.Sévenet N, Lellouch-Tubiana A, Schofield D, et al. Spectrum of hSNF5/INI1 somatic mutations in human cancer and genotype-phenotype correlations. Human Molecular Genetics. 1999;8(13):2359–2368. doi: 10.1093/hmg/8.13.2359. [DOI] [PubMed] [Google Scholar]
- 123.Olsson A, Lind L, Thornell LE, Holmberg M. Myopathy with lactic acidosis is linked to chromosome 12q23.3-24.11 and caused by an intron mutation in the ISCU gene resulting in a splicing defect. Human Molecular Genetics. 2008;17(11):1666–1672. doi: 10.1093/hmg/ddn057. [DOI] [PubMed] [Google Scholar]
- 124.Mochel F, Knight MA, Tong WH, et al. Splice mutation in the Iron-Sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. American Journal of Human Genetics. 2008;82(3):652–660. doi: 10.1016/j.ajhg.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kollberg G, Tulinius M, Melberg A, et al. Clinical manifestation and a new ISCU mutation in ironsulphur cluster deficiency myopathy. Brain. 2009;132(8):2170–2179. doi: 10.1093/brain/awp152. [DOI] [PubMed] [Google Scholar]
- 126.Lucien N, Chiaroni J, Cartron JP, Bailly P. Partial deletion in the JK locus causing a JK(null) phenotype. Blood. 2002;99(3):1079–1081. doi: 10.1182/blood.v99.3.1079. [DOI] [PubMed] [Google Scholar]
- 127.Stucki M, Suormala T, Fowler B, Valle D, Baumgartner MR. Cryptic exon activation by disruption of exon splice enhancer: a novel mechanism causing 3-methyl crotonyl-CoA carboxylase deficiency. The Journal of Biological Chemistry. 2009;284(42):28953–28957. doi: 10.1074/jbc.M109.050674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yamaguchi H, Fujimoto T, Nakamura S, et al. Aberrant splicing of the milk fat globule-EGF factor 8 (MFG-E8) gene in human systemic lupus erythematosus. European Journal of Immunology. 2010;40(6):1778–1785. doi: 10.1002/eji.200940096. [DOI] [PubMed] [Google Scholar]
- 129.Mancini C, Vaula G, Scalzitti L, et al. Megalencephalic leukoencephalopathy with subcortical cysts type 1 (MLC1) due to a homozygous deep intronic splicing mutation (c.895-226T>G) abrogated in vitro using an antisense morpholino oligonucleotide. Neurogenetics. 2012;13(3):205–214. doi: 10.1007/s10048-012-0331-z. [DOI] [PubMed] [Google Scholar]
- 130.Homolova K, Zavadakova P, Doktor TK, Schroeder LD, Kozich V, Andresen BS. The deep intronic c.903+469T > C mutation in the MTRR gene creates an SF2/ASF binding exonic splicing enhancer, which leads to pseudoexon activation and causes the cblE type of homocystinuria. Human Mutation. 2010;31(4):437–444. doi: 10.1002/humu.21206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Vetrini F, Tammaro R, Bondanza S, et al. Aberrant splicing in the ocular albinism type 1 gene (OA1/GPR143) is corrected in vitro by morpholino antisense oligonucleotides. Human Mutation. 2006;27(5):420–426. doi: 10.1002/humu.20303. [DOI] [PubMed] [Google Scholar]
- 132.Mitchell GA, Labuda D, Fontaine G, et al. Splice-mediated insertion of an Alu sequence inactivates ornithine δ-aminotransferase: a role for Alu elements in human mutation. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(3):815–819. doi: 10.1073/pnas.88.3.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Webb TR, Parfitt DA, Gardner JC, et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23) Human Molecular Genetics. 2012;21:3647–3654. doi: 10.1093/hmg/dds194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ogino W, Takeshima Y, Nishiyama A, et al. Mutation analysis of the ornithine transcarbamylase (OTC) gene in five Japanese OTC deficiency patients revealed two known and three novel mutations including a deep intronic mutation. Kobe Journal of Medical Sciences. 2007;53(5):229–240. [PubMed] [Google Scholar]
- 135.Rincón A, Aguado C, Desviat LR, Sánchez-Alcudia R, Ugarte M, Pérez B. Propionic and methylmalonic acidemia: antisense therapeutics for intronic variations causing aberrantly spliced messenger RNA. American Journal of Human Genetics. 2007;81(6):1262–1270. doi: 10.1086/522376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Christie PT, Harding B, Nesbit MA, Whyte MP, Thakker RV. X-linked hypophosphatemia attributable to pseudoexons of the PHEX gene. Journal of Clinical Endocrinology and Metabolism. 2001;86(8):3840–3844. doi: 10.1210/jcem.86.8.7730. [DOI] [PubMed] [Google Scholar]
- 137.Michel-Calemard L, Dijoud F, Till M, et al. Pseudoexon activation in the PKHD1 gene: a French founder intronic mutation IVS46+653A>G causing severe autosomal recessive polycystic kidney disease. Clinical Genetics. 2009;75(2):203–206. doi: 10.1111/j.1399-0004.2008.01106.x. [DOI] [PubMed] [Google Scholar]
- 138.Vega AI, Perez-Cerda C, Desviat LR, Matthijs G, Ugarte M, Pérez B. Functional analysis of three splicing mutations identified in the PMM2 gene: toward a new therapy for congenital disorder of glycosylation type IA. Human Mutation. 2009;30(5):795–803. doi: 10.1002/humu.20960. [DOI] [PubMed] [Google Scholar]
- 139.Schollen E, Keldermans L, Foulquier F, et al. Characterization of two unusual truncating PMM2 mutations in two CDG-Ia patients. Molecular Genetics and Metabolism. 2007;90(4):408–413. doi: 10.1016/j.ymgme.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 140.Frio TR, McGee TL, Wade NM, et al. A single-base substitution within an intronic repetitive element causes dominant retinitis pigmentosa with reduced penetrance. Human Mutation. 2009;30(9):1340–1347. doi: 10.1002/humu.21071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Liu HC, Eng HL, Yang YF, et al. Aberrant RNA splicing in RHD 7-9 exons of DEL individuals in Taiwan: A Mechanism Study. Biochimica et Biophysica Acta. 2010;1800(6):565–573. doi: 10.1016/j.bbagen.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 142.Monnier N, Ferreiro A, Marty I, Labarre-Villa A, Mezin P, Lunardi J. A homozygous splicing mutation causing a depletion of skeletal muscle RYR1 is associated with multi-minicore disease congenital myopathy with ophthalmoplegia. Human Molecular Genetics. 2003;12(10):1171–1178. doi: 10.1093/hmg/ddg121. [DOI] [PubMed] [Google Scholar]
- 143.Nozu K, Iijima K, Nozu Y, et al. A deep intronic mutation in the SLC12A3 gene leads to gitelman syndrome. Pediatric Research. 2009;66(5):590–593. doi: 10.1203/PDR.0b013e3181b9b4d3. [DOI] [PubMed] [Google Scholar]
- 144.Vaché C, Besnard T, le Berre P, et al. Usher syndrome type 2 caused by activation of an USH2A pseudoexon: implications for diagnosis and therapy. Human Mutation. 2012;33(1):104–108. doi: 10.1002/humu.21634. [DOI] [PubMed] [Google Scholar]
- 145.Ruoslahti E. Fibronectin and its receptors. Annual Review of Biochemistry. 1988;57:375–413. doi: 10.1146/annurev.bi.57.070188.002111. [DOI] [PubMed] [Google Scholar]
- 146.White ES, Baralle FE, Muro AF. New insights into form and function of fibronectin splice variants. Journal of Pathology. 2008;216(1):1–14. doi: 10.1002/path.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Vibe-Pedersen K, Magnusson S, Baralle FE. Donor and acceptor splice signals within an exon of the human fibronectin gene: a new type of differential splicing. FEBS Letters. 1986;207(2):287–291. doi: 10.1016/0014-5793(86)81506-x. [DOI] [PubMed] [Google Scholar]
- 148.Hershberger RP, Culp LA. Cell-type-specific expression of alternatively spliced human fibronectin IIICS mRNAs. Molecular and Cellular Biology. 1990;10(2):662–671. doi: 10.1128/mcb.10.2.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Mould AP, Humphries MJ. Identification of a novel recognition sequence for the integrin α4β1 in the COOH-terminal heparin-binding domain of fibronectin. The EMBO Journal. 1991;10(13):4089–4095. doi: 10.1002/j.1460-2075.1991.tb04985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Humphries MJ, Komoriya A, Akiyama SK. Identification of two distinct regions of the type III connecting segment of human plasma fibronectin that promote cell type-specific adhesion. The Journal of Biological Chemistry. 1987;262(14):6886–6892. [PubMed] [Google Scholar]
- 151.Mould AP, Askari JA, Craig SE, Garratt AN, Clements J, Humphries MJ. Integrin α4β1-mediated melanoma cell adhesion and migration on vascular cell adhesion molecule-1 (VCAM-1) and the alternatively spliced IIICS region of fibronectin. The Journal of Biological Chemistry. 1994;269(44):27224–27230. [PubMed] [Google Scholar]
- 152.Kumazaki T, Mitsui Y, Hamada K, Sumida H, Nishiyama M. Detection of alternative splicing of fibronectin mRNA in a single cell. Journal of Cell Science. 1999;112, part 10(10):1449–1453. doi: 10.1242/jcs.112.10.1449. [DOI] [PubMed] [Google Scholar]
- 153.Schofield KP, Humphries MJ. Identification of fibronectin IIICS variants in human bone marrow stroma. Blood. 1999;93(1):410–411. [PubMed] [Google Scholar]
- 154.Trefzer U, Chen Y, Herberth G, et al. The monoclonal antibody SM5-1 recognizes a fibronectin variant which is widely expressed in melanoma. BMC Cancer. 2006;6, article 8 doi: 10.1186/1471-2407-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mardon HJ, Sebastio G. Regulation of alternative splicing in the IIICS region of human fibronectin pre-mRNA encoding cell binding sites CS1 and CS5. Journal of Cell Science. 1992;103, part 2(2):423–433. doi: 10.1242/jcs.103.2.423. [DOI] [PubMed] [Google Scholar]
- 156.Silman I, Sussman JL. Acetylcholinesterase: “Classical” and “non-classical” functions and pharmacology. Current Opinion in Pharmacology. 2005;5(3):293–302. doi: 10.1016/j.coph.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 157.Soreq H, Seidman S. Acetylcholinesterase—new roles for an old actor. Nature Reviews Neuroscience. 2001;2(4):294–302. doi: 10.1038/35067589. [DOI] [PubMed] [Google Scholar]
- 158.Massoulié J. The origin of the molecular diversity and functional anchoring of cholinesterases. NeuroSignals. 2002;11(3):130–143. doi: 10.1159/000065054. [DOI] [PubMed] [Google Scholar]
- 159.Chan RYY, Adatia FA, Krupa AM, Jasmin BJ. Increased expression of acetylcholinesterase T and R transcripts during hematopoietic differentiation is accompanied by parallel elevations in the levels of their respective molecular forms. The Journal of Biological Chemistry. 1998;273(16):9727–9733. doi: 10.1074/jbc.273.16.9727. [DOI] [PubMed] [Google Scholar]
- 160.Perrier NA, Salani M, Falasca C, Bon S, Augusti-Tocco G, Massoulié J. The readthrough variant of acetylcholinesterase remains very minor after heat shock, organophosphate inhibition and stress, in cell culture and in vivo. Journal of Neurochemistry. 2005;94(3):629–638. doi: 10.1111/j.1471-4159.2005.03140.x. [DOI] [PubMed] [Google Scholar]
- 161.Grisaru D, Sternfeld M, Eldor A, Glick D, Soreq H. Structural roles of acetylcholinesterase variants in biology and pathology. European Journal of Biochemistry. 1999;264(3):672–686. doi: 10.1046/j.1432-1327.1999.00693.x. [DOI] [PubMed] [Google Scholar]
- 162.Meshorer E, Soreq H. Virtues and woes of AChE alternative splicing in stress-related neuropathologies. Trends in Neurosciences. 2006;29(4):216–224. doi: 10.1016/j.tins.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 163.Perry C, Sklan EH, Birikh K, et al. Complex regulation of acetylcholinesterase gene expression in human brain tumors. Oncogene. 2002;21(55):8428–8441. doi: 10.1038/sj.onc.1205945. [DOI] [PubMed] [Google Scholar]
- 164.Perry C, Sklan EH, Soreq H. CREB regulates AChE-R-induced proliferation of human glioblastoma cells. Neoplasia. 2004;6(3):279–286. doi: 10.1593/neo.3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Mor I, Bruck T, Greenberg D, et al. Alternate AChE-R variants facilitate cellular metabolic activity and resistance to genotoxic stress through enolase and RACK1 interactions. Chemico-Biological Interactions. 2008;175(1–3):11–21. doi: 10.1016/j.cbi.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 166.Montenegro MF, Ruiz-Espejo F, Campoy FJ, et al. Cholinesterases are down-expressed in human colorectal carcinoma. Cellular and Molecular Life Sciences. 2006;63(18):2175–2182. doi: 10.1007/s00018-006-6231-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Li B, Pan H, Winkelmann JC, Dai W. Thrombopoietin and its alternatively spliced form are expressed in human amygdala and hippocampus. Blood. 1996;87(12):5382–5384. [PubMed] [Google Scholar]
- 168.Sungaran R, Markovic B, Chong BH. Localization and regulation of thrombopoietin mRNA expression in human kidney, liver, bone marrow, and spleen using in situ hybridization. Blood. 1997;89(1):101–107. [PubMed] [Google Scholar]
- 169.Dame C, Wolber EM, Freitag P, Hofmann D, Bartmann P, Fandrey J. Thrombopoietin gene expression in the developing human central nervous system. Developmental Brain Research. 2003;143(2):217–223. doi: 10.1016/s0165-3806(03)00134-2. [DOI] [PubMed] [Google Scholar]
- 170.Marcucci R, Romano M. Thrombopoietin and its splicing variants: structure and functions in thrombopoiesis and beyond. Biochimica et Biophysica Acta. 2008;1782(7-8):427–432. doi: 10.1016/j.bbadis.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 171.Romano M, Marcucci R, Baralle FE. Splicing of constitutive upstream introns is essential for the recognition of intra-exonic suboptimal splice sites in the thrombopoietin gene. Nucleic Acids Research. 2001;29(4):886–894. doi: 10.1093/nar/29.4.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wada T, Nagata Y, Nagahisa H, et al. Characterization of the truncated thrombopoietin variants. Biochemical and Biophysical Research Communications. 1995;213(3):1091–1098. doi: 10.1006/bbrc.1995.2239. [DOI] [PubMed] [Google Scholar]
- 173.Hoffman RC, Andersen H, Walker K, et al. Peptide, disulfide, and glycosylation mapping of recombinant human thrombopoietin from Ser1 to Arg246. Biochemistry. 1996;35(47):14849–14861. doi: 10.1021/bi961075b. [DOI] [PubMed] [Google Scholar]
- 174.Wu XL, Nakayama M, Adamson JW. Identification of five new isoforms of murine thrombopoietin mRNA. Biochemical and Biophysical Research Communications. 2000;276(1):137–143. doi: 10.1006/bbrc.2000.3428. [DOI] [PubMed] [Google Scholar]
- 175.Sasaki Y, Takahashi T, Miyazaki H, et al. Production of thrombopoietin by human carcinomas and its novel isoforms. Blood. 1999;94(6):1952–1960. [PubMed] [Google Scholar]
- 176.Bonnal S, Vigevani L, Valcarcel J. The spliceosome as a target of novel antitumour drugs. Nature Reviews Drug Discovery. 2012;11:847–859. doi: 10.1038/nrd3823. [DOI] [PubMed] [Google Scholar]
- 177.Zaharieva E, Chipman JK, Soller M. Alternative splicing interference by xenobiotics. Toxicology. 2012;296(1–3):1–12. doi: 10.1016/j.tox.2012.01.014. [DOI] [PubMed] [Google Scholar]
- 178.Muntoni F, Wood MJA. Targeting RNA to treat neuromuscular disease. Nature Reviews Drug Discovery. 2011;10(8):621–637. doi: 10.1038/nrd3459. [DOI] [PubMed] [Google Scholar]
- 179.Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nature Reviews Drug Discovery. 2012;11(2):125–140. doi: 10.1038/nrd3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Aartsma-Rus A, Van Ommen GJB. Antisense-mediated exon skipping: a versatile tool with therapeutic and research applications. RNA. 2007;13(10):1609–1624. doi: 10.1261/rna.653607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Dominski Z, Kole R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(18):8673–8677. doi: 10.1073/pnas.90.18.8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kishore S, Stamm S. The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science. 2006;311(5758):230–232. doi: 10.1126/science.1118265. [DOI] [PubMed] [Google Scholar]
- 183.Aartsma-Rus A, van Vliet L, Hirschi M, et al. Guidelines for antisense oligonucleotide design and insight into splice-modulating mechanisms. Molecular Therapy. 2009;17(3):548–553. doi: 10.1038/mt.2008.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Heemskerk H, De Winter CL, Van Ommen GJB, Van Deutekom JCT, Aartsma-Rus A. Development of antisense-mediated exon skipping as a treatment for Duchenne muscular dystrophy. Annals of the New York Academy of Sciences. 2009;1175:71–79. doi: 10.1111/j.1749-6632.2009.04973.x. [DOI] [PubMed] [Google Scholar]
- 185.Kendall GC, Mokhonova EI, Moran M, et al. Dantrolene enhances antisense-mediated exon skipping in human and mouse models of Duchenne muscular dystrophy. Science Translational Medicine. 2012;4(164, article 164ra160) doi: 10.1126/scitranslmed.3005054. [DOI] [PubMed] [Google Scholar]
- 186.Gendron D, Carriero S, Garneau D, et al. Modulation of 5′ splice site selection using tailed oligonucleotides carrying splicing signals. BMC Biotechnology. 2006;6, article 5 doi: 10.1186/1472-6750-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Garcia-Blanco MA, Baraniak AP, Lasda EL. Alternative splicing in disease and therapy. Nature Biotechnology. 2004;22(5):535–546. doi: 10.1038/nbt964. [DOI] [PubMed] [Google Scholar]
- 188.Nissim-Rafinia M, Aviram M, Randell SH, et al. Restoration of the cystic fibrosis transmembrane conductance regulator function by splicing modulation. EMBO Reports. 2004;5(11):1071–1077. doi: 10.1038/sj.embor.7400273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Pros E, Fernández-Rodríguez J, Benito L, et al. Modulation of aberrant NF1 pre-mRNA splicing by kinetin treatment. European Journal of Human Genetics. 2010;18(5):614–617. doi: 10.1038/ejhg.2009.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Cermak T, Doyle EL, Christian M, et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Research. 2011;39(12, article e82) doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]