

Abstract
Hypomethylated, CpG-rich DNA segments (CpG islands, CGIs) are epigenome markers involved in key biological processes. Aberrant methylation is implicated in the appearance of several disorders as cancer, immunodeficiency, or centromere instability. Furthermore, methylation differences at promoter regions between human and chimpanzee strongly associate with genes involved in neurological/psychological disorders and cancers. Therefore, the evolutionary comparative analyses of CGIs can provide insights on the functional role of these epigenome markers in both health and disease. Given the lack of specific tools, we developed CpGislandEVO. Briefly, we first compile a database of statistically significant CGIs for the best assembled mammalian genome sequences available to date. Second, by means of a coupled browser front-end, we focus on the CGIs overlapping orthologous genes extracted from OrthoDB, thus ensuring the comparison between CGIs located on truly homologous genome segments. This allows comparing the main compositional features between homologous CGIs. Finally, to facilitate nucleotide comparisons, we lifted genome coordinates between assemblies from different species, which enables the analysis of sequence divergence by direct count of nucleotide substitutions and indels occurring between homologous CGIs. The resulting CpGislandEVO database, linking together CGIs and single-cytosine DNA methylation data from several mammalian species, is freely available at our website.
1. Introduction
Short stretches of CpG dinucleotides (CpG islands or CGIs) predominantly hypomethylated in healthy tissues [1, 2] are key epigenomic markers in mammalian genomes [3]. Almost all housekeeping genes and a half of the tissue-specific genes are associated to CGIs [4]. DNA methylation plays an important role in the origin as well as in the function of CGIs. Aberrant methylation (mostly hypermethylation) of CGIs can lead to several syndromes, such as cancer [5–10]. Moreover, although it has been shown that certain human diseases may have evolutionary epigenetic origins [11, 12], it remains largely unknown how patterns of DNA methylation differ between closely related species and whether such differences contribute to species-specific phenotypes [11]. Some methylation databases [13–15] and CGI databases [16] have been developed, but, to our knowledge, no existing genome browser addresses specifically the evolutionary relationships between the CGIs from different species. To help describing and understanding the function as well as the mechanisms generating and maintaining CGIs within an evolutionary context, we develop here CpGislandEVO (http://bioinfo2.ugr.es/CpGislandEVO/index.php). The database, coupled to a powerful genome browser, links together experimental and predicted CGIs, as well as single-cytosine-resolution DNA methylation data from different mammalian species.
Early analyses of CGI evolution were based on compositional comparisons between islands from different species but located on homologous gene contexts [17, 18]. Recently, the rapidly increasing number of sequenced genomes enabled evolutionary studies relying on multiple-sequence alignments [19]. Here, we combine both approaches to envisage accurate sequence comparisons between CGIs located on homologous gene contexts.
2. Material and Methods
2.1. Genome Assemblies
Updated chromosome sequences for the best assembled mammalian genomes (Homo sapiens (hg19), Pan troglodytes (panTro3), Gorilla gorilla (gorGor3), Pongo abelii (ponAbe2), Macaca mulatta (rheMac2), Mus musculus (mm10), and Rattus norvegicus (rn5)) were downloaded from the UCSC genome browser [20].
2.2. CGI Predictions
CGIs were predicted by means of an improved version [21] of the CpGcluster algorithm [22]. We used the genome intersection as distance threshold to define the clusters of CpG dinucleotides and a P value threshold of 1E-5 for the statistical significance. For comparison, the database also includes the CGI predictions for hg19 made by a window-based program [23], as well as the UCSC island track for the different species [20].
2.3. Experimental CGIs
Experimental CGI datasets include the 13,277 nonoverlapping promoter regions which are unmethylated in at least one of the two tissues (fibroblast and sperm) analyzed by Weber et al. [24] and the 17,383 CpG-islands experimentally detected in blood cells [25].
2.4. Orthologous Gene-Contexts
To ensure that we are comparing truly homologous genome segments, we focus on the CGIs located around orthologous genes extracted from OrthoDB [26]. The OrthoDB implementation employs a best-reciprocal-hit clustering algorithm based on all-against-all Smith-Waterman [27] protein sequence comparisons. In particular, we take into account all the islands within the gene-body of each of the OrthoDB genes. We defined the gene-body as the region extending from 500 bp upstream from the transcription start site (txStart) to 500 bp downstream the transcription stop site (txEnd).
2.5. Sequence Comparisons
Base level comparisons of homologous CpG-island sequences from different species were carried out by lifting genome coordinates between assemblies by means of the Galaxy implementation [28, 29] of the LiftOver utility, based on the Chain and Net tracks from the UCSC Genome Browser (http://genome.ucsc.edu/cgi-bin/hgLiftOver/). Default parameters were used, except for the “minimum matching region size for the query” which was set to 8 bp, which corresponds to the smallest CGI length.
2.6. Methylation Data
Since the lack of CGI methylation is a very good indicator of function [30], we linked CpGislandEVO to a relevant subset of NGSmethDB [31, 32], where a wide variety of single-cytosine-resolution methylation methylome maps from different tissues and species are available. Methylomes were obtained with NGSmethPipe [33] (http://bioinfo2.ugr.es/NGSmethPipe/) and MethylExtract [34] (http://bioinfo2.ugr.es/MethylExtract/), two tools implementing stringent quality controls to minimize important error sources, as for example sequencing errors, bisulfite failures, clonal reads, or single nucleotide variants. Likewise important, the use of a single bioinformatics protocol homogenizes the database content making the different methylomes comparable among each other even if they are from different studies.
2.7. Database and Genome-Browser Implementation
The orthologous genes were taken from OrthoDB [26], which does not provide information about gene names or coordinates. Therefore, this information is obtained from ensGene (using ensemblIDs as identifiers) and refGene (using gene names) from UCSC [20]. The curated online repository of HGNC-approved gene nomenclature [35] is then used to link names between ensGene and refGene databases.
We implemented an autocomplete function to help the user locate human genes in OrthoDB [26] with at least one orthologous gene in any of the other species (via its gene name or its UniProtID). Once chosen a gene name, the refGene database (214,898 entries) and, if no results are found, the ensGene database (647,600 entries) are searched for this gene. As an output, the chromosome and the start and stop positions of this gene are obtained; a final check for at least one CGI within this gene-body is performed. Then, OrthoDB is queried with the ensemblID of the human gene, and a table with the ensemblIDs of the orthologous genes found in any of the six animal species is generated (Table 1). This table also contains known gene names obtained by converting back ensemblIDs via biomart databases [36].
Table 1.
List of orthologous genes to the human query gene KDM1A for the lysine-specific histone demethylase 1A, offering links to Ensembl and UCSC genome browsers, as well as to a specific island viewer based on JBrowse.
(a) Query gene
| Species | Gene name | Link Ensembl | Link UCSC | Link JBrowseViewer |
|---|---|---|---|---|
| Homo sapiens | KDM1A | hg_KDM1A_ensembl | hg_KDM1A_ucsc | hg_KDM1A_evoDB |
(b) Orthologous genes
| Species | Gene name (EnsemblID) | Link Ensembl | Link UCSC | Link JBrowseViewer |
|---|---|---|---|---|
| Gorilla gorilla | KDM1A (ENSGGOG00000003664) | gorgor_KDM1A_ensembl | gorgor_KDM1A_ucsc | gorgor_KDM1A_evoDB |
| Macaca mulatta | KDM1A (ENSMMUG00000009773) | rhemac_KDM1A_ensembl | rhemac_KDM1A_ucsc | rhemac_KDM1A_evoDB |
| Mus musculus | Kdm1a (ENSMUSG00000036940) | mm_kdm1a_ensembl | mm_kdm1a_ucsc | mm_kdm1a_evoDB |
| Pan troglodytes | KDM1A (ENSPTRG00000000321) | pantro_KDM1A_ensembl | pantro_KDM1A_ucsc | pantro_KDM1A_evoDB |
| Pongo abelii | KDM1A (ENSPPYG00000001747) | ponabe_KDM1A_ensembl | ponabe_KDM1A_ucsc | ponabe_KDM1A_evoDB |
| Rattus norvegicus | Kdm1 (ENSRNOG00000022372) | rn_kdm1a_ensembl | rn_kdm1a_ucsc | rn_kdm1a_evoDB |
When the user introduces a chromosome and an approximate coordinate, the script searches the ensGene table for the closest upstream and/or downstream gene with at least one orthologous gene in orthoDB, then returning the exact chromosome position and gene name. The corresponding ensemblID is then used as above to generate Table 1.
The most recent version (currently 1.9.7) of the cross-platform genome browser JBrowse [37, 38] is used to display genes, CpGislands, LiftOver-mapped tracks, and methylation tracks for the hg19 assembly and to compare it to the other six mammalian species. A pair-wise comparison is performed by means of two frames within a window: the top one is always used to display hg19 tracks and the bottom one for one of the six animal species. Note that, by the moment, both frames are not synchronized. This feature will be implemented as soon as Jbrowse_syn is available (http://gmod.org/w/images/a/aa/ISyIPGMODforComparativeGenomics.pdf, slide 15).
Currently, CpGislandEVO includes the mammalian genomes with comparable genome-wide methylation data (human, chimpanzee, rhesus monkey, and mouse). In this way, the platform allows the user to compare CGIs from these mammalian species. The number of species and methylation datasets will be increased according to the advent of new comparable genome-wide methylation datasets.
2.8. Data Download and Script Availability
The datasets in CpGislandEVO can be freely downloaded by the user from NGSmethDB (http://bioinfo2.ugr.es/NGSmethDB/database.php) in a wide variety of standard data formats: BED, GFF3, bedGraph, Wiggle, and so forth. The Perl script for the most recent version of CpGcluster is also freely available to download from the group webserver (http://bioinfo2.ugr.es/CpGcluster/CpGcluster.zip).
3. Results and Discussion
We first compiled a CGI database (http://bioinfo2.ugr.es/CpGislandEVO/launch.php) for the best assembled mammalian genomes using the CpGcluster algorithm [22] with the genome intersection as distance threshold [21, 22, 39]. This setup is especially appropriate for interspecies comparative genomic studies as (i) the distance threshold is directly obtained from the genome sequence and (ii) a P value is assigned to each CGI. Therefore, exactly the same criterions are used in all species to detect CpG islands. This is not possible when using algorithms based on sliding windows to predict CGIs, as variations in genome G+C content, O/E ratio, or CpG density cannot be easily taken into account to guarantee an unbiased detection [39]. Second, by means of a specifically designed genome browser based on JBrowse [37, 38], we focus on those CGIs located within orthologous gene-contexts [26], thus ensuring that we are comparing CGIs from true homologous sequence segments. Finally, to study sequence divergence at base level between homologous CpG islands, we lifted genome coordinates between assemblies from different species by using the LiftOver utility [20].
The CpGislandEVO platform first offers the possibility to explore the CGI database obtained with the CpGcluster algorithm [21, 22, 39]. After selecting genome assembly and chromosome, the server offers links to (i) CpGcluster predictions, (ii) UCSC genome browser [20], and (iii) single-cytosine methylation data by means of a subset (http://bioinfo2.ugr.es/CpGislandEVO/methylation.php) of NGSmethDB [31, 32]. Summary statistics for the CGI database and CGI distribution in the orthologous gene bodies of the different species are shown on-line: http://bioinfo2.ugr.es/CpGislandEVO/statistics.php. Second, a coupled genome browser allows sequence comparisons between CGIs located on homologous segments from different species. The user can navigate the database in two ways: (1) by directly introducing a human gene/protein reference name or (2) by providing a chromosome and an approximate coordinate (and then the closest upstream and/or downstream human gene with at least one orthologous gene will be shown). The server first returns the orthologous genes (Table 1) for the query gene with links to Ensembl [40] and UCSC [20] genome browsers, as well as to a specific island viewer we have developed on the basis of the JBrowse next-generation browser [37, 38]. The CpGislandEVO viewer allows the comparative genomics of CGIs in different species.
As an example, we focus on the human query gene KDM1A for the lysine-specific histone demethylase 1A. Figure 1 shows the promoter region of this gene and the CGIs and methylation data for PBMC cells. The homologous CGIs from six other species are shown for comparison. The small methylated human CGI is conserved in the three primate species, while the larger unmethylated human CGI is conserved even in the mouse. On the other hand, Figure 2 uses two frames within the same window, to compare the CGIs in the promoter region of the gene KDM1A in human and rhesus monkey. The unmethylated CGI is conserved between the two species, while the small human differentially methylated CGI is missing in the rhesus monkey. In this way, CpGislandEVO put together in the same screen information scattered in diverse sources, or only attainable after running different computer programs, thus allowing evolutionary compositional comparisons as well as accurate sequence analyses between islands from different species, but located on homologous gene contexts.
Figure 1.
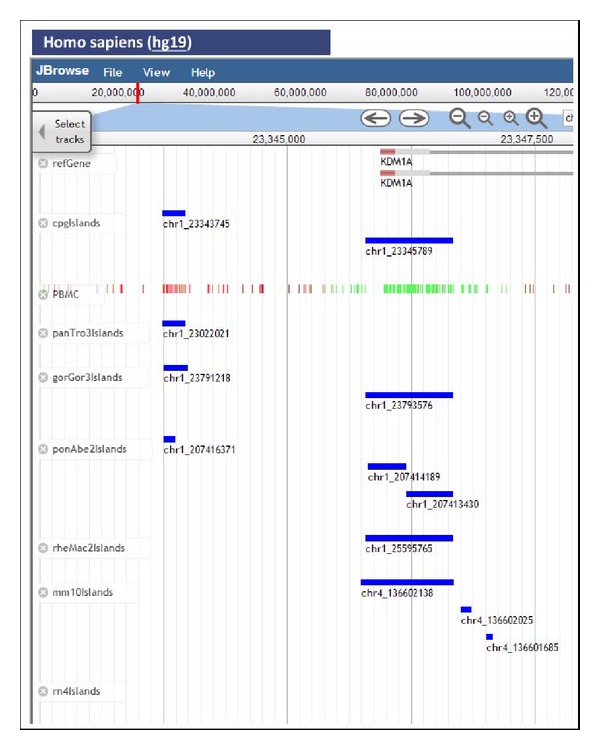
Promoter region of the human gene KDM1A showing CGIs and methylation data for PBMC cells. The lifted homologous CGIs from six other species are shown for comparison. The small methylated human CGI is conserved in three primate species, while the larger unmethylated human CGI is not only conserved in some primates but also in the mouse.
Figure 2.
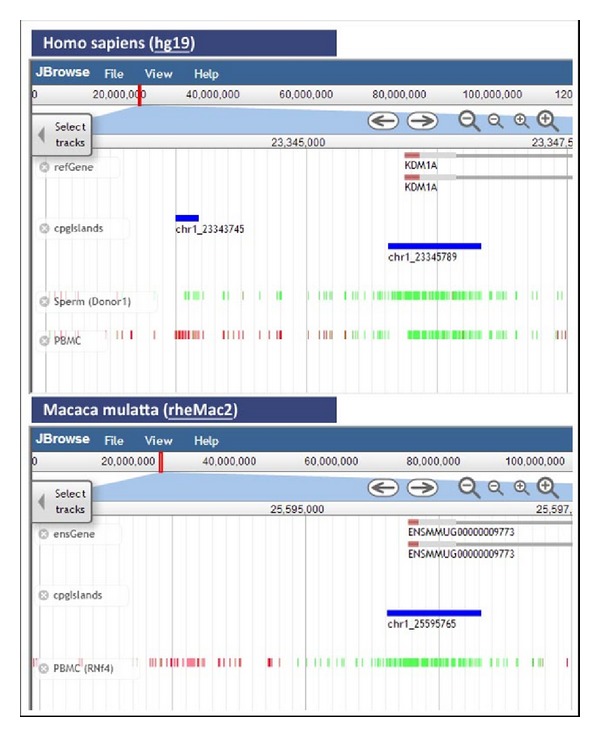
Comparison of the promoter region of the gene KDM1A in human and rhesus monkey using two frames within the same window. The unmethylated CGI is conserved between the two species, while the small human differentially methylated CGI is missing in the rhesus monkey.
4. Conclusions
We have compiled a database of statistically significant CGIs for the best assembled mammalian genomes using an improved version of the CpGcluster algorithm [21, 22, 39]. Then, by means of a specifically designed genome-browser based on JBrowse [37, 38], we focused on those CGIs located within orthologous gene-contexts [26], thus ensuring that we are comparing CGIs from true homologous genome segments. Finally, by lifting genome coordinates between assemblies from different species, the CpGislandEVO platform allows the direct comparison at base level between homologous CpG islands. The evolutionary comparative studies of CGIs can provide insights on their functional role in both health and disease, as well as on the evolutionary mechanisms generating and maintaining these important epigenome markers.
Authors' Contribution
Guillermo Barturen and Stefanie Geisen contributed equally to this work.
Acknowledgments
This work was supported by the Spanish Government [BIO2008-01353 to José L. Oliver and BIO2010-20219 to Michael Hackenberg], Basque country “AE” grant (to Guillermo Barturen) and Erasmus internships (to Stefanie Geisen, Francisco Dios, and E. J. Maarten Hamberg). The authors greatly acknowledge the continuous support by Robert Buels, Lead Developer of JBrowse.
References
- 1.Bird A. DNA methylation patterns and epigenetic memory. Genes and Development. 2002;16(1):6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 2.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321(6067):209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 3.Schubeler D. Molecular biology. Epigenetic islands in a genetic ocean. Science. 2012;338(6108):756–757. doi: 10.1126/science.1227243. [DOI] [PubMed] [Google Scholar]
- 4.Zhu J, He F, Hu S, Yu J. On the nature of human housekeeping genes. Trends in Genetics. 2008;24(10):481–484. doi: 10.1016/j.tig.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 5.Baylin SB, Esteller M, Rountree MR, Bachman KE, Schuebel K, Herman JG. Abberant patterns of DNA methylation, chromatin formation and gene expression in cancer. Human Molecular Genetics. 2001;10(7):687–692. doi: 10.1093/hmg/10.7.687. [DOI] [PubMed] [Google Scholar]
- 6.de Smet C, Lurquin C, Lethé B, Martelange V, Boon T. DNA methylation is the primary silencing mechanism for a set of germ line- and tumor-specific genes with a CpG-rich promoter. Molecular and Cellular Biology. 1999;19(11):7327–7335. doi: 10.1128/mcb.19.11.7327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Research. 2001;61(8):3225–3229. [PubMed] [Google Scholar]
- 8.Issa J-P. CpG island methylator phenotype in cancer. Nature Reviews Cancer. 2004;4(12):988–993. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- 9.Riazalhosseini Y, Hoheisel JD. Do we use the appropriate controls for the identification of informative methylation markers for early cancer detection? Genome Biology. 2008;9(11, article 405) doi: 10.1186/gb-2008-9-11-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krebs AR, Schubeler D. Tracking the evolution of cancer methylomes. Nature Genetics. 2012;44(11):1173–1174. doi: 10.1038/ng.2451. [DOI] [PubMed] [Google Scholar]
- 11.Zeng J, Konopka G, Hunt BG, Preuss TM, Geschwind D, Yi SV. Divergent whole-genome methylation maps of human and chimpanzee brains reveal epigenetic basis of human regulatory evolution. The American Journal of Human Genetics. 2012;91(3):455–465. doi: 10.1016/j.ajhg.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bell CG, Wilson GA, Butcher LM, Roos C, Walter L, Beck S. Human-specific CpG, “beacons” identify loci associated with human-specific traits and disease. Epigenetics. 2012;7(10):1188–1199. doi: 10.4161/epi.22127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Negre V, Grunau C. The MethDB DAS server: adding an epigenetic information layer to the human genome. Epigenetics. 2006;1(2):101–105. doi: 10.4161/epi.1.2.2765. [DOI] [PubMed] [Google Scholar]
- 14.Shi J, Hu J, Zhou Q, Du Y, Jiang C. PEpiD: a prostate epigenetic database in mammals. PLoS One. 2013;8(5) doi: 10.1371/journal.pone.0064289.e64289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu F, Doderer MS, Huang Y-W, et al. CMS: a web-based system for visualization and analysis of genome-wide methylation data of human cancers. PLoS One. 2013;8(4) doi: 10.1371/journal.pone.0060980.e60980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuo H-C, Lin P-Y, Chung T-C, et al. DBCAT: database of CpG islands and analytical tools for identifying comprehensive methylation profiles in cancer cells. Journal of Computational Biology. 2011;18(8):1013–1017. doi: 10.1089/cmb.2010.0038. [DOI] [PubMed] [Google Scholar]
- 17.Aissani B, Bernardi G. CpG islands, genes and isochores in the genomes of vertebrates. Gene. 1991;106(2):185–195. doi: 10.1016/0378-1119(91)90198-k. [DOI] [PubMed] [Google Scholar]
- 18.Jabbari K, Bernardi G. CpG doublets, CpG islands and Alu repeats in long human DNA sequences from different isochore families. Gene. 1998;224(1-2):123–128. doi: 10.1016/s0378-1119(98)00474-0. [DOI] [PubMed] [Google Scholar]
- 19.Cohen NM, Kenigsberg E, Tanay A. Primate CpG islands are maintained by heterogeneous evolutionary regimes involving minimal selection. Cell. 2011;145(5):773–786. doi: 10.1016/j.cell.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 20.Karolchik D, Kuhn RM, Baertsch R, et al. The UCSC genome browser database: 2008 update. Nucleic Acids Research. 2008;36(1):D773–D779. doi: 10.1093/nar/gkm966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hackenberg M, Carpena P, Bernaola-Galván P, Barturen G, Alganza ÁM, Oliver JL. WordCluster: detecting clusters of DNA words and genomic elements. Algorithms for Molecular Biology. 2011;6(1, article 2) doi: 10.1186/1748-7188-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hackenberg M, Previti C, Luque-Escamilla PL, Carpena P, Martínez-Aroza J, Oliver JL. CpGcluster: a distance-based algorithm for CpG-island detection. BMC Bioinformatics. 2006;7(article 446) doi: 10.1186/1471-2105-7-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takai D, Jones PA. The CpG island searcher: a new WWW resource. In Silico Biology. 2003;3(3):235–240. [PubMed] [Google Scholar]
- 24.Weber M, Hellmann I, Stadler MB, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nature Genetics. 2007;39(4):457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 25.Illingworth R, Kerr A, Desousa D, et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biology. 2008;6(1, article e22) doi: 10.1371/journal.pbio.0060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waterhouse RM, Zdobnov EM, Tegenfeldt F, Li J, Kriventseva EV. OrthoDB: the hierarchical catalog of eukaryotic orthologs in 2011. Nucleic Acids Research. 2011;39(1):D283–D288. doi: 10.1093/nar/gkq930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith TF, Waterman MS. Identification of common molecular subsequences. Journal of Molecular Biology. 1981;147(1):195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 28.Giardine B, Riemer C, Hardison RC, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome Research. 2005;15(10):1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biology. 2010;11(8, article R86) doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Illingworth RS, Bird AP. CpG islands—‘a rough guide’. FEBS Letters. 2009;583(11):1713–1720. doi: 10.1016/j.febslet.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 31.Hackenberg M, Barturen G, Oliver JL. NGSmethDB: a database for next-generation sequencing single-cytosine- resolution DNAmethylation data. Nucleic Acids Research. 2011;39(1):D75–D79. doi: 10.1093/nar/gkq942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Geisen S, et al. NGSmethDB: high-quality, single-cytosine resolution methylation maps. doi: 10.1093/nar/gkt1202. submitted to Nucleic Acids Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hackenberg M, Barturen G, Oliver JL. Methylation profiling from high-throughput sequencing data. In: Tatarinova T, Kerton O, editors. DNA Methylation—From Genomics to Technology. In-Tech; 2012. p. p. 27. [Google Scholar]
- 34.Barturen G, Rueda A, Oliver JL, Hackenberg M. MethylExtract: high-quality methylation maps and SNV calling from whole genome bisulfite sequencing data. doi: 10.12688/f1000research.2-217.v1. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gray KA, Daugherty LC, Gordon SM, Seal RL, Wright MW, Bruford EA. Genenames.org: the HGNC resources in 2013. Nucleic Acids Research. 2013;41:D545–D552. doi: 10.1093/nar/gks1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kasprzyk A. BioMart: driving a paradigm change in biological data management. Database. 2011;2011(article bar049) doi: 10.1093/database/bar049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skinner ME, Holmes IH. Setting up the JBrowse genome browser. Current Protocols in Bioinformatics. 2010;(chapter 9):p. unit 9.13. doi: 10.1002/0471250953.bi0913s32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skinner ME, Uzilov AV, Stein LD, Mungall CJ, Holmes IH. JBrowse: a next-generation genome browser. Genome Research. 2009;19(9):1630–1638. doi: 10.1101/gr.094607.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hackenberg M, Barturen G, Carpena P, Luque-Escamilla PL, Previti C, Oliver JL. Prediction of CpG-island function: CpG clustering vs. sliding-window methods. BMC Genomics. 2010;11(1, article 327) doi: 10.1186/1471-2164-11-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hubbard TJ, Aken BL, Ayling S, et al. Ensembl 2009. Nucleic Acids Research. 2009;37(database issue):D690–D697. doi: 10.1093/nar/gkn828. [DOI] [PMC free article] [PubMed] [Google Scholar]