

Abstract
Many protein-protein interactions that play a central role in cellular processes involve α-helical domains. Consequently, there has been great interest in developing strategies for stabilizing short peptides in α-helical conformations toward the inhibition and interrogation of protein-protein interactions. Here, we show that tridentate Hybrid Coordination Motifs (HCMs), which consist of a natural (histidine, His) and an unnatural (8-hydroxyquinoline, Quin) metal binding functionality, can bind divalent metal ions with high affinity and thereby induce/stabilize an α-helical configuration in short peptide sequences. The Quin functionality is readily introduced onto peptide platforms both during or after solid-state peptide synthesis, demonstrating the preparative versatility of HCMs. A systematic study involving a series of HCM-bearing peptides has revealed the critical importance of the length of the linkage between the Quin moiety and the peptide backbone as well as the metal coordination geometry in determining the extent of α-helix induction. Through ZnII coordination or modification with ReI(Quin)(CO)3, the HCM-bearing peptides can be rendered luminescent in the visible region, thus showing that HCMs can be exploited to simultaneously introduce structure and functionality into short peptides.
Introduction
Many natural protein-protein interactions (PPIs) are mediated by α-helical recognition and binding motifs on protein surfaces.1 Because of the central involvement of PPIs in all cellular processes, considerable research activity has focused on designing non-natural,2 protein-3 or peptide-based platforms that structurally and chemically mimic α-helical protein surface motifs.4 A drawback of using small peptidic platforms is that they are generally unstructured and susceptible to proteolytic cleavage.5 To overcome this challenge, a number of strategies have been devised to stabilize them in α-helical conformations. These strategies include the incorporation of α–amino acids with restricted conformation space,6 inclusion of salt-bridging residues in i/i+4 positions,7 crosslinking of side chains through covalent bonds8 or metal-coordination,9, 10, 11, 12 and utilization of hydrogen bond surrogates.13 Each of these platforms has its own set of advantages concerning ease of preparation, the extent of protein modification and α-helix induction, stability, target recognition and in vivo uptake, but none of them simultaneously offer all of these advantages or are universally applicable. Moreover, in most instances, these helix induction motifs solely serve a structural purpose, with little functional value added to the peptide. Therefore, it would be desirable to have access to alternative experimental platforms for helix induction that are easy to incorporate and modify, while simultaneously allowing the peptide to be functionalized and its secondary structure and other physicochemical properties to be easily modified.
Toward this goal, we present here short peptides that display Hybrid Coordination Motifs (HCMs), which are tridentate metal coordination modules consisting of a natural metal-coordinating residue such as histidine (His) at position i and an unnatural, bidentate chelating group such as 8-hydroxyquinoline (Quin) or 1,10-phenanthroline (Phen) covalently attached to the side chain at position i+7 (Figure 1). We first introduced HCMs as high-affinity coordination units on the surface of a folded, four-helix-bundle protein (cytochrome cb562) to mediate protein self-assembly upon metal coordination.14, 15 These studies revealed that the stability of the entire cyt cb562 fold increased by up to 4 kcal/mol upon binding of various divalent transition metal ions (CoII, NiII, CuII and ZnII), suggesting that HCMs might also be employed toward induction of α-helicity in unstructured peptides while allowing the incorporation of metal-based functionalities. Here, we have undertaken a systematic study in which we explored the chemical and physical properties of 10-residue-long peptides decorated with i/i+7 His/Quin HCMs. We describe that the Quin moiety can be readily installed onto peptides either after solid-phase peptide synthesis (SPPS) via coupling to a cysteine (Cys) side chain or in the form of an unnatural amino acid during SPPS. We have found that the induction of α-helicity is critically dependent on the linker length between the Quin moiety and the peptide backbone, and that the extent of α-helicity can be controlled by the choice of the coordinating metal ion. The His/Quin HCMs can be rendered luminescent by through ZnII binding, or alternatively, via coordination to a luminescent and substitution-inert ReI(Quin)(CO)3 moiety following SPPS. Finally, it is shown that His/Quin HCMs can induce considerable α-helicity in a peptide sequence that mimics the BH3 domain of the pro-apoptotic protein Bax. Thus, HCMs represent a versatile, easily implementable platform that allows simultaneous induction/modulation of α-helicity and the introduction of metal-based functionalities into peptide sequences.
Figure 1.

(a) Proposed scheme for α-helix induction through tridentate metal coordination by an HCM. The remaining coordination sites on the metal are likely filled by aquo ligands. (b) Chemical structures of various Quin functionalities. (c) Sequences of peptide constructs prepared in this study. The coloring scheme corresponds to that in (a).
Results and Discussion
Preparation of peptides containing His/Quin HCMs
General
The sequence and basic architecture of the HCM-bearing peptide platforms we have investigated are shown in Figure 1. All constructs are 10-residue, alanine (Ala)-rich peptides that were synthesized with a C-terminal amide and an acylated N-terminus via SPPS using standard Fmoc protecting group chemistry. Complete details on synthesis, peptide modification, purification and chemical analysis can be found in Supporting Information (SI). In general, the peptide sequences contained a His residue at position 2 (i) and a Quin-functionalized sidechain at position 9 (i+7), which would be two turns away in an α-helical configuration. The parent sequence also contained a lysine (Lys) at position 4 and a glutamic acid (Glu) at position 8 to form a potential salt bridge on a second face of the helix to further promote α-helicity and to increase the solubility of the peptide.
Routes for the incorporation of Quin functionality
The Quin functionality was introduced in two different ways: either during SPPS in the form of a pre-synthesized unnatural amino acid or after SPPS through covalent coupling to a Cys residue. For the former strategy, two Quin-bearing, Fmoc-protected amino acids (EQuin and EMeQuin, Figure 1b) were first prepared through the HATU-mediated coupling of 5-amino-8-hydroxyquinoline or 5-aminomethyl-8-hydroxyquinoline to Fmoc-protected L-Glu (see SI for synthetic details). EMeQuin presents the Quin functionality one extra methylene group removed from the peptide backbone compared to EQuin. The third Quin variant, CQuin, was obtained through the covalent coupling of 5-iodoacetamido-8-hydroxyquinoline (IA-Quin) to a Cys at position 9 following SPPS, paralleling our previous strategy to place a Quin moiety on the surface of the cytochrome cb562.14 CQuin, like EMeQuin, is separated from the peptide backbone by five single bonds although two of these bonds are slightly longer thioether linkages. It is important to note that the pre-SPPS preparative route of EQuin and EMeQuin potentially affords flexibility for creating multiply-functionalized HCM-bearing peptides, and eliminates the complications associated with postsynthetic labeling of Cys residues on whole-length peptides. It should further be mentioned that Quin-functionalized L-amino acids such as Sox have been previously reported by Imperiali and colleagues,16, 17 who placed these functionalities within β-turn peptides for ZnII sensing. The short side chains of Sox derivatives, which feature only a single methylene group between the Quin moiety and the α-carbon, are ideal for placement into the interior of a folded peptide, whereas EMeQuin, EQuin, and CQuin functionalities are, by design, long enough to extend over two turns on the surface of an α-helix.
Control peptides
In addition to H-EMeQuin, H-EQuin and H-CQuin (where H stands for the His component of the HCM in the 2 position), we prepared two further sets of peptides to investigate a) the importance of the His component of HCM and b) the necessity of the Lys(4)-Glu(8) salt bridge in the induction of α-helicity. For (a), we prepared the constructs A-CQuin and A-EMeQuin, which have His(2) replaced with a non-coordinating alanine (Ala) residue. For (b), we prepared H-CQuin-A and H-EMeQuin-A, which include an Ala in place of Glu(8).
Metal Binding Properties of His/Quin HCMs
We first studied the binding thermodynamics of CoII, NiII, CuII and ZnII to the three HCM-bearing peptides, H-EMeQuin, H-CQuin and H-EQuin, by monitoring the 20-nm red shift of the π-π* absorption band of Quin (λmax = 244 nm) upon metal coordination (Figure S7). As we previously observed with HCMs installed on the cytochrome cb562 surface,14, 15 the binding affinities of the HCM-bearing peptides were too high to be measured by direct titrations. Therefore, EGTA (ethylene glycol tetraacetic acid) or ADA (N-(2-acetamido)iminodiacetic acid) was included in the titration as a competing ligand. ADA was used in cases where metal-peptide binding was not strong enough to compete with EGTA, specifically in the cases of CoII and ZnII. Under our experimental conditions, which included 10-20 μM peptide, 30-60 μM EGTA or ADA, and 0-60 μM MII, all binding curves were best described using a model that simultaneously took into account a 1:1 peptide:metal binding equilibrium (P:M) and a 2:1 metal-mediated peptide dimerization event (P:M:P) (Figure S8 and Scheme S6). In all cases studied, the dissociation constant (Kd) for metal-mediated peptide dimers was determined to be in the low μM range.
The dissociation constants of the metal-HCM complexes (P:M) range from nanomolar for CoII and ZnII to femtomolar for CuII (Table 1). These Kd’s are two to three orders of magnitude lower than those for the free Quin ligand (Table 1),18 indicating the formation of the intended tridentate coordination mode that we previously observed in the crystal structure of a cytochrome cb562 variant with a surface H-CQuin motif.14, 15 Notably, the metal binding affinities of the HCM-peptides are three to seven orders of magnitude higher than those observed for similar peptides bearing i/i+4 bis-His chelation motifs9, 19 highlighting the dramatic effect of increased denticity on metal affinity. In general, the metal binding affinities of H-EMeQuin, H-CQuin and H-EQuin roughly follow the Irving-Williams series (CoII<NiII<<CuII>>ZnII). The affinity of H-EMeQuin for each metal ion is consistently lower (by one to two orders of magnitude) than those of H-CQuin and H-EQuin, which we attribute to specific interactions between the HCMs and the peptide surface. Quantum mechanics/molecular mechanics (QM/MM) calculations are currently underway to gain structural/energetic insights into H-EMeQuin, H-CQuin and H-EQuin and their metal complexes.
Table 1.
Dissociation constants for various peptide-metal complexes. Number in parentheses correspond to standard deviation in the last reported significant figure.
| Dissociation Constants (M) | ||||
|---|---|---|---|---|
|
| ||||
| H-CQuin | H-EMeQuin | H-EQuin | Quin18 | |
| CoII | 2 (1) × 10−9 | 1.2(5) × 10−8 | 2 (1) × 10−9 | 6.5 × 10−7 |
| NiII | 2 (1) × 10−11 | 1.1(1) × 10−9 | 2(1) × 10−10 | 1.6 × 10−7 |
| CuII | 5.2(4) × 10−14 | 1.4(8) × 10−13 | 6.3(1) × 10−14 | 2.3 × 10−10 |
| ZnII | 6.2(1) × 10−9 | 6.8(3) × 10−9 | 3 (1) × 10−9 | 8.7 × 10−7 |
Metal-Induced α-Helicity in HCM-containing Peptides
Metal-induced changes in the peptide secondary structure were monitored by circular dichroism (CD) spectroscopy (Figure 2, see Figures S9 and S10 for spectra including all metals), initially at 4 °C. In these experiments, a low peptide concentration (10 μM) and a 3-fold excess of CoII, NiII, CuII, or ZnII ensured that metal-induced peptide dimerization was not a concern and that each peptide was fully bound to a metal ion. In the absence of metal binding, H-CQuin, H-EMeQuin, and H-EQuin all displayed a CD signature reflective of a random coil conformation with a minimum at 195 nm. Upon addition of metal ions, the spectrum for H-EQuin showed little to no change from the random coil conformation. In contrast, H-CQuin and H-EMeQuin, whose HCMs are one bond longer than H-EQuin, displayed a significant induction of α-helicity as indicated by the emergence of two characteristic minima at ~206 and 222 nm.20 Of all metal ions tested, CuII induced the greatest extent of helicity in H-CQuin and H-EMeQuin, followed by ZnII, NiII, and CoII in descending order (Figure 2, Figures S9 and S10). In all cases, stoichiometric metal coordination to the Quin functionality was confirmed by full red-shift of the Quin π-π* absorption band from 244 nm to 264 nm.
Figure 2.

Changes in the circular dichroism spectra of HCM-bearing peptides upon binding CuII and ZnII. Spectra were acquired at 4 °C with 10 μM peptide and 30 μM EDTA or metal in 5 mM sodium borate buffer at pH 7.5. Corresponding spectra for CoII and NiII are shown in Figure S9 and S10.
Upon replacement of the His component of the HCMs with an Ala to obtain A-CQuin and A-EMeQuin, metal-induced changes in the CD spectra were abolished (Figure 2, Figures S9 and S10). This finding confirmed that metal binding by the HCMs and the resulting cross-linking of the two-helix turn portion were responsible for α-helix formation. The extent of α-helicities of metal-bound H-CQuin and H-EMeQuin based on mean residue ellipticity at 222 nm are listed in Table S1, although such estimates have been reported to be inaccurate for short helices.21 Therefore, we also obtained the CD spectra of H-CQuin and H-EMeQuin in the presence of 60% trifluoroethanol (TFE), a potent helix inducer.22 Assuming that TFE produces a fully helical conformation, the CuII-complexes of H-CQuin and H-EMeQuin possess ~70% and ~100% helicity, respectively, at 4 °C, based on relative CD intensities at 222 nm. At 25 °C, the CuII complexes still retain significant secondary structure (H-CQuin, 54% helical; H-EMeQuin, 88% helical).
In order to determine whether the Lys(4)-Glu(8) salt bridge is necessary for α-helix formation and the extent of its contribution to the peptide secondary structure, we examined the constructs H-EMeQuin-A and H-CQuin-A. The CD spectra of H-EMeQuin-A and H-CQuin-A showed that metal-induced helix formation was not affected by the elimination of the Lys(4)-Glu(8) salt bridge (Figure 2, Figures S9 and S10): the helicities of both peptides ranged from ~80% in the presence of CuII to ~40% in the presence of CoII (Table S1). In fact, ZnII binding induced more helicity for both peptides relative to the parent constructs containing salt bridges. Taken together, our findings clearly indicate that metal binding by the i/i+7 HCMs alone can induce considerable α-helicity in short helices. Importantly, HCMs occupy only one of the three available faces of an α-helix, meaning that residues facing the other two faces can be used for the incorporation of other functionalities and for target recognition.
Functionalization of HCMs
Having shown that H-CQuin and H-EMeQuin motifs tightly bind divalent transition metal ions and induce an α-helical peptide conformation in doing so, we next investigated whether they can be simultaneously exploited for incorporating metal-based functionalities. In a rare previous example where this has been achieved, the Ball group has employed dirhodium complexes to induce helicity in short, unstructured peptides upon coordination to i/i+3 and i/i+4 Glu or Asp pairs;11 the catalytic activity of these dirhodium-peptide conjugates were reported in separate studies.23 In our case, we initially set out to take advantage of the Quin functionality, whose derivatives have long been employed in various analytical applications owing to their intense fluorescence that is activated in selective response to metal ions,24 in particular to ZnII.25, 26 Indeed, H-CQuin displayed a ~5-fold increase in fluorescence intensity at 540 nm upon ZnII coordination, but not in the presence of CoII, NiII, or CuII (Figure 3a). As calculated with reference to a quinine sulfate standard, the quantum yield (ϕ540nm) of ZnII-H-CQuin is 0.27%, which is comparable to that of the ZnII complex of free 8-hydroxy-2-methylquinoline (0.4%).26 The amine group directly attached to the quinoline ring in H-CQuin likely quenches fluorescence by photoinduced electron transfer and is therefore partially responsible for the low quantum yield.27 In accordance with this proposal, H-EMeQuin, in which the amine group is one methylene unit removed from the aromatic ring, showed a ~20-fold fluorescence enhancement upon ZnII binding over background (Figure 3b), with an improved ϕ540nm of 0.64%. Further, regardless of His coordination, peptides tested containing a methylene spacer (A-EMeQuin, H-EMeQuin), consistently showed higher fluorescent intensity than those peptides with the amine group directly attached to the quinoline ring (H-CQuin, A-CQuin, H-EQuin) (Figure 3b).
Figure 3.

(a) H-EMeQuin fluorescence upon metal addition (λexc = 385 nm). (b) Normalized fluorescence intensity (F/F0) of each peptide when bound to ZnII as compared to the metal-free peptide.
Aside from coordinating aquated transition metal ions, the tripodal coordination motif of HCMs should also stably accommodate metal complexes that possess the appropriate geometry and could be used to endow the peptide scaffolds with additional functionalities. In particular, complexes of second- and third-row transition metals (ReI, RuII, OsII, IrIII and RhIII) display rich photophysical properties such as long lifetimes and high photostabilities28 that have been exploited in biological imaging,29, 30 sometimes in the form of metallopeptide conjugates.31, 32 The direct integration of such metal complexes into a peptide crosslinking moiety would obviate the need for additional functional groups (i.e., fluorophores), which have been shown to influence the cellular localization of peptides.33 Additionally, incorporation of these capped, substitution-inert metal complexes would eliminate the possibility of undesired metal-mediated peptide dimerization and yield kinetically stable conjugates that could persist in the intracellular environment, which is deprived of free metal ions.
To demonstrate the ability of HCMs to anchor functional metal complexes, we synthesized the luminescent ReI(IA-Quin)(CO)3X compound,34 which could be directly coupled to Cys(9) following SPPS with >90% yield (Figure 4a). The product, purified by HPLC, had a mass of 1453.80 amu (theoretical mass = 1490.10 amu), which is consistent with the loss of the chloride ligand either during the MS experiment or earlier during purification through substitution by a solvento species. Because ReI is substitution-inert, we first heated the resulting Re-peptide conjugate to 65 °C in order to promote His(2)-Re coordination, then cooled it down to room temperature, while monitoring the α-helicity of the peptide by CD spectroscopy at 222 nm (Figure 4 a). Upon the completion of the heating-cooling cycle, the α-helicity of the sample significantly increased, suggesting that the ReI-HCM was successfully formed (Figure 4b, 4c). The product displayed a visible band at ~410 nm (Figure S13), whose excitation produced two luminescence bands with maxima at 550 nm and 680 nm (Figure 4d; see Figure S14 for changes in the luminescence spectrum upon heating). These spectral features accord very well with those of the model complex, ReI(Quin)(CO)3(pyridine), which was reported to have an absorption band centered at 420-430 nm, a fluorescence band at ~530 nm and a phosphorescence band 670-690 nm.35 The IR-spectrum of the Re-HCM peptide was also very similar to that of the model complex, with three maxima at 1888, 1903, and 2016 cm−1 corresponding to the three carbonyl groups in a facial arrangement (Figure 4e). These results confirm the intended coordination geometry of the ReI-HCM complex and establish that His-Quin HCMs can be used to harbor functional metal complexes on an α-helical peptide platform.
Figure 4.

(a) Proposed scheme for the formation of the Re(Quin)(CO)3(His) HCM upon heating and the subsequent formation of the α-helix upon cooling. (b) Observed changes in the CD spectrum upon the heating and subsequent cooling of the Re-HCM peptide. (c) Changes in the CD signal (222 nm) during heating and cooling. (d) The emission spectrum of the Re-HCM peptide obtained after the heating/cooling cycle (λexc = 410 nm). (e) The IR spectrum of the Re-HCM peptide. The peptide sample was first lyophilized and then combined with KBr to make a pellet.
HCM-mediated helicity in non-Ala-rich peptide sequences
Although the HCM-peptide sequences presented thus far all a display random coil signatures in the absence of metal coordination, it is well documented that Ala-rich sequences have high propensity for α-helix formation.36 Therefore, we sought to determine whether our HCM strategy could be employed to induce helicity also in non-Ala-rich peptide sequences that may have downstream applications. To this end, we synthesized an HCM-bearing peptide sequence (Bax-HCM) based on the helical BH3 (Bcl-2 homology domain 3) motif of the pro-apoptotic protein Bax. Because BH3 domains play a central role in the interactions between Bcl-2 family proteins involved in the regulation of cell death, structural mimics of BH3 domains have received considerable attention as potential cancer therapeutics37 and therefore constituted an interesting proof-of-principle target for our strategy. Bax-HCM, a 26-residue sequence, contained the entire helical BH3 domain (residues 55-80, Figure 5) of Bax which is necessary and sufficient for interaction with Bcl-2.38 The only two variations from the wildtype protein sequence were Ser72→His and Met79→EMeQuin for the construction of the i/i+7 HCM motif on the face of the BH3 helix that does not interact with the target protein. CD experiments indicated that Bax-HCM (10 μM in concentration) displayed little–if any–helicity in the absence of metals (Figure 5). Upon addition of metal ions, we observed a clear emergence of an α-helical signature, the effect being largest with CuII, followed by ZnII, CoII and NiII (Figures 5, S9 and S10), roughly following the same trend observed with the 10-mer sequences. Notably, in contrast to these short Ala-rich peptides, Bax-HCM displayed no appreciable difference in helicity between 4 °C and 25 °C (Figure 5), which may be attributable to its longer length.
Figure 5.
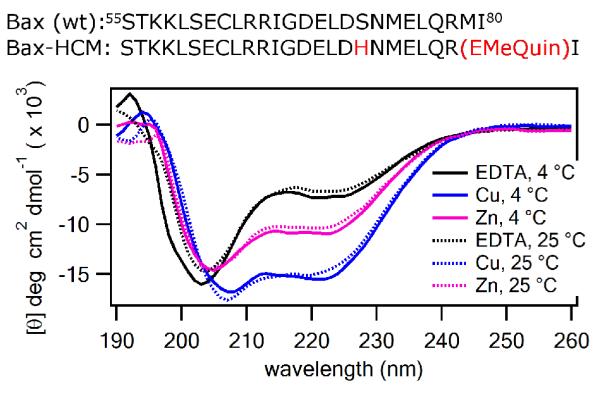
Sequences of wild-type and HCM-modified Bax BH3 domain peptides (top). Far-UV CD spectra of the Bax-HCM in the presence and absence of CuII (blue) and ZnII (magenta) acquired at 4 °C (solid lines) or 25 °C (dashed lines). The samples contained 10 μM peptide and 30 μM EDTA or metal ion in a 5 mM sodium borate buffer solution at pH 7.5.
Conclusions
In summary, we have presented here the synthesis and characterization of various Quin-based HCM-bearing peptides and their metal-dependent structural and photophysical properties. There is rich and extensive literature on metal-induced α-helicity in peptides,9, 10, 39 metal-peptide conjugates,32, 40 and functional metallopeptides.16, 41 Our strategy of building HCMs on peptide sequences borrows from all of these somewhat non-overlapping efforts to provide a unique combination of advantages for creating functional, α-helical structures. HCM-bearing peptides are readily prepared via multiple routes with high yield in a few synthetic steps from commercially available components. HCMs are modular in the sense that they can be interchangeably (i.e., reversibly) complexed with various divalent transition metal ions or metal complexes whose different coordination properties can be utilized to control the α-helicity of the peptide scaffold. Though not explored here, HCMs could also allow the control of peptide conformation through changes in solution pH or redox potential (as both of these factors can modulate inner-sphere metal coordination) so as to construct stimuli-responsive peptide platforms. Based on the two sets of peptides we studied here (the 10mer, Ala-rich sequences and the 26mer Bax BH3 domain), HCMs appear to be generally applicable for helix induction, with the added advantage that the “helix staple” can now carry an intrinsic and useful functionality such as luminescence.
On a final note, given the tight regulation and scarcity of uncomplexed transition metal ions in the intracellular environment, the use of α-helical HCM peptides containing labile metal ions (e.g., CoII, NiII, CuII and ZnII) may be more useful for in vitro experiments or for targeting extracellular proteins such as cell surface receptors. Nevertheless, as we have demonstrated with a ReI-carbonyl compound, HCM peptides are readily modified with substitution-inert metal complexes, which should not only yield stable α-helical peptides, but also may lend their intrinsic physical properties and chemical reactivities toward diagnostic and therapeutic in vivo applications.29
Supplementary Material
Acknowledgements
We thank Professor Seth Cohen for use of a hydrogenator, Professor Nathan Gianneschi for use of a peptide synthesizer, Professor Elizabeth Komives for use of a peptide synthesizer and HPLC and Dr. Yongxuan Su for help with mass spectrometry and CD. We also thank Steven Chabolla for assistance with synthetic analysis and discussion. This work was supported by the US Department of Energy (DOE) (Division of Materials Sciences, Office of Basic Energy Sciences, Award DE-FG02-10ER46677 to F.A.T., peptide synthesis), the National Science Foundation (CHE-0908115 to F.A.T.), and the National Institutes of Health (Molecular Biophysics traineeship to S.J.S.)
Footnotes
Electronic Supplementary Information (ESI) available: [Experimental details, figures and tables]. See DOI: 10.1039/b000000x/
Notes and References
- 1.Bullock BN, Jochim AL, Arora PS. J. Am. Chem. Soc. 2011;133:14220. doi: 10.1021/ja206074j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orner BP, Ernst JT, Hamilton AD. J. Am. Chem. Soc. 2001;123:5382. doi: 10.1021/ja0025548. [DOI] [PubMed] [Google Scholar]; Seebach D, Matthews JL. Chem. Commun. 1997:2015. 0. [Google Scholar]; Gellman SH. Acc. Chem. Res. 1998;31:173. [Google Scholar]; Cheng RP, DeGrado WF. J. Am. Chem. Soc. 2001;123:5162. doi: 10.1021/ja010438e. [DOI] [PubMed] [Google Scholar]; Kritzer JA, Lear JD, Hodsdon ME, Schepartz A. J. Am. Chem. Soc. 2004;126:9468. doi: 10.1021/ja031625a. [DOI] [PubMed] [Google Scholar]; Fowler SA, Blackwell HE. Org. Biomol. Chem. 2009;7:1508. doi: 10.1039/b817980h. [DOI] [PMC free article] [PubMed] [Google Scholar]; Appella DH, Christianson LA, Klein DA, Powell DR, Huang X, Barchi JJ, Gellman SH. Nature. 1997;387:381. doi: 10.1038/387381a0. [DOI] [PubMed] [Google Scholar]
- 3.Chin JW, Schepartz A. J. Am. Chem. Soc. 2001;123:2929. doi: 10.1021/ja0056668. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zondlo NJ, Schepartz A. J. Am. Chem. Soc. 1999;121:6938. [Google Scholar]
- 4.Azzarito V, Long K, Murphy NS, Wilson AJ. Nat. Chem. 2013;5:161. doi: 10.1038/nchem.1568. [DOI] [PubMed] [Google Scholar]; Estieu-Gionnet K, Guichard G. Expert Opin. Drug Del. 2011;6:937. doi: 10.1517/17460441.2011.603723. [DOI] [PubMed] [Google Scholar]
- 5.Sato AK, Viswanathan M, Kent RB, Wood CR. Curr. Opin. Biotechnol. 2006;17:638. doi: 10.1016/j.copbio.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Toniolo C, Crisma M, Formaggio F, Peggion C. Biopolymers. 2001;60:396. doi: 10.1002/1097-0282(2001)60:6<396::AID-BIP10184>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]; Bavoso A, Benedetti E, Di Blasio B, Pavone V, Pedone C, Toniolo C, Bonora GM. Proc. Natl. Acad. Sci. U.S.A. 1986;83:1988. doi: 10.1073/pnas.83.7.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marqusee S, Baldwin RL. Proc. Natl. Acad. Sci. U.S.A. 1987;84:8898. doi: 10.1073/pnas.84.24.8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujimoto K, Kajino M, Inouye M. Chem. Eur. J. 2008;14:857. doi: 10.1002/chem.200700843. [DOI] [PubMed] [Google Scholar]; Schafmeister CE, Po J, Verdine GL. J. Am. Chem. Soc. 2000;122:5891. [Google Scholar]; Blackwell HE, Grubbs RH. Angew. Chem. Int. Ed. 1998;37:3281. doi: 10.1002/(SICI)1521-3773(19981217)37:23<3281::AID-ANIE3281>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]; Jackson DY, King DS, Chmielewski J, Singh S, Schultz PG. J. Am. Chem. Soc. 1991;113:9391. [Google Scholar]; Phelan JC, Skelton NJ, Braisted AC, McDowell RS. J. Am. Chem. Soc. 1997;119:455. [Google Scholar]; Zhang F, Sadovski O, Xin SJ, Woolley GA. J. Am. Chem. Soc. 2007;129:14154. doi: 10.1021/ja075829t. [DOI] [PubMed] [Google Scholar]; Taylor JW. Biopolymers. 2002;66:49. doi: 10.1002/bip.10203. [DOI] [PubMed] [Google Scholar]; Pellegrini M, Royo M, Chorev M, Mierke DF. J. Pept. Res. 1997;49:404. doi: 10.1111/j.1399-3011.1997.tb00892.x. [DOI] [PubMed] [Google Scholar]; Harrison RS, Shepherd NE, Hoang HN, Ruiz-Gomez G, Hill TA, Driver RW, Desai VS, Young PR, Abbenante G, Fairlie DP. Proc. Natl. Acad. Sci. U.S.A. 2010;107:11686. doi: 10.1073/pnas.1002498107. [DOI] [PMC free article] [PubMed] [Google Scholar]; Shepherd NE, Hoang HN, Abbenante G, Fairlie DP. J. Am. Chem. Soc. 2005;127:2974. doi: 10.1021/ja0456003. [DOI] [PubMed] [Google Scholar]
- 9.Ghadiri MR, Choi C. J. Am. Chem. Soc. 1990;112:1630. [Google Scholar]
- 10.Ruan F, Chen Y, Hopkins PB. J. Am. Chem. Soc. 1990;1990:9403. [Google Scholar]; Kharenko OA, Ogawa MY. J. Inorg. Biochem. 2004;98:1971. doi: 10.1016/j.jinorgbio.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 11.Zaykov AN, Popp BV, Ball ZT. Chem. Eur. J. 2010;16:6651. doi: 10.1002/chem.200903092. [DOI] [PubMed] [Google Scholar]
- 12.Ma MT, Hoang HN, Scully CCG, Appleton TG, Fairlie DP. J. Am. Chem. Soc. 2009;131:4505. doi: 10.1021/ja900047w. [DOI] [PubMed] [Google Scholar]; Ghadiri MR, Fernholz AK. J. Am. Chem. Soc. 1990;112:9633. [Google Scholar]
- 13.Chapman RN, Dimartino G, Arora PS. J. Am. Chem. Soc. 2004;126:12252. doi: 10.1021/ja0466659. [DOI] [PubMed] [Google Scholar]; Cabezas E, Satterthwait AC. J. Am. Chem. Soc. 1999;121:3862. [Google Scholar]
- 14.Radford RJ, Nguyen PC, Ditri TB, Figueroa JS, Tezcan FA. Inorg. Chem. 2010;49:4362. doi: 10.1021/ic100534y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Radford RJ, Nguyen PC, Tezcan FA. Inorg. Chem. 2010;2010:7106. doi: 10.1021/ic100926g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shults MD, Pearce DA, Imperiali B. J. Am. Chem. Soc. 2003;125:10591. doi: 10.1021/ja0355980. [DOI] [PubMed] [Google Scholar]
- 17.Walkup GK, Imperiali B. J. Am. Chem. Soc. 1997;119:3443. [Google Scholar]
- 18.Martell AE, Smith RM. Critical Stability Constants. Plenum Press; New York: 1974. [Google Scholar]
- 19.Krantz BA, Sosnick TR. Nat. Struct. Mol. Biol. 2001;8:1042. doi: 10.1038/nsb723. [DOI] [PubMed] [Google Scholar]
- 20.Chin D-H, Woody RW, Rohl CA, Baldwin RL. Proc. Natl. Acad. Sci. U.S.A. 2002;99:15416. doi: 10.1073/pnas.232591399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patgiri A, Jochim AL, Arora PS. Acc. Chem. Res. 2008;41:1289. doi: 10.1021/ar700264k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo P, Baldwin RL. Biochemistry. 1997;36:8413. doi: 10.1021/bi9707133. [DOI] [PubMed] [Google Scholar]
- 23.Popp BV, Ball ZT. J. Am. Chem. Soc. 2010;132:6660. doi: 10.1021/ja101456c. [DOI] [PubMed] [Google Scholar]; Chen Z, Vohidov F, Coughlin JM, Stagg LJ, Arold ST, Ladbury JE, Ball ZT. J. Am. Chem. Soc. 2012;134:10138. doi: 10.1021/ja302284p. [DOI] [PubMed] [Google Scholar]
- 24.Bhatnagar DC, Forster LS. Spectrochim. Acta. 1965;21:1803. [Google Scholar]; Zhang H, Han L-F, Zachariasse KA, Jiang Y-B. Org. Lett. 2005;7:4217. doi: 10.1021/ol051614h. [DOI] [PubMed] [Google Scholar]
- 25.Fahrni CJ, O’Halloran TV. J. Am. Chem. Soc. 1999;121:11448. [Google Scholar]; Jotterand N, Pearce DA, Imperiali B. J. Org. Chem. 2001;66:3224. doi: 10.1021/jo001681j. [DOI] [PubMed] [Google Scholar]; Nolan EM, Jaworski J, Okamoto K-I, Hayashi Y, Sheng M, Lippard SJ. J. Am. Chem. Soc. 2005;127:16812. doi: 10.1021/ja052184t. [DOI] [PMC free article] [PubMed] [Google Scholar]; Jiang P, Guo Z. Coord. Chem. Rev. 2004;248:205. [Google Scholar]
- 26.Pearce DA, Jotterand N, Carrico IS, Imperiali B. J. Am. Chem. Soc. 2001;123:5160. doi: 10.1021/ja0039839. [DOI] [PubMed] [Google Scholar]
- 27.Su J, Xu T, Chen K, Tian H. Dyes Pigm. 2000;44:87. [Google Scholar]; Bronson RT, Montalti M, Prodi L, Zaccheroni N, Lamb RD, Dalley NK, Izatt RM, Bradshaw JS, Savage PB. Tetrahedron. 2004;60:11139. [Google Scholar]
- 28.Lloyd D, Coogan M, Pope SA. In: Reviews in Fluorescence. Geddes CD, editor. vol. 2010. Springer; New York: 2010. 2010. p. 15. [Google Scholar]
- 29.Zhao Q, Huang C, Li F. Chem. Soc. Rev. 2011;40:2508. doi: 10.1039/c0cs00114g. [DOI] [PubMed] [Google Scholar]; Fernandez-Moreira V, Thorp-Greenwood FL, Coogan MP. Chem. Commun. 2010;46:186. doi: 10.1039/b917757d. [DOI] [PubMed] [Google Scholar]
- 30.Lo KK-W, Zhang KY, Li SP-Y. Eur. J. Inorg. Chem. 2011;2011:3551. [Google Scholar]; Amoroso AJ, Coogan MP, Dunne JE, Fernandez-Moreira V, Hess JB, Hayes AJ, Lloyd D, Millet C, Pope SJA, Williams C. Chem. Commun. 2007;2007:3066. doi: 10.1039/b706657k. [DOI] [PubMed] [Google Scholar]; Stephenson KA, Banerjee SR, Besanger T, Sogbein OO, Levadala MK, McFarlane N, Lemon JA, Boreham DR, Maresca KP, Brennan JD, Babich JW, Zubieta J, Valliant JF. J. Am. Chem. Soc. 2004;126:8598. doi: 10.1021/ja047751b. [DOI] [PubMed] [Google Scholar]; Raszeja L, Maghnouj A, Hahn S, Metzler-Nolte N. ChemBioChem. 2011;12:371. doi: 10.1002/cbic.201000576. [DOI] [PubMed] [Google Scholar]
- 31.Dirscherl G, König B. Eur. J. Org. Chem. 2008;2008:597. [Google Scholar]
- 32.Metzler-Nolte N. Chimia. 2007;61:736. [Google Scholar]
- 33.Szeto HH, Schiller PW, Zhao KS, Luo GX. Faseb J. 2005;19:118. doi: 10.1096/fj.04-1982fje. [DOI] [PubMed] [Google Scholar]; Puckett CA, Barton JK. J. Am. Chem. Soc. 2009;131:8738. doi: 10.1021/ja9025165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dattelbaum JD, Abugo OO, Lakowicz JR, Kunkely H, Vogler A. Bioconjugate Chem. Inorg. Chem. Commun. 2000;1998;111:533, 398. doi: 10.1021/bc990174+. [DOI] [PMC free article] [PubMed] [Google Scholar]; Vogler A, Kunkely H. Coord. Chem. Rev. 2000;200–202:991. [Google Scholar]
- 35.Czerwieniec R, Kapturkiewicz A, Anulewicz-Ostrowska R, Nowacki J. J. Chem. Soc., Dalton Trans. 2001:2756. [Google Scholar]
- 36.Marqusee S, Robbins VH, Baldwin RL. Proc. Natl. Acad. Sci. U.S.A. 1989;86:5286. doi: 10.1073/pnas.86.14.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Science. 2004;305:1466. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang D, Liao W, Arora PS. Angew. Chem. Int. Ed. 2005;44:6525. doi: 10.1002/anie.200501603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ku B, Liang C, Jung JU, Oh B-H. Cell Res. 2011;21:627. doi: 10.1038/cr.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]; Shangary S, Johnson DE. Biochemistry. 2002;41:9485. doi: 10.1021/bi025605h. [DOI] [PubMed] [Google Scholar]
- 39.Siedlecka M, Goch G, Ejchart A, Sticht H, Bierzyǹski A. Proc. Natl. Acad. Sci. U.S.A. 1999;96:903. doi: 10.1073/pnas.96.3.903. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kelso MJ, Beyer RL, Hoang HN, Lakdawala AS, Snyder JP, Oliver WV, Robertson TA, Appleton TG, Fairlie DP. J. Am. Chem. Soc. 2004;126:4828. doi: 10.1021/ja037980i. [DOI] [PubMed] [Google Scholar]; Nicoll AJ, Miller DJ, Fütterer K, Ravelli R, Allemann RK. J. Am. Chem. Soc. 2006;128:9187. doi: 10.1021/ja061513u. [DOI] [PubMed] [Google Scholar]; Kelso MJ, Hoang HN, Appleton TG, Fairlie DP. J. Am. Chem. Soc. 2000;122:10488. [Google Scholar]; Signarvic RS, DeGrado WF. J. Am. Chem. Soc. 2009;131:3377. doi: 10.1021/ja809580b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kozłowski H, Bal W, Dyba M, Kowalik-Jankowska T. Coord. Chem. Rev. 1999;184:319. [Google Scholar]; Curran TP, Grant AL, Lucht RA, Carter JC, Affonso J. Org. Lett. 2002;4:2917. doi: 10.1021/ol026298a. [DOI] [PubMed] [Google Scholar]; Vairaprakash P, Ueki H, Tashiro K, Yaghi OM. J. Am. Chem. Soc. 2010;133:759. doi: 10.1021/ja1097644. [DOI] [PubMed] [Google Scholar]; Myers CP, Williams ME. Coord. Chem. Rev. 2010;254:2416. [Google Scholar]
- 41.Copeland KD, Fitzsimons MP, Houser RP, Barton JK. Biochemistry. 2001;41:343. doi: 10.1021/bi011793k. [DOI] [PubMed] [Google Scholar]; Ball ZT. Acc. Chem. Res. 2012;46:560. doi: 10.1021/ar300261h. [DOI] [PubMed] [Google Scholar]; Jain A, Buchko GW, Reback ML, O’Hagan M, Ginovska-Pangovska B, Linehan JC, Shaw WJ. ACS Catal. 2012;2:2114. [Google Scholar]; Ghosh D, Pecoraro VL. Inorg. Chem. 2004;43:7902. doi: 10.1021/ic048939z. [DOI] [PubMed] [Google Scholar]; Xing G, DeRose VJ. Curr. Opin. Chem. Bio. 2001;5:196. doi: 10.1016/s1367-5931(00)00190-3. [DOI] [PubMed] [Google Scholar]; Franklin SJ. Curr. Opin. Chem. Biol. 2001;5:201. doi: 10.1016/s1367-5931(00)00191-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.