

Abstract
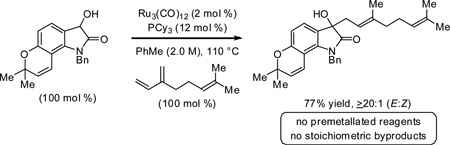
The direct conversion of secondary to tertiary alcohols via ruthenium(0) catalyzed C-C coupling of substituted 3-hydroxy-2-oxindoles with various dienes is described. Coupling occurs in a completely regioselective manner in the absence of stoichiometric byproducts.
In the course of exploring hydrogen-mediated C-C couplings outside of hydroformylation,1 our laboratory has developed metal catalysts that promote the transfer of hydrogen from primary alcohols to various π-unsaturated compounds resulting in pairwise generation of aldehydeorganometallic nucleophile-electrophile pairs that combine to form products of aldehyde addition. These processes merge redox and C-C bond construction events,2 bypassing both discrete alcohol-to-aldehyde oxidation and the stoichiometric use of premetallated reagents. Although conditions for the C-C coupling of primary alcohols to dienes 3, 4, 5 and other π-unsaturated reactants (allenes, enynes, alkynes and allylic acetates)1 have been developed, corresponding couplings of secondary alcohols have proven more challenging.
Recently, it was found that ruthenium(0) complexes derived from Ru3(CO)12 and phosphine ligands catalyze C-C coupling of mandelic esters with 2-substituted dienes at the C4-position to form prenylated and geranylated compounds from isoprene and myrcene, respectively.4e As corroborated by mechanistic studies, this regioselectivity is a consequence of a catalytic mechanism wherein alcohol dehydrogenation drives oxidative coupling between the diene and a transient α-oxoester.6 To our knowledge, such C4 regioselectivity is unique for intermolecular reductive couplings of 2-substituted dienes to carbonyl partners.7,8,9 Given the structural homology of substituted mandelic esters and 3-hydroxy-2-oxindoles, which represent important natural product substructures, the latter compounds were explored as partners for redox triggered C-C coupling with dienes.10 Here, we report that exposure of 3-hydroxy-2-oxindoles to isoprene or myrcene and the ruthenium(0) catalyst derived from Ru3(CO)12 and tricyclohexylphosphine, PCy3, results in direct, secondary carbinol C-H prenylation and geranylation, respectively, in the absence of stoichiometric byproducts. Further, we find that dienes beyond isoprene or myrcene participate in such secondary alcohol C-C couplings.
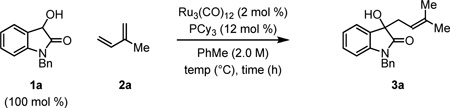
To probe the feasibility of isatin prenylation from the alcohol oxidation level, 3-hydroxy-2-oxindole 1a was exposed to conditions optimized for the prenylation of ethyl mandelate.4e Gratifyingly, upon exposure of 1a (100 mol %) to isoprene 2a (500 mol %), Ru3(CO)12 (2 mol %) and PCy3 (12 mol %) at 130 °C in toluene (2.0 M) for 48 hours, the product of prenylation 3a was generated in 76% isolated yield (Table 1, entry 2). Extended reaction times (72 h) did not enhance the isolated yield of 3a (Table 1, entry 1), and for reactions conducted over a 24 hour period comparable isolated yields of 3a were obtained (Table 1, entry 3). The isolated yield of 3a was not adversely effected at lower loadings of isoprene (300 mol %) (Table 1, entry 4) even when equimolar quantities of 3-hydroxy-2-oxindole 1a and isoprene 2a were employed (Table 1, entry 5). When the reaction was conducted at 110 °C the isolated yield of 3a improved (Table 1, entry 6), however, any further decrease in reaction temperature led to a significant decrease in conversion (Table 1, entry 7).
Table 1.
Selected Optimization Experiments in the Ruthenium Catalyzed C-C Coupling of 3-Hydroxy-2-oxindole 1a and Isoprene 2a.a
 | ||||
|---|---|---|---|---|
| entry | 2a (mol %) | temp | time (h) | yield % |
| 1 | 500 | 130 | 72 | 71 |
| 2 | 500 | 130 | 48 | 76 |
| 3 | 500 | 130 | 24 | 73 |
| 4 | 300 | 130 | 24 | 74 |
| 5 | 100 | 130 | 24 | 76 |
| 6 | 100 | 110 | 24 | 83 |
| 7 | 100 | 90 | 24 | 57 |
Yields are of material isolated by silica gel chromatography. See Supporting Information for further details.
Optimal conditions identified for the n-prenylation of 3-hydroxy-2-oxindole 1a (Table 1, entry 6) were applied to substituted 3-hydroxy-2-oxindoles 1b-1f. The desired products of prenylation 3b-3f were obtained in excellent isolated yields and in each case single regioisomers were obtained (Table 2). Similarly, exposure of 3-hydroxy-2-oxindoles 1a-1f to myrcene 2b provides the products of n-geranylation as single regioisomers with complete control of olefin geometry (Table 3). Thus, direct prenylation and geranylation of secondary carbinol C-H bonds is achieved in the absence of stoichiometric byproducts or premetallated reagents.
Table 2.
Ruthenium Catalyzed C-C Coupling of 3-Hydroxy-2-oxindoles 1a-1f and Isoprene 2a to Form Products of Prenylation 3a–3f.a
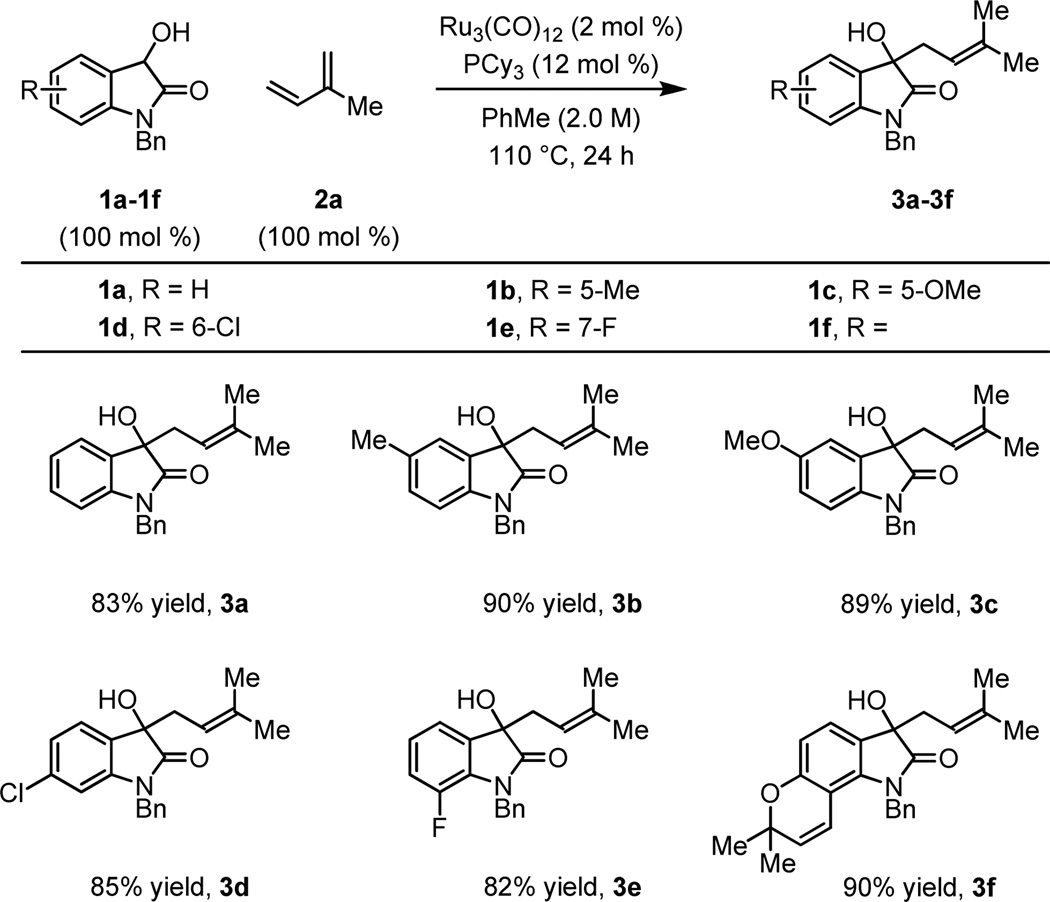 |
Yields are of material isolated by silica gel chromatography. See Supporting Information for further details.
Table 3.
Ruthenium Catalyzed Coupling of 3-Hydroxy-2-oxindoles 1a-1f and Myrcene 2b to Form Products of Geranylation 4a-4fa
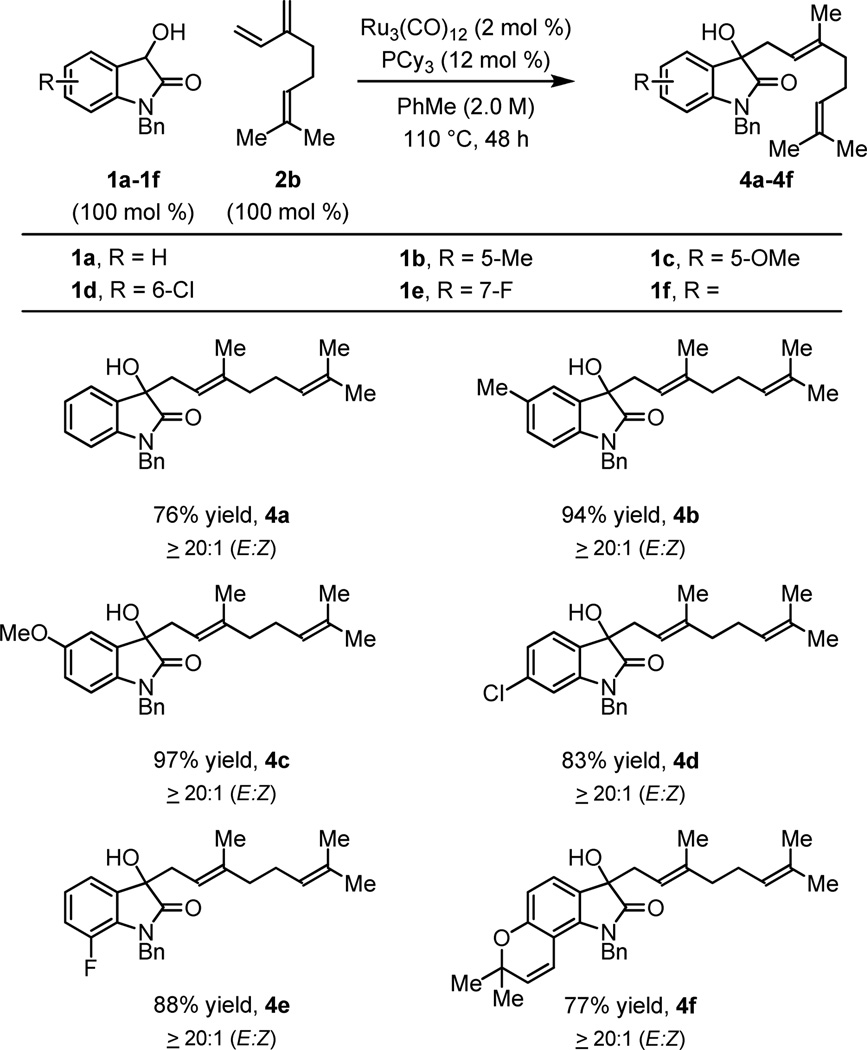 |
Yields are of material isolated by silica gel chromatography. See Supporting Information for further details.
To further evaluate scope, the redox triggered C-C coupling of N-benzyl 3-hydroxy-2-oxindole 1a with dienes 2c-2f was investigated. The coupling of 1a with butadiene 2c provided the product of (Z)-2-butenylation 5a as a single geometrical isomer. As will be discussed, the (Z)-olefin geometry of 5a is indicative of a catalytic mechanism involving diene-carbonyl oxidative coupling.4e As illustrated in the conversion of 1,3-cyclohexadiene 2d to the product of 2-cyclohexenylation 5b, cyclic dienes also participate in efficient coupling. In a similar manner, the 2-silyl substituted butadiene 2e and pentadiene 2f are converted to adducts 5c and 5d, respectively, as single olefin geometrical isomers (Scheme 1).
Scheme 1.

Ruthenium Catalyzed C-C Coupling of 2-Hydroxy-3-oxindoles 1a and Dienes 2c-2f.a
aYields are of material isolated by silica gel chromatography. See Supporting Information for further details.
A plausible catalytic mechanism accounting for the linear regioselectivity of diene C-C coupling and (Z)-stereoselectivity observed in the formation of the butadiene adduct 5a is as follows (Scheme 2). Exposure of Ru3(CO)12 to chelating phosphine ligands provides complexes of the type Ru(CO)3(diphosphine). 11 Although the present catalytic system employs a non-chelating mono-phosphine ligand, intervention of a discrete, mono-metallic catalyst is anticipated based on this precedent. As implicated in related Pauson-Khand type reactions of 1,2-diones,6 along with subsequent studies from our laboratory,4e ruthenium(0) mediated diene-carbonyl oxidative coupling delivers the ruthenium(II) oxametallacycle I, which isomerizes to oxametallacycle II, the presumably more stable primary σ-allyl haptomer. The (Z)-stereoselectivity observed in the formation of adduct 5a ultimately is defined by the olefin geometry evident in oxametallacycle II. Oxindole 1a protonates oxametallacycle II to form the ruthenium(II) alkoxide III, which upon β-hydride elimination delivers the isatin oxo-1a and the allylruthenium(II) hydride IV. Related β-hydride eliminations of σ-hydroxy amides employing catalysts derived from Ru3(CO)12 have been reported.12 Reductive elimination of the allylruthenium hydride IV delivers the product 5a returns ruthenium to its zero-valent form. The reductive elimination event must occur at a faster rate than π-facial interconversion of the allyl moiety, which would otherwise erode the control of olefin geometry (Scheme 2).
Scheme 2.

Plausible catalytic mechanism for the redox triggered C-C coupling of oxindole-1a and butadiene 2c accounting for the observed olefin (Z)-stereoselectivity.
In summary, the present ruthenium(0) catalyst system has proven unique in its ability to mechanistically link alcohol dehydrogenation to diene-carbonyl oxidative C-C coupling. Based on this novel pattern of reactivity, a new transformation of the ubiquitous hydroxyl functional group has been availed: the direct prenylation and geranylation of secondary carbinol C-H bonds in the absence of stoichiometric byproducts employing isoprene 2a and myrcene 2b as prenyl and geranyl donors, respectively. Whereas in prior work, secondary alcohols in the form of mandelic esters were shown to participate in such C-C bond forming transfer hydrogenations,4e the present study demonstrates that oxindoles 1a-1f readily engage in analogous transformations. These studies contribute to a departure from the stoichiometric use of premetallated reagents in organic synthesis.
Supplementary Material
Acknowledgments
Acknowledgment is made to the Robert A. Welch Foundation (F-0038), the NIH-NIGMS (RO1-GM093905) and the Center for Green Chemistry and Catalysis for partial support of this research.
Footnotes
Supporting Information Available. Spectral data for all new compounds (1H NMR, 13C NMR, IR, HRMS). This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1. For selected reviews on C-C bond forming hydrogenation and transfer hydrogenation, see: Bower JF, Krische MJ. Top. Organomet. Chem. 2011;43:107. doi: 10.1007/978-3-642-15334-1_5. Hassan A, Krische MJ. Org. Proc. Res. Devel. 2011;15:1236. doi: 10.1021/op200195m. Moran J, Krische MJ. Pure Appl. Chem. 2012;84:1729. doi: 10.1351/PAC-CON-11-10-18.
- 2. For a review on the topic of redox economy, see: Baran PS, Hoffmann RW, Burns NZ. Angew. Chem. Int. Ed. 2009;48:2854. doi: 10.1002/anie.200806086.
- 3. For iridium catalyzed transfer hydrogenative coupling of alcohols to dienes, see: Bower JF, Patman RL, Krische MJ. Org. Lett. 2008;10:1033. doi: 10.1021/ol800159w. Zbieg JR, Fukuzumi T, Krische MJ. Adv. Synth. Catal. 2010;352:2416. doi: 10.1002/adsc.201000599.
- 4. For ruthenium catalyzed transfer hydrogenative coupling of alcohols to dienes, see: Shibahara F, Bower JF, Krische MJ. J. Am. Chem. Soc. 2008;130:6338. doi: 10.1021/ja801213x. Han H, Krische MJ. Org. Lett. 2010;12:2844. doi: 10.1021/ol101077v. Zbieg JR, Moran J, Krische MJ. J. Am. Chem. Soc. 2011;133:10582. doi: 10.1021/ja2046028. Zbieg JR, Yamaguchi E, McInturff EL, Krische MJ. Science. 2012;336:324. doi: 10.1126/science.1219274. Leung JC, Geary LM, Chen T-Y, Zbieg JR, Krische MJ. J. Am. Chem. Soc. 2012;134:15700. doi: 10.1021/ja3075049. Geary LM, Glasspoole BW, Kim MM, Krische MJ. J. Am. Chem. Soc. 2013;134:3796. doi: 10.1021/ja400691t. Also, see reference 5.
- 5. For ruthenium catalyzed transfer hydrogenative reductive coupling of dienes to paraformaldehyde, see: Smejkal T, Han H, Breit B, Krische MJ. J. Am. Chem. Soc. 2009;131:10366. doi: 10.1021/ja904124b.
- 6. A related oxidative coupling pathway is observed in ruthenium(0) catalyzed Pauson-Khand type reactions of 1,2-diones: Chatani N, Tobisu M, Asaumi T, Fukumoto Y, Murai S. J. Am. Chem. Soc. 1999;121:7160. Tobisu M, Chatani N, Asaumi T, Amako K, Ie Y, Fukumoto Y, Murai S. J. Am. Chem. Soc. 2000;122:12663.
- 7. For nickel catalyzed intermolecular diene-aldehyde reductive coupling, see: Kimura M, Ezoe A, Shibata K, Tamaru Y. J. Am. Chem. Soc. 1998;120:4033. Takimoto M, Hiraga Y, Sato Y, Mori M. Tetrahedron Lett. 1998;39:4543. Kimura M, Fujimatsu H, Ezoe A, Shibata K, Shimizu M, Matsumoto S, Tamaru Y. Angew. Chem. Int. Ed. 1999;38:397. doi: 10.1002/(SICI)1521-3773(19990201)38:3<397::AID-ANIE397>3.0.CO;2-Y. Kimura M, Shibata K, Koudahashi Y, Tamaru Y. Tetrahedron Lett. 2000;41:6789. Kimura M, Ezoe A, Tanaka S, Tamaru Y. Angew. Chem. Int. Ed. 2001;40:3600. doi: 10.1002/1521-3773(20011001)40:19<3600::aid-anie3600>3.0.co;2-n. Loh T-P, Song H-Y, Zhou Y. Org. Lett. 2002;4:2715. doi: 10.1021/ol026216i. Sato Y, Sawaki R, Saito N, Mori M. J. Org. Chem. 2002;67:656. doi: 10.1021/jo0106086. Kimura M, Ezoe A, Mori M, Iwata K, Tamaru Y. J. Am. Chem. Soc. 2006;128:8559. doi: 10.1021/ja0608904. Yang Y, Zhu S-F, Duan H-F, Zhou C-Y, Wang L-X, Zhou Q-L. J. Am. Chem. Soc. 2007;129:2248. doi: 10.1021/ja0693183. Sato Y, Hinata Y, Seki R, Oonishi Y, Saito N. Org. Lett. 2007;9:5597. doi: 10.1021/ol702543m. Köpfer A, Sam B, Breit B, Krische MJ. Chem. Sci. 2013;4:1876.
- 8. For rhodium catalyzed intermolecular diene-aldehyde reductive coupling, see: Jang H-Y, Huddleston RR, Krische MJ. Angew. Chem. Int. Ed. 2003;42:4074. doi: 10.1002/anie.200351986. Kimura M, Nojiri D, Fukushima M, Oi S, Sonoda Y, Inoue Y. Org. Lett. 2009;11:3794. doi: 10.1021/ol901527b.
- 9. For titanium catalyzed intermolecular diene-aldehyde reductive coupling, see: Bareille L, Le Gendre P, Moïse C. Chem. Commun. 2005;775 doi: 10.1039/b414322a.
- 10. For related allylations of isatins under the conditions of iridium catalyzed transfer hydrogenation, see: Itoh J, Han SB, Krische MJ. Angew. Chem. Int. Ed. 2009;48:6313. doi: 10.1002/anie.200902328.
- 11. For example, exposure of Ru3(CO)12 to dppe in benzene solvent provides Ru(CO)3(dppe): Sanchez-Delgado RA, Bradley JS, Wilkinson G. J. Chem. Soc. Dalton Trans. 1976:399.
- 12. For mechanistically related Ru3(CO)12 catalyzed secondary alcohol amination via alcohol mediated hydrogen transfer, see: Baehn S, Tillack A, Imm S, Mevius K, Michalik D, Hollmann D, Neubert L, Beller M. ChemSusChem. 2009;2:551. doi: 10.1002/cssc.200900034. Pingen D, Müller C, Vogt D. Angew. Chem. Int. Ed. 2010;49:8130. doi: 10.1002/anie.201002583. Zhang M, Imm S, Bahn S, Neumann H, Beller M. Angew. Chem. Int. Ed. 2011;50:11197. doi: 10.1002/anie.201104309.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.