

Abstract
Design of smart polymeric therapeutics
We designed and synthesized trigger-responsive chain-shattering polymeric therapeutics (CSPTs) via condensation polymerization of a UV-or hydrogen peroxide-responsive domain and a bisfunctional drug as co-monomers. CSPTs have precisely controlled molecular composition and unique chain-shattering type of drug release mechanism. Drug release kinetics can be precisely controlled by means of the trigger treatment. Chemotherapeutic-containing CSPTs showed trigger-responsive in vitro and in vivo antitumor efficacy.
Keywords: polymer-drug conjugate, trigger-responsive polymer, controlled release, nanomedicine, smart polymer, cancer therapy
Polymer–drug conjugate is an important polymeric therapeutic (PT) platform[1] with drug molecules being attached via cleavable linkages to the pendant functional groups of linear, branched, brushed polymers[2] that are typically synthesized prior to drug conjugation.[3] The synthesis and conjugation processes developed to date, however, may not provide precise control over the composition and the structure of the conjugates.[4] When polymer with a large number of conjugation-amenable, functional side groups is used, for example, the site of conjugation usually cannot be controlled.[5] As such, batch-to-batch variations of drug loading and release profiles are often observed with polymer–drug conjugates, and these variations may present a key bottleneck to the clinical translation of PTs.[6]
To address these challenges, we recently reported drug-initiated ring-opening polymerization of lactide and other cyclic esters in the presence of a zinc catalyst, a technique that can provide excellent control over drug loading.[7] Hydroxyl-containing drugs are conjugated to polyesters or polycarbonates via an ester linkage, and drug loading can be controlled by tuning the monomer/initiator ratio. Although this technique provides excellent control over drug loading and affords polymer–drug conjugates with controlled structures and compositions, the ability to control drug release from the resulting conjugates is limited. Drug molecules are released by means of hydrolysis or enzymatic cleavage of the ester linkage.[7a] Incorporating a linker that permits trigger-responsive, active release of the terminally conjugated drug remains synthetically challenging.
To develop a new PT with precise control of both drug loading and release, we attempted to incorporate a trigger-responsive domain (TRD) into PT, aiming to achieve specific PT structure and to use the TRD to precisely control drug release. One feasible approach would be using drug and TRD as monomers to construct an A/B (TRD/drug) type of condensation polymer. The resulting PT would have specific repeating unit, and therefore specific molecular structure and composition. Drug release would be precisely controlled by the TRD. Application of an external trigger would activate the TRD, which would subsequently induce chain-shattering type of degradation of the polymer and release the neighboring drug molecules (Scheme 1). Here, we report the use of this approach for the design of chain-shattering polymeric therapeutics (CSPTs) and demonstrate the trigger-induced anticancer activity of CSPT in vitro and in vivo.
Scheme 1.
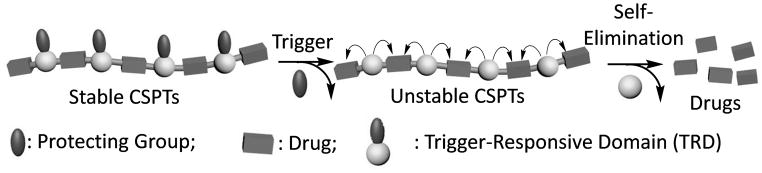
Chain-Shattering Polymeric Therapeutics.
The TRD needs to meet two requirements. First, it should be difunctional and allow for the formation of TRD–drug linkages that are stable under untreated condition but become instantaneously unstable when the trigger is applied. Second, the TRD–drug linkage should degrade rapidly on both sides of the TRD to facilitate chain-shattering type of depolymerization and release of drug molecules in its original form. Because (4-aminophenyl)methanol has been used in the design of trigger-responsive carbonate or urethane linkages that can release the conjugated drug molecules via a 1,6-elimination reaction once the protecting group is removed from the aniline moiety (Scheme 2a), we reasoned that 2,6-bis(hydroxymethyl)aniline (1, Scheme 2b)[8] would likely be condensed with a diol drug to form a PT with trigger-responsive carbonate bonds. Once the protecting group was removed from 1, the PT (two repeating units shown in Scheme 2b) should undergo a 1,4-elimination followed by a 1,8-elimination, leading to chain shattering and the release of the constituent drug molecules.
Scheme 2.

(a) Degradation of (4-aminophenyl)methanol carbonates and carbamates. (b) Degradation of 2,6-bis(hydroxymethyl)aniline (1) carbonate units in a CSPT via 1,4-and 1,8-elimination reactions (two repeating units shown in the scheme). (c) Synthesis of UV-responsive model drug conjugate CPT-1a-CPT.
To determine whether 1 underwent the anticipated elimination reactions, we prepared CPT-1a-CPT (Scheme 2c), a conjugate consisting of 1 protected with a UV-sensitive O-nitrobenzyloxyl-carbonyl group and attached to two camptothecin (CPT) molecules via carbonate linkages (Scheme 2c, Figures S7 and S8). When CPT-1a-CPT was dissolved in acetonitrile/water (9:1, v/v), CPT release was found to be negligible. However, when the conjugate solution was irradiated with UV light (365 nm, 40 mW/cm2) for just 2 min, more than 93 ± 5% of CPT was released (Figures 1a and S10), substantiating the expected 1,4- and 1,8-elimination reactions and the feasibility of using 1 and related analogues for the design of CSPTs.
Figure 1.

(a) Release of CPT from CPT-1a-CPT with or without UV irradiation. (b) Gel permeation chromatographic analysis of CSPT(1a/HCPT) (i) before and (ii) after UV irradiation (365 nm, 40 mW/cm2, 20 min). (c) Release of HCPT and ACPT from CSPT(1a/HCPT) and CSPT(1a/ACPT), respectively, with continuous UV irradiation (+UV) for 15 min or without UV irradiation (−UV). (d) Pulsatile release of HCPT and ACPT from CSPT(1a/HCPT) and CSPT(1a/ACPT), respectively, in response to periodic (every 60 min) UV irradiation for 1 min.
Next, we used 10-hydroxycamptothecin (HCPT) as a model diol drug and synthesized CSPT(1a/HCPT) with a molecular weight (Mn) of 4,200 g/mol and a polydispersity index (PDI) of 1.48 through condensation polymerization (Scheme 3a). To study its UV-triggered degradation, we monitored the change of its Mn value in dimethylformamide (DMF) by means of gel permeation chromatography (GPC). In the absence of UV irradiation, the Mn of CSPT(1a/HCPT) remained unchanged in DMF over a long period of time. In contrast, when CSPT(1a/HCPT) was irradiated with UV light (365 nm, 40 mW/cm2) for 20 min, its Mn changed drastically (from 4,200 to 800 g/mol), and CSPT(1a/HCPT) was almost completely degraded (Figure 1b). We then investigated the release of HCPT from CSPT(1a/HCPT) in DMF/water (9:1, v/v). Without UV irradiation, the proportion of HCPT released from CSPT(1a/HCPT) was negligible (Figure 1c). In contrast, when CSPT(1a/HCPT) was irradiated with UV light for just 2 min, 40% of the HCPT was burst-released in its original form (Figures 1c, S12, and S13). Up to 92% of HCPT was released from CSPT(1a/HCPT) when the CSPT(1a/HCPT) solution was exposed to UV light for an additional 13 min. The drastic decrease in the MW of CSPT(1a/HCPT) and the rapid release of HCPT suggest that the polymer was degraded by means of a chain-shattering mechanism.
Scheme 3.

(a) Synthesis of UV- and H2O2-Responsive CSPTs. (b) Suggested Chain-Shattering Degradation and Release of Drugs from UV-Responsive CSPTs upon UV Irradiation.
Degradation of the CSPT(1a/HCPT) backbone should occur only at 1a residues from which the O-nitrobenzyloxyl-1-carbonyl protecting group has been removed. Once the UV irradiation is stopped, depletion of the protecting group should also stop immediately, resulting in a pause in backbone degradation and HCPT release. The degradation and release should not resume until the trigger (UV light) is reapplied. To verify this expected release behavior, we monitored the release of HCPT from CSPT(1a/HCPT) in DMF/water (9:1, v/v) in response to periodic UV irradiation. As expected, when irradiation was turned on for 1 min and then off for 60 min, pulsatile release of HCPT was observed during the 1-min UV-on periods, and minimal drug release was observed during the 60-min UV-off periods (Figure 1d). This pulsatile HCPT release pattern in response to periodic UV irradiation further substantiates the remarkable responsiveness of this class of CSPT.
Because the amine group is another common functional group amenable to conjugation in natural product–based therapeutics, we next determined whether we could apply the CSPT design strategy to amine-containing therapeutics. We selected 9-aminocamptothecin (ACPT) as the monomer for the synthesis of CSPT(1a/ACPT), and studied its UV responsiveness (Scheme 3a, Figures S5, S15, and S16). ACPT was incorporated to the CSPT(1a/ACPT) backbone via one carbonate bond and one urethane bond. The UV responsiveness of and drug release from CSPT(1a/ACPT) were similar to those of CSPT(1a/HCPT) in DMF/water (9:1, v/v; Figures 1c–d, S14, and S15). Without UV irradiation, ACPT was released from CSPT(1a/ACPT) very slowly (Figure 1c). In contrast, when CSPT(1a/ACPT) was irradiated with UV light (365 nm, 40 mW/cm2) for 2 min, 30% of the ACPT underwent burst release. Up to 88% of ACPT was released when the CSPT(1a/ACPT) solution was exposed to UV light for 15 min (Figures 1c). A pulsatile ACPT release pattern was also observed in response to periodic UV irradiation (Figure 1d).
We next determined whether other triggers could be used to control the degradation of CSPTs and the release of the constituent drug molecules. 4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl-(2,6-bis(hydroxylmethyl)phenyl)carbamate (1b, Scheme 3), an analogue of 1 with a redox-sensitive protecting group, was synthesized and co-condensed with ACPT (Figure S6). CSPT(1b/ACPT) showed the expected H2O2-triggered degradation and rapid ACPT release in DMF/water (9:1, v/v; Scheme S9, Figures S17 and S18).
We further investigated whether CSPTs could be used for formulation of nanoparticle (NP)–based delivery systems with on-demand release profiles. By co-precipitating CSPTs with poly(ethylene glycol)-block-poly(L-lactide) (PEG113-b-PLLA18 or PEL) in water (Figure 2a), we obtained the CSPTs/PEL NPs with diameter below 150 nm, very high drug loading (> 48 wt. %) and very high loading efficiency (> 92%) (Table S1). CSPTs/PEL NPs showed appropriate particle size and drug loading for drug delivery applications. On the contrary, CPT, HCPT, ACPT, and CPT-1a-CPT loaded NPs prepared similarly by co-precipitating with PEL in water afforded particles with very large particle size (> 1 μm), low drug loading and very low loading efficiency (< 10%) (Table S1). CSPT(1a/HCPT)/PEL NPs showed excellent responsiveness to triggered-induced drug release. Without UV irradiation, the proportion of HCPT released from the NPs in phosphate buffered saline (PBS) solution was nearly negligible (Figure 2b). However, when the NPs were irradiated with UV light for 10 min, 59% of the HCPT was released (Figure 2b). Pulsatile release of HCPT from CSPT(1a/HCPT)/PEL NPs was also observed with periodic UV irradiation (Figure 2c). UV-responsive CSPT(1a/ACPT)/PEL NPs were similarly prepared, and they also showed burst release and pulsatile release of ACPT in response to UV irradiation (Figure 2b–c).
Figure 2.

(a) Preparation of CSPT/PEL NPs by a nanoprecipitation method, disassembly of the NPs in response to trigger-induced CSPT degradation, and drug release from the NPs. (b) Release of HCPT and ACPT from CSPT(1a/HCPT)/PEL and CSPT(1a/ACPT)/PEL NPs, respectively, with continuous UV irradiation or without UV irradiation. (c) Pulsatile release of HCPT and ACPT from CSPT(1a/HCPT)/PEL and CSPT(1a/ACPT)/PEL NPs, respectively, in response to periodic (every 60 min) 1-min UV irradiation.
We next evaluated the cytotoxicity of CSPTs/PEL NPs using microculture tetrazolium (MTT) assay. Without UV treatment, CSPT(1a/HCPT)/PEL and CSPT(1a/ACPT)/PEL NPs showed low cytotoxicity in HeLa cells with IC50’s of 1230 nM and 1687 nM respectively (Figure 3a, b). Upon UV treatment, the IC50 values decreased substantially to 97 nM and 109 nM for CSPT(1a/HCPT)/PEL and CSPT(1a/ACPT)/PEL NPs, respectively, suggesting that the cytotoxicity of CSPTs/PEL NPs can be well controlled by external stimulations. Similarly, CSPT(1b/ACPT)/PEL NPs showed drastically higher cytotoxicity in the presence of H2O2 (IC50 = 113 nM) than the untreated CSPT(1b/ACPT)/PEL NPs (IC50 = 1436 nM) (Figure 3a–b). Degradation species from the control polymer (poly(1a/3), Scheme S7) without anticancer drugs did not show any cytotoxicity to the same cells (Figure S20).
Figure 3.

(a, b) Cytotoxicity of CSPTs/PEL NPs in HeLa cells with or without trigger treatment was analyzed by microculture tetrazolium (MTT) assay. Triggering conditions: UV treatment (360 nm, 20 mW/cm2, 10 min) for CSPT(1a/HCPT)/PEL NPs and CSPT(1a/ACPT)/PEL NPs; H2O2 treatment (1 mM) for CSPT(1b/ACPT)/PEL NPs. The half maximal inhibitory concentration (IC50) values were determined by half-cell viability concentration from the MTT assay and summarized in the table. (c, d) BALB/c mice bearing subcutaneous 4T1 tumors received a single intratumoral injection of phosphate buffered saline (PBS), H2O2, ACPT or CSPT(1b/ACPT)/PEL NPs (0.5 mg ACPT equiv/tumor) with or without H2O2 (10 mM, 100 μL/tumor). H2O2 was administered intratumorally 1h after the injection of CSPT(1b/ACPT)/PEL NPs. The mice were sacrificed 48 h post injection. The 4T1 tumors were collected, sectioned and stained with deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end (TUNEL) for apoptosis analysis. Representative images (c) and quantification by ImageJ (d) of TUNEL stains are shown. Scale bar: 50 μm. The apoptosis index was determined as the ratio of apoptotic cell number (TUNEL, green) to the total cell number (4′,6-diamidino-2-phenylindole (DAPI), blue) (20 tissue sections were counted per tumor; n = 4; data are represented as average ± SEM and analyzed by One-way ANOVA (Fisher) (*p < 0.05; n.s. = not significant)).
To further demonstrate the therapeutic efficacy of the CSPTs in vivo, we evaluated the triggered cell apoptosis in subcutaneous 4T1 tumors in BALB/c mice treated with the CSPTs/PEL NPs (Figure 3c, d; Figure S21). Tumors that were injected intratumorally with CSPT(1b/ACPT)/PEL NPs followed by H2O2 treatment showed 2.5 fold higher apoptosis index (69.6 ± 5.0%) compared to those without H2O2 treatment (27.3 ± 2.7%). To exclude the possibility that cell apoptosis were induced by H2O2, the mice were treated intratumorally with H2O2 (10 mM, 100 μL/tumor) alone; no significant cell apoptosis (with apoptosis index of 18.3 ± 1.7%) was observed as compared to PBS (1 ×, 100 μL) negative control group (with apoptosis index of 15.2 ± 4.8%) (Figure 3c–d). Therefore, the trigger-responsive CSPT(1b/ACPT)/PEL NPs markedly improved the antitumor efficacy by inducing higher apoptosis index in tumors with elevated level of reactive oxygen species, including H2O2, which is one of the characteristics of tumor tissues.[9]
The development of PTs for personalized medicine requires precise control over drug release; the payload ideally is retained in the delivery vehicle during circulation, tissue distribution, and cellular trafficking processes and then burst released when the delivery vehicle reaches the target cells or intracellular compartments. In this study, we designed 2,6-bis(hydroxymethyl)anilines with UV- and redox-sensitive protecting groups and used these anilines as monomers for condensation with bisfunctional drugs to create CSPTs and as TRDs for controlling the complete drug release on a chain-shattering manner upon exposure to external triggers. Pulsatile drug release from the CSPTs was observed in response to periodically applied triggers. The trigger-responsive cytotoxicity and in vivo antitumor efficacy of CSPTs were demonstrated by applying external stimulations. This class of CSPTs showed precise control over drug release and may become important building blocks for the preparation of controlled release devices and nanomedicines for in vitro and in vivo applications.
Supplementary Material
Acknowledgments
This work is supported by Dow Chemical Company. We thank Dr. Liang Hong and Dr. Keith Harris for helpful discussion. Q.Y. was funded at UIUC from NIH National Cancer Institute Alliance for Nanotechnology in Cancer ‘Midwest Cancer Nanotechnology Training Center’ Grant R25 CA154015A.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Peer D, Karp JM, Hong S, FaroKHzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]; b) Pack DW, Hoffman AS, Pun S, Stayton PS. Nat Rev Drug Discovery. 2005;4:581–593. doi: 10.1038/nrd1775. [DOI] [PubMed] [Google Scholar]; c) Bagalkot V, Farokhzad OC, Langer R, Jon S. Angew Chem. 2006;118:8329–8332. doi: 10.1002/anie.200602251. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:8149–8152. doi: 10.1002/anie.200602251. [DOI] [PubMed] [Google Scholar]; d) Johnson JA, Lu YY, Burts AO, Lim YH, Finn MG, Koberstein JT, Turro NJ, Tirrell DA, Grubbs RH. J Am Chem Soc. 2011;133:559–566. doi: 10.1021/ja108441d. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Mammen M, Choi SK, Whitesides GM. Angew Chem. 1998;110:2908–2953. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 1998;37:2755–2794. [Google Scholar]; f) Henne WA, Doorneweerd DD, Hilgenbrink AR, Kularatne SA, Low PS. Bioorg Med Chem Lett. 2006;16:5350–5355. doi: 10.1016/j.bmcl.2006.07.076. [DOI] [PubMed] [Google Scholar]; g) Qiu LY, Bae YH. Pharm Res. 2006;23:1–30. doi: 10.1007/s11095-005-9046-2. [DOI] [PubMed] [Google Scholar]; h) Fox ME, Szoka FC, Frechet JMJ. Acc Chem Res. 2009;42:1141–1151. doi: 10.1021/ar900035f. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Venkataraman S, Hedrick JL, Ong ZY, Yang C, Ee PLR, Hammond PT, Yang YY. Adv Drug Deliv Rev. 2011;63:1228–1246. doi: 10.1016/j.addr.2011.06.016. [DOI] [PubMed] [Google Scholar]; j) Ihre HR, De Jesus OLP, Szoka FC, Frechet JMJ. Bioconjugate Chem. 2002;13:443–452. doi: 10.1021/bc010102u. [DOI] [PubMed] [Google Scholar]
- 2.a) Erdmann L, Uhrich KE. Biomaterials. 2000;21:1941–1946. doi: 10.1016/s0142-9612(00)00073-9. [DOI] [PubMed] [Google Scholar]; b) Duncan R. Nat Rev Cancer. 2006;6:688–701. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]; c) Li C, Wallace S. Adv Drug Deliv Rev. 2008;60:886–898. doi: 10.1016/j.addr.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ulbrich K, Subr V. Adv Drug Deliv Rev. 2004;56:1023–1050. doi: 10.1016/j.addr.2003.10.040. [DOI] [PubMed] [Google Scholar]; e) Zhou Z, Shen Y, Tang J, Fan M, Van Kirk EA, Murdoch WJ, Radosz M. Adv Funct Mater. 2009;19:3580–3589. [Google Scholar]; f) Tong R, Christian DA, Tang L, Cabral H, Baker JR, Kataoka K, Discher DE, Cheng J. MRS Bulletin. 2009;34:422–431. [Google Scholar]
- 3.a) MacKay JA, Chen MN, McDaniel JR, Liu W, Simnick AJ, Chilkoti A. Nat Mater. 2009;8:993–999. doi: 10.1038/nmat2569. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Canal F, Sanchis J, Vicent MJ. Curr Opin Biotech. 2011;22:894–900. doi: 10.1016/j.copbio.2011.06.003. [DOI] [PubMed] [Google Scholar]; c) Duncan R. Curr Opin Biotech. 2011;22:492–501. doi: 10.1016/j.copbio.2011.05.507. [DOI] [PubMed] [Google Scholar]; d) Aryal S, Hu CMJ, Zhang L. Acs Nano. 2010;4:251–258. doi: 10.1021/nn9014032. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Su J, Chen F, Cryns VL, Messersmith PB. J Am Chem Soc. 2011;133:11850–11853. doi: 10.1021/ja203077x. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Bae Y, Nishiyama N, Fukushima S, Koyama H, Yasuhiro M, Kataoka K. Bioconjugate Chem. 2005;16:122–130. doi: 10.1021/bc0498166. [DOI] [PubMed] [Google Scholar]; g) Zhao H, Lee C, Sai PK, Choe YH, Boro M, Pendri A, Guan SY, Greenwald RB. J Org Chem. 2000;65:4601–4606. doi: 10.1021/jo000221n. [DOI] [PubMed] [Google Scholar]
- 4.a) Lammers T. Adv Drug Deliv Rev. 2010;62:203–230. doi: 10.1016/j.addr.2009.11.028. [DOI] [PubMed] [Google Scholar]; b) Chytil P, Etrych T, Konak C, Sirova M, Mrkvan T, Boucek J, Rihova B, Ulbrich K. J Control Release. 2008;127:121–130. doi: 10.1016/j.jconrel.2008.01.007. [DOI] [PubMed] [Google Scholar]; c) Nori A, Kopecek J. Adv Drug Deliv Rev. 2005;57:609–636. doi: 10.1016/j.addr.2004.10.006. [DOI] [PubMed] [Google Scholar]; d) Parrott MC, Finniss M, Luft JC, Pandya A, Gullapalli A, Napier ME, Desimone JM. J Am Chem Soc. 2012;134:7978–7982. doi: 10.1021/ja301710z. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Liu T, Li X, Qian Y, Hu X, Liu S. Biomaterials. 2012;33:2521–2531. doi: 10.1016/j.biomaterials.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 5.a) Christie RJ, Grainger DW. Adv Drug Deliv Rev. 2003;55:421–437. doi: 10.1016/s0169-409x(02)00229-6. [DOI] [PubMed] [Google Scholar]; b) Duncan R, Vicent MJ. Adv Drug Deliv Rev. 2010;62:272–282. doi: 10.1016/j.addr.2009.12.005. [DOI] [PubMed] [Google Scholar]; c) Miller K, Erez R, Segal E, Shabat D, Satchi-Fainaro R. Angew Chem. 2009;121:2993–2998. doi: 10.1002/anie.200805133. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:2949–2954. doi: 10.1002/anie.200805133. [DOI] [PubMed] [Google Scholar]; d) Satchi-Fainaro R, Hailu H, Davies JW, Summerford C, Duncan R. Bioconjugate Chem. 2003;14:797–804. doi: 10.1021/bc020091k. [DOI] [PubMed] [Google Scholar]
- 6.a) Erez R, Segal E, Miller K, Satchi-Fainaro R, Shabat D. Bioorgan Med Chem. 2009;17:4327–4335. doi: 10.1016/j.bmc.2009.05.028. [DOI] [PubMed] [Google Scholar]; b) Sirova M, Strohalm J, Subr V, Plocova D, Rossmann P, Mrkvan T, Ulbrich K, Rihova B. Cancer Immunol Immun. 2007;56:35–47. doi: 10.1007/s00262-006-0168-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kovar L, Etrych T, Kabesova M, Subr V, Vetvicka D, Hovorka O, Strohalm J, Sklenar J, Chytil P, Ulbrich K, Rihova B. Tumor Biol. 2010;31:233–242. doi: 10.1007/s13277-010-0019-7. [DOI] [PubMed] [Google Scholar]; d) Caiolfa VR, Zamai M, Fiorino A, Frigerio E, Pellizzoni C, d’Argy R, Ghiglieri A, Castelli MG, Farao M, Pesenti E, Gigli M, Angelucci F, Suarato A. J Control Release. 2000;65:105–119. doi: 10.1016/s0168-3659(99)00243-6. [DOI] [PubMed] [Google Scholar]
- 7.a) Tong R. J Cheng, Angew Chem. 2008;120:4908–2912. [Google Scholar]; Angew Chem Int Ed. 2008;47:4830–4834. doi: 10.1002/anie.200800491. [DOI] [PubMed] [Google Scholar]; b) Tong R, Cheng J. J Am Chem Soc. 2009;131:4744–4754. doi: 10.1021/ja8084675. [DOI] [PubMed] [Google Scholar]; c) Tong R, Yala LD, Fan TM, Cheng J. Biomaterials. 2010;31:3043–3053. doi: 10.1016/j.biomaterials.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Tong R, Cheng J. Bioconjugate Chem. 2010;21:111–121. doi: 10.1021/bc900356g. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Tong R, Cheng J. Macromolecules. 2012;45:2225–2232. doi: 10.1021/ma202581d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y, Ma L, Deng X, Cheng J. Polym Chem. 2013;4:224–228. [Google Scholar]
- 9.a) Bechtel W, Bauer G. Anticancer Res. 2009;29:4559–4570. [PubMed] [Google Scholar]; b) Mantovani G, Maccio A, Madeddu C, Mura L, Massa E, Gramignano G, Lusso MR, Murgia V, Camboni P, Ferreli L. J Cell Mol Med. 2002;6:570–582. doi: 10.1111/j.1582-4934.2002.tb00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sies H. Angew Chem. 1986;98:1061–1078. [Google Scholar]; Angew Chem Int Ed. 1986;25:1058–1071. [Google Scholar]; d) Yoo D, Jeong H, Preihs C, Choi JS, Shin TH, Sessler JL, Cheon J. Angew Chem. 2012;124:12650–12653. doi: 10.1002/anie.201206400. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2012;51:12482–12485. doi: 10.1002/anie.201206400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.