

Significance
Many proteins undergo various posttranslational modifications for proper functions. One such modification is methylation carried out by enzyme–cofactor pairs of protein methyltransferases (PMTs) and S-adenosyl-L-methionine (SAM). Identification of methylated proteins is quite challenging because of the small size and chemical inertness of the methyl group. To address this challenge, we have synthesized SAM surrogates by replacing SAM’s methyl group with bulky, chemically active functionalities and demonstrated their utility as alternative cofactors of engineered PMTs for substrate labeling. Proteins modified with such chemical moieties are amenable to bioorthogonal reactions for subsequent enrichment and identification. An engineered enzyme–cofactor pair has been successfully used to reveal numerous methylated proteins.
Keywords: epigenetic, bump-hole, posttranslation, proteomics
Abstract
Protein methyltransferase (PMT)-mediated posttranslational modification of histone and nonhistone substrates modulates stability, localization, and interacting partners of target proteins in diverse cellular contexts. These events play critical roles in normal biological processes and are frequently deregulated in human diseases. In the course of identifying substrates of individual PMTs, bioorthogonal profiling of protein methylation (BPPM) has demonstrated its merits. In this approach, specific PMTs are engineered to process S-adenosyl-L-methionine (SAM) analogs as cofactor surrogates and label their substrates with distinct chemical modifications for target elucidation. Despite the proof-of-concept advancement of BPPM, few efforts have been made to explore its generality. With two cancer-relevant PMTs, EuHMT1 (GLP1/KMT1D) and EuHMT2 (G9a/KMT1C), as models, we defined the key structural features of engineered PMTs and matched SAM analogs that can render the orthogonal enzyme–cofactor pairs for efficient catalysis. Here we have demonstrated that the presence of sulfonium-β-sp2 carbon and flexible, medium-sized sulfonium-δ-substituents are crucial for SAM analogs as BPPM reagents. The bulky cofactors can be accommodated by tailoring the conserved Y1211/Y1154 residues and nearby hydrophobic cavities of EuHMT1/2. Profiling proteome-wide substrates with BPPM allowed identification of >500 targets of EuHMT1/2 with representative targets validated using native EuHMT1/2 and SAM. This finding indicates that EuHMT1/2 may regulate many cellular events previously unrecognized to be modulated by methylation. The present work, therefore, paves the way to a broader application of the BPPM technology to profile methylomes of diverse PMTs and elucidate their downstream functions.
Histone modifications through protein methyltransferases (PMTs) play essential roles in gene transcription, cellular differentiation, and organismal development (1). Accumulated evidence also shows that the physiological functions of PMT-mediated methylation go beyond chromatin biology and can act through diverse nonhistone targets (2). Protein methylation has attracted a lot of attention for its role in modulating protein–protein interactions in cellular interactome dynamics (3). PMT-mediated methylation can alter physical properties, stability, and localization of target proteins, and create docking sites for binding partners (3, 4). The downstream functions of individual PMTs are thus closely associated with their methylomes and interactomes (proteome-wide methylation targets and their binding partners, respectively) (3). Aberrant levels of PMTs and their gain/loss-of-function mutations can disrupt these interacting networks and are often implicated in cancer and other disorders (3, 5).
Efficient methods to elucidate proteome-wide substrates of individual PMTs are valuable for understanding biological functions of PMT-catalyzed methylation. Given that the human genome encodes more than 60 PMTs, most of which use highly conserved catalytic domains to process the methylation cofactor S-adenosyl-L-methionine (SAM) 1 (6, 7) and often act via multimeric complexes in cellular milieu (8), it is a considerable challenge to profile methylomes of individual PMTs in the native state without the interference of other PMTs. To identify novel substrates of a specific PMT, prior approaches relied on truncated PMTs to screen substrate candidates such as peptide/protein arrays or PMT-knockout proteome (9, 10). SAM analogs containing terminal alkyne were also reported to be active for certain native PMTs. These cofactor surrogates can therefore be used for target identification (10) when coupled with copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC/“Click Chemistry”) (11, 12). The utility of terminal alkyne/azide-containing SAM analogs was further expanded by the bioorthogonal profiling of protein methylation (BPPM) technology (13), in which PMTs are engineered to accommodate bulky SAM analogs (the “bump-hole” approach as applied for kinases) (14) for substrate labeling and subsequent target identification (SI Appendix, Fig. S1). Despite the proof-of-concept advancement and promising application of the BPPM technology, little is known about key structural features of extended SAM analogs and PMT variants that can render such orthogonal activities.
In the present work, we focused on two human PMTs, EuHMT1 (GLP1/KMT1D) and EuHMT2 (G9a/KMT1C). The two SET-domain-containing PMTs were previously characterized to methylate histone H3 lysine 9 (H3K9) and some nonhistone substrates such as tumor suppressor p53, DNA cytosine methyltransferase 3A, ATF-α-associated modulator, CCAAT/enhancer-binding protein-β, chromatin-remodeling factor Reptin, and myogenic regulatory factor MyoD (15–21). Among implicated functions of EuHMT1/2 are establishing de novo DNA methylation, silencing proviral/tumor suppressor genes, and regulating neurons/skeletal muscle differentiation (19, 22). With these PMTs as a model, we systematically leveraged structural and biochemical evaluation on a set of SAM analogs 2−7 as cofactor surrogates (Fig. 1 and SI Appendix, Fig. S1). Our biochemical characterization concludes that the existence of the sulfonium-β-sp2 carbon and the structurally matched sulfonium-δ-substituents are essential for SAM analogs as active BPPM cofactors. Structural analysis further revealed that replacing EuHMT1/2’s bulky Y1211/Y1154 with a smaller Ala not only extends the SAM-binding pockets of the parent enzymes but also makes preexisting hydrophobic cavities accessible for the bulky SAM analogs. We therefore defined a set of SAM analogs as privileged methyl surrogate tagging (m-Tag) reagents and a general principle to engineer SET-domain-containing PMTs for BPPM application. The BPPM uncovered >500 potential nonhistone substrates of EuHMT1/2, including many proteins characterized previously and unique targets readily validated here. The broad scope of the nonhistone substrates of EuHMT1/2 suggests that the PMTs may participate in a number of cellular processes that were not previously recognized to be modulated by protein methylation. The BPPM approach is therefore expected to be generally applicable to decipher methylomes and annotate biological functions of diverse PMTs.
Fig. 1.

Heat-map analysis of the extent of modification (percentage) of H3K9 peptide by SAM or SAM analogs with native and mutated EuHMT1. Sites of mutation are highlighted for EuHMT1 (PDB ID code 2RFI). The results of EuHMT1 are similar to those of EuHMT2 (SI Appendix, Fig. S2).
Results
Screening SAM Analogs As Potential Cofactors for BPPM.
Multiple SAM analogs have been reported as active cofactors for native and engineered PMTs (10, 13, 23–26). Most of these compounds contain a characteristic sulfonium-β-sp2 carbon to promote the enzymatic transalkylation reaction (10). To rationalize the generality of the cofactors as m-Tag reagents for BPPM, we synthesized and evaluated a panel of such SAM analogs 2–7 (Fig. 1 and SI Appendix, Figs. S1 and S2). These SAM analogs are featured by the sulfonium-β-sp2 moiety but vary at their δ-substituents. Here we evaluated these SAM derivatives as cofactor surrogates against a panel of previously examined PMT variants including EuHMT1’s Y1211A and EuHMT2’s Y1154A mutants (Fig. 1 and SI Appendix, Fig. S2, respectively), which were shown to be active toward bulky (E)-hex-2-en-5-ynyl SAM (Hey-SAM) 6 and 4-azidobut-2-enyl SAM (Ab-SAM) (13, 25). The panel of SAM analogs and PMT variants were screened in a combinatorial manner and their activities were quantified by the extent of modification (percentage) of the substrate using MALDI-MS as a primary assay (Fig. 1 and SI Appendix, Figs. S2–S4).
All of the SAM derivatives examined here were shown to be active toward EuHMT-1’s Y1211A and EuHMT-2’s Y1154A mutants, albeit to different extents, confirming the importance of the sulfonium-β-sp2 moiety in the enzymatic transalkylation reactions (13, 25). Among the SAM analogs, the smallest SAM 1 and the largest homo Hey-SAM 7 are least active, whereas the medium-sized SAM analogs such as trans-butene-SAM 3, trans-pentene-SAM 4, and Hey-SAM 6 are most active toward EuHMT-1/2’s Y1211A/Y1154A mutants (Fig. 1 and SI Appendix, Figs. S2–S4). Enyn-SAM 5, as an outlier, displayed much lower activity than the size-comparable SAM analogs 4 and 6, likely owing to the rigidity of the former’s enyne moiety (Discussion). In contrast, the panel of bulky SAM analogs displayed either low or undetectable activities toward native EuHMT1/2 and other PMT variants (EuHMT-1/2’s Y1124A/Y1067A, Y1142A/Y1085A, F1144A/F1087A, F1195A/F1138A, F1215A/F1158A, and W1216A/W1159A; Fig. 1 and SI Appendix, Fig. S2). Although most of the EuHMT-1/2 variants are inactive toward bulky SAM analogs, F1209A/F1152A and Y1211A/Y1154A mutants displayed robust activity (Fig. 1 and SI Appendix, Fig. S2). The gain-of-function character of Y1211A/Y1154A mutations is consistent with our previous observation that the two PMT variants can accommodate sulfonium-β-sp2–containing bulky cofactors (13). However, the 100-fold alteration of activity among these compounds indicates that certain structural features of m-Tag SAM analogs have profound effects on their efficiency as cofactors.
Structural Characterization for Recognition of EuHMT1/2’s Variants on Bulky Cofactors.
To elucidate the structural basis of the promiscuous recognition of EuHMT1/2’s Y1211A/Y1154A variants on bulky SAM analogs, we determined the ternary structure of Y1211A in complex with S-adenosyl-L-homocysteine (SAH) and H3K9 Nε-allyl peptide (27) (an enzyme-product complex of the transalkylation reaction between allyl-SAM 2 and H3K9 peptide, PDB ID code 4H4H; SI Appendix, Table S1). Given the robust activity of EuHMT1’s Y1211A mutant on bulky SAM analogs, we first focused on the Y1211 region and its interaction with the allyl moiety in the enzyme–product complex. Despite the overall similarity between native EuHMT1 and Y1211A variant in their SAH-binding sites (discussed below), Y1211A mutant is distinct from the native enzyme at an entrance site of a preexisting hydrophobic cavity formed by the side chains of H1170/F1209/F1215/W1216 and the backbones of I1168/N1169. Given that the allyl moiety of the ternary Y1211A–product complex is pointed toward the hydrophobic cavity, this cavity is expected to accommodate the bulky side chains of the SAM analogs (Fig. 2A). However, the access to the hydrophobic cavity is blocked in native EuHMT1 by the bulky Y1211 residue acting as a gatekeeper (Fig. 2 A and B). The Y1211A mutation discharges the gatekeeper residue and thus leads to a widely open hydrophobic pocket. This pocket was shown to be occupied partially by the allyl moiety of the H3K9-allylated peptide and likely the corresponding sulfonium alkyl substitutions of active SAM analogs.
Fig. 2.

Structural characterization of EuHMT1-Y1211A mutant in complex with SAH and the allylated histone H3K9 peptide. (A) Allyl modification on substrate peptide is accommodated by the hydrophobic cavity formed by I1168/N1169/H1170/F1209/F1215/W1216 EuHMT1’s Y1211A variant (PDB ID code 4H4H). (B) Superimposed structures of EuHMT1 (PDB ID code 2RFI) and its Y1211A mutant (PDB ID code 4H4H). Y1211 residue of native EuHMT1 acts as a gatekeeper residue.
The overlaid structures of native EuHMT1 and its Y1211A mutant further revealed that the terminal sp2 carbon of the allyl moiety of allylated H3K9 peptide (and likely allyl-SAM) resides in the vicinity of the side chain of the Y1211 residue (Fig. 2B). Such proximity suggests a potential steric clash between bulkier SAM analogs 3–7 and the Y1211 residue of native EuHMT1, and thus prevents native EuHMT1 from binding these SAM analogs. In contrast, replacing the Y1211 residue with Ala not only avoids such steric crowding but also allows the extended alkyl moieties of SAM analogs to gain additional hydrophobic interaction with the preexisting pocket. This recognition pattern is expected to extend to other SET-domain-containing PMTs (SI Appendix, Fig. S5), given their structural similarity with EuHMT1/2 (28, 29) (Discussion).
The structure of the ternary enzyme–product complex (Y1211A mutant, allylated H3K9 peptide, and SAH) is superimposable with that of native EuHMT1 in complex with the dimethylated peptide and SAH (PDB ID code 2RFI) within 0.24-Å backbone deviation. This excellent overlay argues that the Y1211A mutant maintains the overall structural integrity of native EuHMT1 (Fig. 2B and SI Appendix, Fig. S6). Except for the region adjacent to the mutated Y1211 residue (a slightly kinked δ-carbon of the substrate Lys9 and the loss of the side-chain hydrogen bond of Y1211), the Y1211A mutant with the allylated H3K9 peptide and SAH recapitulates most of the interactions of native EuHMT1 with the dimethylated H3K9 peptide and SAH (SI Appendix, Fig. S7). Such consistency therefore rationalizes the previous observation (25) that the Y1211A mutant behaves like the native enzyme in terms of substrate recognition by solely acting on the H3K9 site among over 50 lysine residues of the histone octamer (13, 25, 30). The overall structural comparison between native EuHMT1 and its Y1211A mutant thus provides the rationale that the single Y1211A mutation enhances the cofactor promiscuity of the PMT without altering its substrate specificity.
Quantitative Determination of Kinetic Parameters for Active Cofactor–Mutant Pairs.
To further explore the structure–activity relationship of the panel of cofactors for BPPM, we determined their apparent Km and kcat with native EuHMT1/2 and the most active Y1211A/Y1154A variants (Fig. 3 and SI Appendix, Figs. S8 and S9). Here the smallest SAM 1, the sterically rigid SAM analog 5, and the largest cofactor 7 showed slightly higher Km (low affinity) and much smaller kcat toward Y1211A/Y1154A mutants (slow enzymatic turnovers as reflected by 10- to 100-fold smaller kcat/Km) than those of EuHMT1/2 on SAM (Fig. 3 and SI Appendix, Fig. S9). For EuHMT1/2, a slightly increased size of the sulfonium-δ-substituents of SAM analogs can dramatically disrupt their activities as cofactors, as shown by ∼20-fold loss of Km and 2- to 20-fold decrease of kcat from SAM 1 to allyl-SAM 2 and no detectable activity for bulkier SAM analogs 3–7. The biochemical analysis further revealed that with the increased sizes of the sulfonium-δ-substituents of the SAM analogs from the hydrogen in 2 to methyl/ethyl/propargyl in 3, 4, and 6, the corresponding Km and kcat of the cofactors toward Y1211A/Y1154A mutants were improved to the range comparable to those of SAM toward native EuHMT1/2 (Fig. 3 and SI Appendix, Fig. S9). These kinetic experiments thus provided a quantitative correlation between the δ-substituents of sulfonium-β-sp2–containing SAM analogs and their cofactor activities. Here the cofactors 3, 4, and 6, when paired with EuHMT1/2’s Y1211A/Y1154A mutants, displayed higher catalytic efficiency (larger kcat/Km). Among the synthetic cofactors examined here, highly reactive Hey-SAM 6 stands as an ideal BPPM reagent, given the presence of the terminal alkyne moiety for CuAAC (11).
Fig. 3.
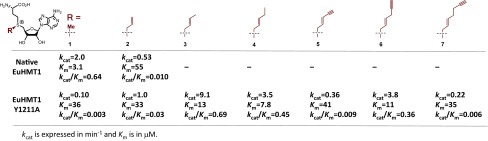
Steady-state kinetic analysis of native EuHMT1 and its Y1211A mutant with SAM 1 and its analogs 2–7 as cofactors. Apparent kcat and Km were obtained with the fixed concentration of H3K9 peptide substrate (25 μM) and the varied concentration of cofactors.
Application of Efficient Mutant–Cofactor Pairs to Profile EuHMT1/2 Nonhistone Targets.
The concept of BPPM was demonstrated previously with Ab-SAM as a SAM surrogate (13). However, the size of Ab-SAM is similar to that of 7, whose activity toward Y1211A/Y1154A is ∼100-fold lower than that of SAM toward EuHMT1/2 (results in Fig. 3 and SI Appendix, Fig. S9). Because of the compromised activity, Ab-SAM might not be an ideal cofactor to reveal the full spectrum of EuHMT1/2’s targets via BPPM (Discussion). Given that the cofactor–mutant pairs of 6 and Y1211A/Y1154A show in vitro kcat and Km comparable to native SAM-EuHMT1/2 pairs, we were intrigued to use Hey-SAM 6 to identify the targets of EuHMT1/2 (Fig. 4A). Here, human embryonic kidney (HEK293T) cells were transfected with the plasmids containing full-length Y1211A/Y1154A mutants or an empty vector as control. The proteome-wide targets of EuHMT1/2 were first visualized by treating the cell lysates with 6 followed by CuAAC with an azido-containing fluorescent probe azo-Rho (31). In-gel fluorescence showed substantial labeling of proteins as putative targets of EuHMT1/2 (SI Appendix, Fig. S10). Barely detectable fluorescence signals in the control further indicate that Hey-SAM 6 cannot be processed efficiently by endogenous PMTs of HEK293T cells.
Fig. 4.

Labeling and MS identification of proteome-wide substrates of EuHMT1/2. (A) Schematic description of labeling and MS-based proteomic analysis via BPPM. (B) Venn diagram of the BPPM-revealed substrates of EuHMT1/2 via the direct comparison of the LC-MS/MS data or the quantitive comparison of TMT MS/MS data. (C and D) Correlation analysis of the EuHMT1/2 targets of high confidence. The criteria to select these proteins: present in the mutant-transfected cell samples but absent in the empty vector-transfected cell samples for direct comparison of LC-MS/MS data and >1.5-fold enrichment in mutant-transfected over empty vector-transfected cell lysates in TMT-based proteomics. The abundance ratios of the BPPM-revealed substrates of EuHMT1/2 versus controls were plotted against x and y axis, respectively (C). The abundance ratios of the BPPM-revealed substrates of EuHMT1 versus EuHMT2 were plotted against the unique target ID (D). A significant part of the targets displayed >1.5-fold enrichment in the sample of EuHMT1 in comparison with that of EuHMT2.
To identify the putative targets of EuHMT1/2, cell lysates containing full-length Y1211A/Y1154A mutants or empty vector were treated with 6 followed by CuAAC-mediated conjugation with an azide-biotin probe. This probe is embellished with a cleavable azobenzene linker, which is susceptible to the treatment of sodium dithionite (32). After affinity pulldown of biotin-conjugated proteins with streptavidin beads, the enriched targets were released with sodium dithionite (32) and then subject to liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) analysis (SI Appendix). The direct MS results revealed 1,324 and 1,648 proteins (these proteins are not present in the control) as potential targets of EuHMT1 and EuHMT2, respectively (SI Appendix, Fig. S10 and Tables S2–S4). With the less-quantitative spectral counts of LC-MS/MS (SI Appendix, Tables S2 andS3), 799 proteins (36% overlap) were found to be shared by EuHMT1 and EuHMT2.
Target Validation via Quantitative Amine-Reactive Isobaric Tandem Mass Tagging and with Native Enzyme–Cofactor Pairs.
We then implemented amine-reactive isobaric tandem mass tagging (TMT) for simultaneous labeling and analysis of multiple samples in a quantitative manner. Here the tryptic peptides pulled down from empty vector- and EuHMT1/2-tarnsfected cells were treated with the isobaric reagents containing the reporter ions with m/z 126, 128, and 131, respectively (Fig. 4A). Samples were then mixed and subjected to MS/MS with protein identities uncovered by MS-decoded peptide sequences, whereas the reporter ions are dissociated from tryptic peptides with respective abundance reflected through the relative intensities of the reporter ions. A total of 1,145 proteins were uncovered in the TMT-based proteome as EuHMT1/2 substrates (SI Appendix, Table S5). Cross-analysis between results of the direct LC-MS/MS (SI Appendix, Fig. S10 and Tables S2–S4) and the quantitive TMT LC-MS/MS allowed the identification of 774 overlaid targets (Fig. 4B), which we designated as EuHMT1/2 substrates of high confidence.
Quantitative analysis of these high-confidence proteins indicates that they were significantly enriched in the enzyme-transfected cell lysates compared with control cells (Fig. 4C) with the improved overlay between EuHMT1 and EuHMT2 (Fig. 4D) compared with the direct LC-MS/MS analysis (SI Appendix, Fig. S10). The BPPM-revealed proteins (solely present or significantly enriched in the mutant-transfected cell lysates) include a set of known nonhistone substrates of EuHMT1/2 (SI Appendix, Table S6). Such consistency thus implied the robustness of the BPPM approach to tag, enrich, and identify the substrates of designated PMTs from complex cellular milieu. Here 774 distinct proteins were identified as the substrates of EuHMT1/2 with Hey-SAM 6. In contrast, only 128 proteins were identified previously with Ab-SAM (13). A substantial portion of the previously obtained targets was included in the current dataset revealed with Hey-SAM 6 as the BPPM probe. The sixfold increase in target identification is consistent with the higher activity of Hey-SAM 6 (Fig. 3 and SI Appendix, Fig. S9) and presents this SAM analog as a desired BPPM regent for EuHMT1/2.
To validate the BPPM-revealed proteins as authentic EuHMT1/2 targets, a diverse, representative set of commercially available proteins, such as nucleolin, EEF1A1, POLR2A, HAT1, PARP1, PKIM1, HNPRK, TARS, ACLY, PRMT5, and IDH1, were subject to in vitro assays with native EuHMT1/2 and radiolabeled SAM (3H-SAM), followed by autoradiographic analysis (SI Appendix, Fig. S11). The robust methylation was readily detected for EuHMT1, EuHTM2, or both, to various degrees and with the efficiency of many substrates comparable to histone H3 (e.g., POLR2A, PRMT5, IDH1, and nucelolin). MS-based identification of methylation sites in these nonhistone substrates would be the next step to define the sequence specificity of EuHMT1/2. The ready validation of these proteins as EuHMT1/2 targets thus demonstrates the merit of the BPPM technology to uncover PMT targets from complex cellular milieu. The current in vitro data showed slightly preferential methylation by EuHMT1 (SI Appendix, Fig. S11), consistent with the quantitive comparison of the targets of EuHMT1/2 (Fig. 4D). However, given the reported higher activities of the full-length EuHMT1/2 in cellular contexts (13), the in vitro data likely only recapitulate EuHMT1/2’s partial abilities for substrate methylation.
Subcellular Localization and Functional Annotation of EuHMT1/2’s Targets.
Among the BPPM-revealed targets of EuHMT1/2 are histones (e.g., H1.4/H3), transcription factors (e.g., P53), cytoskeletal proteins (e.g., myosin/ARP2/3), chromatin modifying/remodeling enzymes (e.g., DNMT/CHD3), RNA helicases/splicing factors (e.g., DHX29/PRP16), components of nuclear pore complex (e.g., Nup93/Nup155), and the proteins involved in cellular division, stress, and metabolism (e.g., Cdc42/BUB3/CDK2/HSP70/HSP90). Given that only a handful of nuclear proteins have been reported previously as the substrates of EuHMT1/2 (15–19), we evaluated the subcellular localization of the BPPM-derived EuHMT1/2 substrates (SI Appendix, Fig. S12A and Table S7). Around 30% of the EuHMT1/2 targets are nuclear proteins, consistent with the preferential nuclear localization of EuHMT1/2 (15–19). However, a significant portion of the newly revealed EuHMT1/2 targets are cytosolic, membrane-bound, and mitochondrial proteins, likely revealing hitherto unappreciated roles of EuHMT1/2 in cellular processes.
A survey of biological functions of the nonhistone targets of EuHMT1/2 by IPA (Ingenuity Systems, www.ingenuity.com) indicates that these proteins participate in diverse cellular and molecular processes such as cell death and survival, gene expression, and RNA posttranscriptional modification (SI Appendix, Fig. S12B and Table S8). Further functional annotation revealed that EuHMT1/2-mediated nonhistone methylation plays important physiological roles such as organismal survival and embryonic and tissue development (SI Appendix, Fig. S12C and Table S8). Such analysis thus suggests that PMTs may exert important functions in eucaryotic biology through nonhistone methylation.
Discussion
Structural Features of Engineered EuHMT1 to Recognize Bulky SAM Analogs.
Our previous work demonstrated that EuHMT1 and its closely related homolog EuHMT2 can be engineered to process bulky SAM analogs (13, 25). The current structural analysis provides molecular-level understanding of how the engineered EuHMT1 (and likely other related PMTs) recognizes the SAM analog cofactors. Apparently, EuHMT1’s Y1211A mutation can generate extra space at the active site by replacing the bulky Tyr with a smaller Ala. More importantly, Y1211 was further characterized as a gatekeeper to block bulky SAM analogs to exploit the preexisting hydrophobic cavity formed by its I1168/N1169/H1170/F1209/F1215/W1216 residues. The Y1211A mutation, which does not alter the overall substrate-binding pattern of native EuHMT1, removes the gatekeeper, thus allowing the bulky cofactors to fully access the preexisting hydrophobic cavity (Fig. 2 and SI Appendix, Fig. S13A).
The structural model is consistent with our biochemical results that native EuHMT1 is active toward SAM with Km = 3.1 μM, less active on ally-SAM 2 with Km = 54.5 μM (20-fold loss of Km), and completely inert toward other bulkier SAM analogs 3–7 (Fig. 3 and SI Appendix, Fig. S9). In contrast, the Y1211A mutant showed promiscuous activities toward SAM analogs 2–7 with comparable Km of 7–35 μM. A similar trend is also observed for EuHMT1’s homolog EuHMT2 (Km = 4.0 μM for SAM; Km = 68 μM for allyl-SAM; no activity for 3–7) and its Y1154A mutant (Km of 12–46 μM for SAM and SAM analogs 2–7; Fig. 3 and SI Appendix, Fig. S9). The two- to fivefold improved Km of 3, 4, and 6 versus 1 and 2 toward Y1211A/Y1154A mutants (Fig. 3 and SI Appendix, Fig. S9) suggests that the structures of these cofactors fit better in the active sites of the EuHMT1/2 variants, likely via the extra hydrophobic interaction between the extended sulfonium-δ-substituents of 3, 4, and 6 and the preexisting hydrophobic cavity (SI Appendix, Fig. S13A).
Structural Features of SAM Analogs Suitable for BPPM.
Although several previous efforts show that the existence of sulfonium-β-sp2 carbon is crucial for the SAM analogs to be active cofactors of PMT-mediated transalkylation (native PMTs or their variants) (25, 33), the current structural and biochemical studies shed light on how such unsaturated moiety can play a role in enzyme catalysis. Prior structural analysis on native PMTs suggests that the highly conserved Tyr residues in SET-domain-containing PMTs (e.g., EuHMT1/2’s Y1211/Y1154) facilitate the transmethylation reaction through the formation of nonclassical C–H…O hydrogen bonds with the transferred methyl moiety (SI Appendix, Fig. S13B) (34, 35). Disruption of this interaction with Ala mutation causes 300-fold decrease of the catalytic efficiency (Fig. 3 and SI Appendix, Fig. S9). Remarkably, although SAM and allyl-SAM showed similar affinity for Y1211A/Y1154A mutants as reflected by their comparable Km of 30–40 μM, kcat of allyl-SAM is at least 10-fold higher than that of SAM (Fig. 3 and SI Appendix, Fig. S9). Because neighboring unsaturated substituents can stabilize the transition state of a linear transalkylation SN2 reaction (33), the allyl moiety of 2 is expected to play a similar role in the enzyme catalysis (SI Appendix, Fig. S13 C and D). In a similar manner, the loss of stabilizing C–H…O interaction in the transition state is partially compensated by the transition-state participation of the sulfonium-β-sp2 allyl group of 2–7 (SI Appendix, Fig. S13 C and D).
In addition to the sulfonium-β-sp2 moiety, the δ-substituents of the SAM analogs proved to be a major determinant in enzymatic turnover (kcat). Remarkably, despite dramatic size differences in the δ-substituents of 2–7, these compounds show comparable affinity to Y1211A/Y1154A mutants as reflected by the less than fivefold fluctuation of their Km (8–40 μM and 12–80 μM for Y1211A/Y1154A mutants, respectively; Fig. 3 and SI Appendix, Fig. S9). This observation suggests that the preexisting hydrophobic cavity is spacious and flexible enough to accommodate the structurally diverse sulfonium-δ-substituents in 2–7.
In contrast to the small variation of Km of 1–7 against Y1211A/Y1154A mutants, the corresponding kcat values can alter by 100-fold with the fast turnover of 3.5–10 min−1 for 3, 4, and 6 versus the slow turnover of 0.08–1 min−1 for 1, 2, 5, and 7 (Fig. 3 and SI Appendix, Fig. S9). Strikingly, the trend of the kcat mimics that of the Km with the strong preference of the flexible medium-sized, sulfonium-δ-substituents as the most active SAM analogs. The combined effects make 3, 4, and 6 around 10- to 400-fold better than 1, 2, 5, and 7 toward Y1211A/Y1154A mutants (kcat/Km = 0.3–0.69 min−1⋅μM−1 versus 0.03–0.002 min−1⋅μM−1, Fig. 3 and SI Appendix, Fig. S9) and comparable to the native SAM-EuHMT1/2 pairs (kcat/Km = 0.64 min−1⋅μM−1). Here we reason that the optimal binding of 3, 4, and 6 at the active sites of Y1211A/Y1154A mutants may facilitate a productive transition state by aligning all of the reaction centers in a linear fashion (SI Appendix, Fig. S13C). In contrast, weaker binding of 2, 5, and 7 may result in a compromised transition state structure leading to inefficient enzyme catalysis (SI Appendix, Fig. S13D). In the context of Y1211A/Y1154A mutants, we therefore defined two key structural features for m-tag SAM analogs: (i) sulfonium-β-sp2 moiety for transition-state stabilization and (ii) suitable δ-substituents for optimal cofactor binding (SI Appendix, Fig. S13 C and D).
Generality of Substrate Labeling with SAM Analogs and Structurally Matched PMT Variants.
Structure-based sequence alignment of multiple SET-domain-containing PMTs reveals that EuHMT1/2’s Y1211/Y1154, the gatekeeper residues, are highly conserved among the family of PMTs (SI Appendix, Figs. S5 and S14). More importantly, the preexisting hydrophobic cavity, which is constructed by EuHMT1’s I1168/N1169/H1170/F1209/F1215/W1216 residues, is well conserved in SET-domain-containing PMTs (SI Appendix, Figs. S5 and S14). We envision that multiple SET-domain-containing PMTs can be tailored to process the SAM analogs as cofactors through mutating the gatekeeper Tyr alone or in combination with the residues of the nearby preexisting hydrophobic pockets. With the structurally diverse SAM analogs (e.g., 2–7) as cofactor candidates, the matched mutant-cofactor pairs can be readily identified for BPPM.
Nonhistone Substrates Revealed by BPPM Technology.
Several prior methods, which relied on peptide/protein arrays or PMT-knockout proteome as substrate candidates, allowed identification of some nuclear proteins as nonhistone targets of EuHMT1/2 (15–19). In contrast, a single set of experiments with our BPPM approach revealed hundreds of EuHMT1/2 targets. Given that the C-terminal catalytic domains of EuHMT1/2, although active (15, 28, 36), show less optimal activity (13), we reasoned that one merit of the BPPM approach lies in its ability to implement full-length enzymes to profile their activities. In addition, EuHMT1/2 was shown to function via multimeric complexes with SUV39H1 and SETDB1 (8). Overexpressing or down-regulating a single component (e.g., SUV39H1 or EuHMT1/2) may alter the stability and thus the activity of multimeric complexes (8). These context-dependent challenges in profiling activities of PMTs can be well addressed by the BPPM approach with engineered full-length PMT variants and matched SAM analogs. The BPPM-derived proteomic data indicate that EuHMT1/2’s targets are not restricted to the nuclear proteins, as reported previously (15–19), but indeed cover a wide range of nuclear, cytosolic, and membrane-bound proteins (SI Appendix, Fig. S12A). Functional annotation of these targets suggests that many canonical pathways may be regulated via EuHMT1/2-mediated methylation (SI Appendix, Fig. S12B). This work thus serves as a starting point for validating the newly revealed targets with accurate biological models and defining the downstream functions of these methylation events.
Materials and Methods
New compounds were fully characterized according to standard practices. Compound synthesis and characterization, biochemical assays, crystallization, structure determination, and proteomic analysis are provided in SI Appendix. The atomic coordinates and structure factors of EuHMT1’s Y1211A mutant in complex with SAH and allylated H3K9 peptide have been deposited in the Protein Data Bank (PDB code 4H4H).
Supplementary Material
Acknowledgments
The authors thank Drs. J. Huang, M. Walsh, and H. Lin for EuHMT1/2 plasmids, POLR2A CTD, and PARP1 proteins; Dr. Dhirendra K. Simanshu for help in preparing structure figures; and Drs. G. Sukenick and H. Liu for NMR and mass analyses. This work was supported by National Institute of General Medical Sciences Grant 1R01GM096056 (to M.L.) and National Institutes of Health Director’s New Innovator Award Program Grant 1DP2-OD007335 (to M.L.), the V Foundation for Cancer Research (M.L.), March of Dimes Foundation (M.L.), Starr Cancer Consortium (M.L. and H.D.), the Alfred W. Bressler Scholars Endowment Fund (M.L.), and the Structural Genomics Consortium (J.M.), which is a registered charity (1097737) that receives funds from AbbVie, Boehringer Ingelheim, the Canada Foundation for Innovation, the Canadian Institutes for Health Research, Genome Canada through the Ontario Genomics Institute (OGI-055), GlaxoSmithKline, Janssen, Lilly Canada, the Novartis Research Foundation, the Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and the Wellcome Trust (092809/Z/10/Z). Use of the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, under Contract DE-AC02-06CH11357.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: Crystallography, atomic coordinates, and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 4H4H).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1216365110/-/DCSupplemental.
References
- 1.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6(11):838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 2.Zhang X, Wen H, Shi X. Lysine methylation: Beyond histones. Acta Biochim Biophys Sin (Shanghai) 2012;44(1):14–27. doi: 10.1093/abbs/gmr100. [DOI] [PubMed] [Google Scholar]
- 3.Erce MA, Pang CN, Hart-Smith G, Wilkins MR. The methylproteome and the intracellular methylation network. Proteomics. 2012;12(4-5):564–586. doi: 10.1002/pmic.201100397. [DOI] [PubMed] [Google Scholar]
- 4.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14(11):1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis. Nat Struct Mol Biol. 2007;14(11):1008–1016. doi: 10.1038/nsmb1337. [DOI] [PubMed] [Google Scholar]
- 6.Fontecave M, Atta M, Mulliez E. S-adenosylmethionine: Nothing goes to waste. Trends Biochem Sci. 2004;29(5):243–249. doi: 10.1016/j.tibs.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 7.Dillon SC, Zhang X, Trievel RC, Cheng X. The SET-domain protein superfamily: Protein lysine methyltransferases. Genome Biol. 2005;6(8):227. doi: 10.1186/gb-2005-6-8-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fritsch L, et al. A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol Cell. 2010;37(1):46–56. doi: 10.1016/j.molcel.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 9.Rathert P, Dhayalan A, Ma HM, Jeltsch A. Specificity of protein lysine methyltransferases and methods for detection of lysine methylation of non-histone proteins. Mol Biosyst. 2008;4(12):1186–1190. doi: 10.1039/b811673c. [DOI] [PubMed] [Google Scholar]
- 10.Luo M. Current chemical biology approaches to interrogate protein methyltransferases. ACS Chem Biol. 2012;7(3):443–463. doi: 10.1021/cb200519y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demko ZP, Sharpless KB. A click chemistry approach to tetrazoles by Huisgen 1,3-dipolar cycloaddition: Synthesis of 5-sulfonyl tetrazoles from azides and sulfonyl cyanides. Angew Chem Int Ed Engl. 2002;41(12):2110–2113. [PubMed] [Google Scholar]
- 12.Tornøe CW, Christensen C, Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J Org Chem. 2002;67(9):3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 13.Islam K, et al. Bioorthogonal profiling of protein methylation using azido derivative of S-adenosyl-L-methionine. J Am Chem Soc. 2012;134(13):5909–5915. doi: 10.1021/ja2118333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bishop AC, Buzko O, Shokat KM. Magic bullets for protein kinases. Trends Cell Biol. 2001;11(4):167–172. doi: 10.1016/s0962-8924(01)01928-6. [DOI] [PubMed] [Google Scholar]
- 15.Rathert P, et al. Protein lysine methyltransferase G9a acts on non-histone targets. Nat Chem Biol. 2008;4(6):344–346. doi: 10.1038/nchembio.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang J, et al. G9a and Glp methylate lysine 373 in the tumor suppressor p53. J Biol Chem. 2010;285(13):9636–9641. doi: 10.1074/jbc.M109.062588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JS, et al. Negative regulation of hypoxic responses via induced Reptin methylation. Mol Cell. 2010;39(1):71–85. doi: 10.1016/j.molcel.2010.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pless O, et al. G9a-mediated lysine methylation alters the function of CCAAT/enhancer-binding protein-beta. J Biol Chem. 2008;283(39):26357–26363. doi: 10.1074/jbc.M802132200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ling BM, et al. Lysine methyltransferase G9a methylates the transcription factor MyoD and regulates skeletal muscle differentiation. Proc Natl Acad Sci USA. 2012;109(3):841–846. doi: 10.1073/pnas.1111628109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang Y, et al. MPP8 mediates the interactions between DNA methyltransferase Dnmt3a and H3K9 methyltransferase GLP/G9a. Nat Commun. 2011;2:533. doi: 10.1038/ncomms1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chin HG, et al. Automethylation of G9a and its implication in wider substrate specificity and HP1 binding. Nucleic Acids Res. 2007;35(21):7313–7323. doi: 10.1093/nar/gkm726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev. 2011;25(8):781–788. doi: 10.1101/gad.2027411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Binda O, et al. A chemical method for labeling lysine methyltransferase substrates. ChemBioChem. 2011;12(2):330–334. doi: 10.1002/cbic.201000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peters W, et al. Enzymatic site-specific functionalization of protein methyltransferase substrates with alkynes for click labeling. Angew Chem Int Ed Engl. 2010;49(30):5170–5173. doi: 10.1002/anie.201001240. [DOI] [PubMed] [Google Scholar]
- 25.Islam K, Zheng W, Yu H, Deng H, Luo M. Expanding cofactor repertoire of protein lysine methyltransferase for substrate labeling. ACS Chem Biol. 2011;6(7):679–684. doi: 10.1021/cb2000567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang R, Zheng W, Yu H, Deng H, Luo M. Labeling substrates of protein arginine methyltransferase with engineered enzymes and matched S-adenosyl-L-methionine analogues. J Am Chem Soc. 2011;133(20):7648–7651. doi: 10.1021/ja2006719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chakraborty D, Islam K, Luo M. Facile synthesis and altered ionization efficiency of diverse Nε-alkyllysine-containing peptides. Chem Commun (Camb) 2012;48(10):1514–1516. doi: 10.1039/c1cc14711k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu H, et al. Structural biology of human H3K9 methyltransferases. PLoS ONE. 2010;5(1):e8570. doi: 10.1371/journal.pone.0008570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng XD, Collins RE, Zhang X. Structural and sequence motifs of protein (histone) methylation enzymes. Annu Rev Biophys Biomol Struct. 2005;34:267–294. doi: 10.1146/annurev.biophys.34.040204.144452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kouzarides T. SnapShot: Histone-modifying enzymes. Cell. 2007;128(4):802–803. doi: 10.1016/j.cell.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 31.Charron G, et al. Robust fluorescent detection of protein fatty-acylation with chemical reporters. J Am Chem Soc. 2009;131(13):4967–4975. doi: 10.1021/ja810122f. [DOI] [PubMed] [Google Scholar]
- 32.Yang YY, Grammel M, Raghavan AS, Charron G, Hang HC. Comparative analysis of cleavable azobenzene-based affinity tags for bioorthogonal chemical proteomics. Chem Biol. 2010;17(11):1212–1222. doi: 10.1016/j.chembiol.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dalhoff C, Lukinavicius G, Klimasăuskas S, Weinhold E. Direct transfer of extended groups from synthetic cofactors by DNA methyltransferases. Nat Chem Biol. 2006;2(1):31–32. doi: 10.1038/nchembio754. [DOI] [PubMed] [Google Scholar]
- 34.Horowitz S, Yesselman JD, Al-Hashimi HM, Trievel RC. Direct evidence for methyl group coordination by carbon-oxygen hydrogen bonds in the lysine methyltransferase SET7/9. J Biol Chem. 2011;286(21):18658–18663. doi: 10.1074/jbc.M111.232876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Couture JF, Hauk G, Thompson MJ, Blackburn GM, Trievel RC. Catalytic roles for carbon-oxygen hydrogen bonding in SET domain lysine methyltransferases. J Biol Chem. 2006;281(28):19280–19287. doi: 10.1074/jbc.M602257200. [DOI] [PubMed] [Google Scholar]
- 36.Estève PO, et al. Functional analysis of the N- and C-terminus of mammalian G9a histone H3 methyltransferase. Nucleic Acids Res. 2005;33(10):3211–3223. doi: 10.1093/nar/gki635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.