

Significance
Myofibroblasts are contractile smooth muscle-like cells that control tissue repair and remodeling. Inappropriate myofibroblast differentiation can cause organ fibrosis or inefficient wound healing. In this article, we demonstrate that myocardin-related transcription factor (MRTF)-A is necessary for myofibroblast differentiation. We also identify an isoxazole ring-containing small molecule that stimulates MRTF-A–dependent gene expression, myofibroblast differentiation, and wound healing. These findings suggest that targeting MRTFs pharmacologically may prove useful in treating fibrotic diseases.
Keywords: fibrosis, dermal injury, MAP kinase signaling, granulation tissue
Abstract
Myocardin-related transcription factors (MRTFs) regulate cellular contractility and motility by associating with serum response factor (SRF) and activating genes involved in cytoskeletal dynamics. We reported previously that MRTF-A contributes to pathological cardiac remodeling by promoting differentiation of fibroblasts to myofibroblasts following myocardial infarction. Here, we show that forced expression of MRTF-A in dermal fibroblasts stimulates contraction of a collagen matrix, whereas contractility of MRTF-A null fibroblasts is impaired under basal conditions and in response to TGF–β1 stimulation. We also identify an isoxazole ring-containing small molecule, previously shown to induce smooth muscle α-actin gene expression in cardiac progenitor cells, as an agonist of myofibroblast differentiation. Isoxazole stimulates myofibroblast differentiation via induction of MRTF-A–dependent gene expression. The MRTF-SRF signaling axis is activated in response to skin injury, and treatment of dermal wounds with isoxazole accelerates wound closure and suppresses the inflammatory response. These results reveal an important role for MRTF-SRF signaling in dermal myofibroblast differentiation and wound healing and suggest that targeting MRTFs pharmacologically may prove useful in treating diseases associated with inappropriate myofibroblast activity.
Fibroblasts proliferate in response to biomechanical tension or local stress signals, ultimately differentiating into smooth muscle cell (SMC)-like, contractile myofibroblasts (1–4). Myofibroblast differentiation represents a normal step in the healing process and is essential for the repair of a wide range of traumas including cutaneous injury and myocardial infarction (4–6). Defective myofibroblast differentiation can result in abnormal or inefficient wound healing whereas persistent myofibroblast activation is associated with pathological fibrosis and scarring (7). Strategies for modulating myofibroblast number and phenotype may have therapeutic applications for the acute treatment of wounds or fibrosis-associated pathologies; however, the molecular mechanisms that regulate these processes are not well understood.
The SMC phenotype is at least partially dependent upon the activity of serum response factor (SRF), which promotes cell growth or differentiation, depending upon association with distinct transcriptional coactivators (8). Interaction with myocardin leads to the activation of genes encoding SMC contractile proteins and the differentiated phenotype (9–14). Mitogen-activated protein kinase signaling displaces myocardin from SRF by the ternary complex factor family of Ets domain proteins and activates the immediate early gene response and cell proliferation (15).
Although myocardin is specifically expressed in cardiomyocytes and SMCs and is constitutively localized to the nucleus, myocardin-related transcription factor-A (MRTF-A, MKL1, MAL, BSAC) and -B (MRTF-B, MKL2) are broadly expressed and sequestered in the cytoplasm via interactions with G-actin (8, 11, 12, 16–18). Situations of stress that locally alter actin dynamics and promote F-actin polymerization promote nuclear accumulation of MRTFs (18–24). SRF/MRTF complexes bind to consensus CArG elements [CC(A/T)6GG] within the promoters of contractile and SMC-specific target genes, most notably smooth muscle α-actin (SMA, ACTA2) and transgelin (TAGLN, SM22) (25). MRTFs are thus critical regulators of the smooth muscle contractile phenotype in response to stress.
Previously, we found that MRTF-A promotes differentiation of cardiac fibroblasts to a myofibroblast phenotype and production of stress fibers (26). Deletion of MRTF-A in mice attenuates myofibroblast responses following cardiac injury and reduces cardiac fibrosis and scarring (26). In this study, we demonstrate that MRTF-A stimulates fibroblast contractility, whereas MRTF-A–null fibroblasts fail to induce the SMC contractile program in response to stress signals. An isoxazole (ISX) ring-containing small molecule previously shown to induce SMA expression in cardiac progenitor cells (27) activates fibroblast differentiation via the induction of MRTF-A activity. Our findings implicate the MRTF-SRF gene regulatory axis as a key mediator of myofibroblast differentiation and wound healing and suggest that therapeutic manipulation of this pathway may provide benefit in diseases associated with inappropriate myofibroblast differentiation.
Results
MRTF-A Contributes to Myofibroblast Differentiation and Contractility.
MRTF-A is broadly expressed and promotes the expression of genes encoding components of the actin cytoskeleton. Using cultured human dermal fibroblasts (BR5) suspended in a collagen solution, we sought to probe the function of MRTF-A in fibroblast contractility and myofibroblast differentiation. The fibroblast phenotype varies depending upon whether cells are grown in a collagen matrix that is floating or attached. Contraction of floating matrices results in mechanically relaxed tissue with cells that display the morphological and proliferative features of dermis (28). Contraction can be stimulated by addition of growth factors such as FBS or PDGF (28). Overexpression of MRTF-A by adenoviral-delivery stimulated matrix contraction of BR5 fibroblasts compared with uninfected cells or cells infected with GFP-expressing adenovirus as a control (Fig. 1 A and C). The extent of contraction in response to MRTF-A expression was approximately equivalent to that observed upon stimulation with FBS or PDGF (Fig. 1 A and C).
Fig. 1.

MRTF-A promotes fibroblast contractility. BR5 human dermal fibroblasts were infected with 25 MOI adenovirus expressing flag-MRTF-A or GFP control and cultured in a collagen matrix overnight. Floating (A) or stressed (B) collagen gels were imaged after stimulation with growth factor (FBS or PDGF) or BSA control for 1 h. (C and D) Quantification of gel diameter (in A and B) following indicated treatments. Data represent three independent experiments. **P < 0.01; ***P < 0.001; ****P < 0.0001.
Stressed matrix contraction reflects active shortening by cells that are spread, exhibit stress fibers, and are well-adhered to the restrained matrix, in a process that can be stimulated by FBS, but not by PDGF. Adhered collagen matrices develop into a stressed tissue resembling that of granulation tissue. Cell shortening was potently stimulated by forced expression of MRTF-A under all conditions tested (Fig. 1 B and D). These data suggest that MRTF-A stimulates the myofibroblast phenotype, enhancing constitutive matrix remodeling and cell shortening.
We next asked whether MRTF-A is required for the process of myofibroblast differentiation, using tail tip fibroblasts (TTFs) isolated from MRTF-A knockout (KO) mice and WT littermates. When grown in culture, fibroblasts respond to TGF-β1 stimulation by adopting an elongated morphology and producing SMA-positive stress fibers (Fig. 2A). MRTF-A deficient fibroblasts did not display an enrichment of SMA-positive stress fibers as observed in WT fibroblasts treated with TGF-β1 (Fig. 2A). Furthermore, the expression of genes encoding SMC contractile proteins (SMA and SM22) in response to TGF-β1 stimulation was blunted in MRTF-A–deficient TTFs as demonstrated by quantitative RT-PCR (qRT-PCR) and Western blot (Fig. 2 B–D). Because MRTF-A KO fibroblasts continue to express MRTF-B, it is likely that the residual expression of SMA in KO cells reflects MRTF-B redundancy or MRTF independent pathways.
Fig. 2.

MRTF-A is required for TGF-β1–induced contractile gene expression and stress fiber formation. Tail-tip dermal fibroblasts isolated from MRTF-A KO mice and WT littermates were serum-starved before treatment with TGF-β1 or vehicle for 48 h. (A) SMA immunostaining visualized by confocal microscopy at 40× magnification. (Scale bar, 50 μm.) (B) qRT-PCR and (C) Western blot analysis of SMA and SM22 expression, as indicated, normalized to GAPDH. (D) Quantification of Western blot data. ***P < 0.001; ****P < 0.0001.
Isoxazole Promotes MRTF-A–Dependent SMA Reporter Activation.
To examine the potential to pharmacologically manipulate myofibroblast differentiation, we sought to identify molecules capable of modulating MRTF-A–dependent SMA expression as a surrogate marker of the contractile phenotype. We used a luciferase reporter controlled by ∼125 nucleotides of the SMA promoter (SMA-luc) in NIH/3T3 fibroblasts, which provided a faithful readout of CArG-dependent MRTF-A activity (Fig. 3A). Using this approach, we screened ∼300 compounds previously shown to promote cardiac and neural progenitor cell differentiation in vitro (27, 29). N-cyclopropyl-5-(thiophen-2-yl)-isoxazole-3-carboxamide (ISX) displayed a dose-dependent stimulation of SMA-luc upon forced expression of a minimal amount of MRTF-A (Fig. 3B). Isoxazole consistently stimulated reporter activity ∼10–50 fold (at 20 μM), compared with MRTF-A alone, and enhanced basal SMA-luc expression to an extent similar to or greater than that of TGF-β1 (Fig. 3C).
Fig. 3.

ISX stimulates MRTF-A–dependent promoter activity. NIH/3T3 mouse fibroblasts were cotransfected with (A) 0–100 ng of MRTF-A or empty vector and 100 ng of SMA promoter-controlled luciferase construct to confirm promoter activation. (B) To determine the effect of ISX on MRTF-dependent SMC reporter activation, cells were cotransfected with a low dose of MRTF-A (10 ng) and SMA-luc (100 ng) and then serum-starved for 24 h before treatment with 5 or 20 μM ISX. (C) The effect of TGF-β1 (10 ng/mL) or ISX (20 µM) treatment on basal SMA-luc activity in the absence of exogenous MRTF-A. (D) ISX (20 μM) enhances the activation of multiple CArG-containing reporters [SMA, SM22, skeletal muscle actin (SKA), or atrial natriuretic factor (ANF)] by an intermediate dose of MRTF-A (25 ng). Mutations in the indicated CArG boxes in the ANF (E) or SMA (F) promoters ablate responsiveness to ISX. Data are normalized to β-gal internal control and expressed relative to empty vector (A, B, D, E, F) or vehicle (C). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To directly analyze the influence of isoxazole on MRTF-A activity and myofibroblast differentiation, we examined various CArG-dependent reporter constructs. In addition to SMA-luc, isoxazole stimulated MRTF-A–dependent luciferase reporters controlled by the SM22, skeletal α-actin, and atrial natriuretic factor (ANF) promoters (Fig. 3D) in the presence of an intermediate dose of exogenous MRTF-A. Point mutations in the CArG elements of the ANF and SMA promoters abrogated their responsiveness to isoxazole (Fig. 3 E and F). These data suggest the induction of smooth muscle contractile gene expression by isoxazole occurs via a CArG– and SRF/MRTF-A–dependent mechanism.
Isoxazole Induces Endogenous MRTF-A Targets and the Myofibroblast Phenotype.
Because isoxazole displayed such robust activity in our reporter assay, we examined its effect on endogenous gene expression. Indeed, isoxazole treatment of BR5 fibroblasts induced morphological changes and the generation of SMA-positive stress fibers to a similar extent to that of TGF-β1 (Fig. 4A). Likewise, isoxazole resulted in a significant enrichment of SMA and SM22 mRNA levels as demonstrated by qRT-PCR (Fig. 4B), and in an increase in SMA protein (Fig. 4C).
Fig. 4.

ISX induces endogenous MRTF targets. BR5 dermal fibroblasts infected with 10 MOI adenovirus expressing flag-MRTF-A were serum-starved and then treated with ISX (20 μM), TGF-β1, or vehicle for 48 h, as indicated. (A) SMA-positive stress fibers were visualized by confocal microscopy at 40× magnification. (Scale bar, 50 μm.) (B) Expression of SMA and SM22 was analyzed by qRT-PCR. Data are normalized to GAPDH. (C) Levels of SMA protein were analyzed by Western blot. (D) Levels of SMA were examined by Western blot in MRTF-A WT and KO tail-tip dermal fibroblasts treated with vehicle or ISX. (E) Quantification of SMA protein levels normalized to GAPDH. *P < 0.05. **P < 0.01; ****P < 0.0001.
To determine whether MRTF-A was required for isoxazole-dependent SMA enrichment, we isolated TTFs from MRTF-A KO and WT littermates. Isoxazole treatment stimulated the expression of SMA protein in WT but not in MRTF-A KO fibroblasts (Fig. 4 D and E). These observations reveal a physiological response to MRTF-A and isoxazole in stimulation of the SMC contractile program and dermal myofibroblast differentiation.
Isoxazole Regulates MRTF-A/SRF Stability and Activity.
To further probe the mechanism whereby isoxazole promotes MRTF-A target gene expression, we examined the possible effects of this compound on MRTF-A protein function. Because MRTF-A activity is regulated at the level of nuclear localization, we first used an epitope-tagged MRTF-A construct to examine subcellular localization in response to isoxazole treatment. We observed a striking accumulation of MRTF-A protein in the presence of isoxazole, which was accompanied by an increase in the number of cells displaying nuclear MRTF-A (Fig. 5 A and B and Fig. S1). Nuclear accumulation under these conditions appeared to stem from increased levels of MRTF-A protein, as we did not observe an increased number of cells displaying MRTF-A exclusively in the nucleus (Fig. 5B). Western analysis revealed the enrichment of both MRTF-A (Fig. 5C) and SRF (Fig. 5D) in isoxazole treated cells, and transcript levels were not altered (Fig. S2). In contrast, isoxazole did not increase the level of the pan-fibroblast marker fibroblast-specific protein-1 (Fsp1, S100a4) and vimentin (Fig. 5D), suggesting a relatively specific effect on SRF/MRTF protein.
Fig. 5.

ISX promotes MRTF-A enrichment and nuclear accumulation. NIH/3T3 fibroblasts were infected with 25 MOI adenovirus expressing HA-tagged MRTF-A and treated with ISX (20 μM) or vehicle. (A) ISX promotes the enrichment and nuclear accumulation of MRTF-A (green) as visualized by HA immunostaining and confocal microscopy at 60× magnification. SMA (red) is enriched in stress fibers of MRTF-A–expressing cells. (Scale bar, 50 μm.) (B) Quantification of cells with HA–MRTF-A in nucleus only, cytoplasm only, or in both. Data represent three independent experiments. (C) Forced expression of flag-MRTF-A (10 MOI) demonstrates enrichment in ISX-treated cells. (D) SRF protein levels are enriched with ISX treatment, whereas pan-fibroblast markers (Fsp1 and vimentin) are not. (E) ISX treatment leads to phosphorylation of Erk1/2, but not Smad2/3, as demonstrated by Western blot. ****P < 0.0001.
We next examined the potential influence of isoxazole on signal transduction pathways that affect the fibroblast phenotype and might impinge upon SRF/MRTF activity. Isoxazole treatment did not affect the phosphorylation of Smad2/3 (Fig. 5E), further suggesting that isoxazole does not promote canonical TGF-β–Smad signaling. Isoxazole treatment did, however, lead to the rapid and transient phosphorylation of extracellular-signal–regulated kinases Erk1/2 (Fig. 5E).
Isoxazole Promotes Wound Healing in Vivo.
Given the role of myofibroblasts in wound healing and the potent stimulation of MRTF-A activity and myofibroblast gene expression by isoxazole, we examined whether isoxazole could promote wound healing in vivo. To validate the potential involvement of the MRTF-SRF axis in healing wounds, we evaluated MRTF-A and SMC contractile gene expression in biopsies from WT mice 7 d postinjury. Dermal wounds displayed enrichment of the myofibroblast marker SMA and various collagens (COL1A1, COL1A2, and COL3A1) as well as MRTF-A compared with dermis from uninjured controls (Fig. 6A).
Fig. 6.

ISX promotes wound closure in vivo. WT mice were subjected to full-thickness cutaneous punch biopsies (n = 4). (A) Expression of MRTF-A and myofibroblast markers was analyzed 7 d postinjury by qRT-PCR. Data are normalized to GAPDH. (B) H&E (a, d) and Masson’s trichrome staining (b, c, e, f) of ISX- or vehicle-treated wounds (7 d postinjury) depicting the migrating epidermal sheets (black arrow) and granulation tissue area [4× (a, b, d, e) and 40× (c, f) magnification]. [Scale bar: 500 μm (4×) or 50 μm (40×).] (C) Quantification of distance between epithelial sheets or granulation tissue layers in the healing wound. n = 4 (day 7) or 8 (day 5). (D) Immunofluorescent detection of SMA in ISX- or vehicle-treated cutaneous wounds. Dashed line denotes border of wound, and boxed area identifies area of granulation tissue shown in b and d. [Scale bar, 200 μm (a, c) and 50 μm (b, d).] (E) Quantification of the depth of SMA-positive granulation tissue. E, epidermis; D, dermis; GT, granulation tissue. *P < 0.05; **P < 0.01.
To determine if isoxazole could promote wound healing, WT mice were subjected to two full-thickness cutaneous wounds; one wound was treated with isoxazole (20 μM) twice per day for 7 d, and the second wound was treated with vehicle, serving as an internal control to limit intermouse variability. Isoxazole-treated wounds were smaller (Fig. S3) and displayed a blunted immune response as measured by the reduction of the proinflammatory cytokine TNF-α (Fig. S4). Histological analysis 7 d after injury revealed a much more compact wound with expanded and more cellularized granulation tissue upon isoxazole treatment, which was associated with a significant narrowing between the epithelial sheets (Fig. 6B). Quantification of the wound diameter demonstrated accelerated closure of the granulation tissue layer by 5 d after injury and a significantly smaller wound by 7 d (Fig. 6C). Furthermore, immunofluorescent detection of SMA revealed qualitatively more staining in the granulation tissue of isoxazole-treated wounds (Fig. 6D), and the depth of SMA positive granulation tissue was significantly greater than that of control wounds (Fig. 6E), although we did not observe alterations in cell proliferation 7 d postinjury (Fig. S5). Taken together, these findings reveal that isoxazole promotes MRTF/SRF–dependent myofibroblast differentiation in vitro and leads to the improvement of cutaneous wound healing.
Discussion
Although it is well-accepted that resident fibroblasts contribute to the healing process in nearly every tissue examined, the molecular mechanisms that govern the phenotypic transformation of a quiescent fibroblast into a contractile myofibroblast and the precise role of this cell population in wound repair are poorly understood (30). In the current study, we demonstrate that MRTF-A is a potent agonist of the myofibroblast phenotype. MRTF-A activates the smooth muscle contractile gene program, promotes SMA-positive stress fiber formation, and enhances fibroblast contractility in a collagen matrix. Furthermore, fibroblasts from MRTF-A KO mice display defective myofibroblast differentiation, demonstrating the involvement of MRTF-A in this process and confirming results of a previous study using a siRNA knockdown approach (31).
Using a SMA-luciferase reporter, we devised a strategy to screen for molecules that influence myofibroblast activation. We identified an isoxazole-ring containing analog, which stimulated the SMA promoter and induced the myofibroblast phenotype in an MRTF-A–dependent manner (Fig. 7). This method could potentially also identify MRTF-A–independent pathways, because the luciferase reporter construct used in this assay contains multiple regulatory elements in addition to the two CArG boxes, including a TGF-β/Smad response element and AP2 site (32–34). Based on our finding that isoxazole does not affect Smad2/3 phosphorylation, and mutation of CArG boxes abolishes responsiveness to isoxazole, the phenotypic change elicited by isoxazole likely occurs via the promotion of MRTF/SRF activity. Importantly, a recent study using a similar SMA-luc–based screen identified the TRPC6 Ca2+ channel as a key agonist of TGF-β1/SRF–dependent myofibroblast differentiation and wound healing (35). Isoxazole has been shown to activate the G-protein–coupled receptor GPR68, leading to Ca2+ signaling (36), which could potentially couple to this pathway. Together, these studies support the conclusion that the MRTF-SRF regulatory axis plays a central role in myofibroblast differentiation.
Fig. 7.
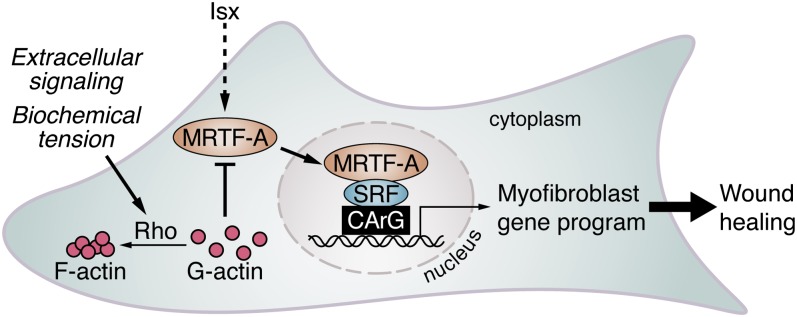
ISX stimulates MRTF-mediated myofibroblast differentiation. Simplified view of the actions of MRTF-A and ISX during fibroblast differentiation. Mechanical stress or extracellular signals that stimulate Rho-ROCK–dependent F-actin polymerization lead to MRTF-A nuclear translocation and activation of SRF target genes. ISX treatment leads to the accumulation of MRTF-A protein and stimulation of SRF target genes that promote the myofibroblast phenotype.
Interestingly, isoxazole induced the myofibroblast phenotype at least in part via stimulation of MRTF-A activity, likely reflecting the enrichment of MRTF-A protein. Levels of MRTF-A were significantly higher in isoxazole-treated fibroblasts, which led to a greater proportion of cells displaying nuclear accumulation of MRTF-A. A previous study reported regulation of MRTF-A protein stability by the proteasome degradation pathway, suggesting the possibility that isoxazole might inhibit the ubiquitin-proteasome system (19).
We also observed stimulation of Erk1/2 phosphorylation by isoxazole, implying the potential regulation of both the SRF-dependent growth response and the contractile gene program (37). Indeed, phosphorylation of MRTF-A by Erk1/2 inhibits its nuclear localization (38), potentially explaining our observation that the majority of the stabilized MRTF-A remains in the cytoplasm.
Recently, isoxazole analogs have been used to stimulate differentiation of adult cardiac progenitors and neural stem cells (27, 29) and have been shown to enhance insulin production by pancreatic β-cells (39). It is not yet clear whether a common mechanism underlies these findings, although each response is associated with increased activity of key transcription factors. Isoxazole enhanced the phosphorylation and nuclear export of histone deacetylase 5 (HDAC5) in neural stem cells, thereby promoting transcriptional activation of myocyte enhancer factor-2 target genes (29) and also stimulated p300 and cAMP response element binding protein acetyl-transferase activity in pancreatic β-cells (39). Of note, myocardin family proteins bind to and are stimulated by p300 and are repressed by HDAC activity (40).
A mouse model of cutaneous wound healing was used in this study as a means of testing the therapeutic efficacy of promoting myofibroblast differentiation with isoxazole in vivo. Isoxazole treatment increased the accumulation of granulation tissue and stimulated wound closure after 7 d. If the beneficial effect of isoxazole was primarily due to MRTF-A activation, MRTF-A KO mice would be expected to display deficient wound closure. Surprisingly, however, MRTF-A deletion did not significantly impact wound healing over the course of 11 d (Fig. S6). We postulate that MRTF-B, which is expressed in the wounds of MRTF-A–null mice (Fig. S7), plays a redundant role to that of MRTF-A either in fibroblasts or in keratinocytes. Indeed, SRF-deficient keratinocytes display reduced cell–cell and cell–matrix contacts and loss of epidermal homeostasis (41). Isoxazole may also act independently of MRTF-A in the setting of a healing wound such that stimulation of Erk1/2 signaling by isoxazole contributes to wound closure by promoting the growth response. Alternatively, suppression of the inflammatory response, as demonstrated by reduced TNF-α expression, might contribute to the accumulation of granulation tissue in the isoxazole-treated wound (42, 43). To fully elucidate the role of MRTFs in wound healing, deletion of MRTF-A and -B with both fibroblast- and keratinocyte-specific Cre drivers will likely be necessary.
Fibroblast activation is invariably associated with the healing process, contributing to injury repair in settings as diverse as cutaneous wound healing, heart disease, renal obstruction, pulmonary and liver fibrosis, and scleroderma (2, 4). Therefore, insight gained using one model of tissue remodeling is likely to be applicable to a wide range of pathologies that are associated with inappropriate myofibroblast differentiation. One example of excessive fibroblast differentiation is keloid formation, a type of benign, but sometimes debilitating, fibrotic tumor that expands following injury. Both keloid formation and the MRTF-SRF axis are promoted by mechanical tension and focal adhesion kinase (44), potentially linking MRTFs to abnormal scar formation. The current finding of an MRTF-regulated pathway in dermal myofibroblast differentiation should accelerate the development of therapeutic strategies directed towards various fibrotic diseases including keloid formation.
Materials and Methods
Cell Culture and Transfection.
NIH/3T3 mouse embryonic fibroblasts, BR5 human foreskin fibroblasts, and primary mouse TTFs isolated from 6- to 8-wk-old MRTF-A KO or WT littermates were cultured as described in the SI Materials and Methods. Reporter and expression constructs were introduced using Fugene6 (Roche) transfection or adenoviral transduction. For treatments, cells were serum-starved for 24 h, followed by incubation in serum-free media containing TGF-β1 (10 ng/mL, R&D Systems) or isoxazole (20 μM) for an additional 24–48 h.
RNA Isolation and Analysis.
Total RNA was isolated from cell cultures or tissues using TRIzol reagent (Invitrogen). cDNA was generated using iScript cDNA Synthesis Kit (BioRad) and analyzed by standard RT-PCR or qRT-PCR with iQ SYBR Green (BioRad). Primer sequences and PCR conditions are listed in Table S1.
Collagen Gel Contraction Assay.
BR5 cells (2 × 105) were infected with adenovirus encoding flag-MRTF-A or GFP as control at a multiplicity of infection (MOI) of 25 and seeded in 1.5 mg/mL rat tail collagen (BD Bioscience), as described in SI Materials and Methods. Upon release from the tissue culture plastic, the reduction of gel diameter in response to MRTF-A expression, BSA control (5 mg/mL), FBS [10% (vol/vol)], or PDGF (50 ng/mL) was calculated as a measurement of cell contractility.
Immunocytochemistry.
Indirect immunofluorescence was performed using a mouse monoclonal Cy3-conjugated anti-SMA antibody (Sigma, clone 1A4, 1:200), or a rabbit monoclonal anti-HA antibody (Cell Signaling, 1:300) as described in SI Materials and Methods. All images were captured using an Olympus IX81 confocal microscope.
Western Blot Analysis.
Whole-cell extracts were subjected to SDS/PAGE and immunoblotted onto PVDF membranes (Millipore) with the antibodies listed in Table S2 at 4 °C overnight. HRP-conjugated secondary antibody (Bio-Rad) was incubated for 1 h at room temperature and developed with luminol reagent (Santa Cruz).
Mouse Lines and Wound-Healing Assay.
All experiments using animals were previously approved by the University Committee on Animal Resources at the University of Rochester and the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center. The MRTF-A−/− mouse line used in this study and the wound-healing assay are described in SI Materials and Methods. Briefly, two full-thickness biopsies (4 mm) were created on the dorsal surface of each 6- to 8-wk-old mouse. ISX (20 μM in PBS) was applied to one wound, and an equal concentration of DMSO was applied to the second internal control wound twice per day in an ∼50-μL volume for 7 d.
Histology.
All samples were fixed in 4% paraformaldehyde and processed for 5-μm sectioning and staining as described in the SI Materials and Methods. Sections were incubated with the following antibodies at 4 °C overnight: anti-SMA-Cy3 antibody (Sigma, 1:200) and Ki-67 (DAKO, clone TEC-3, 1:500). Immunofluorescent images were taken on an Olympus IX81 confocal microscope and wound dimensions were analyzed with National Institutes of Health ImageJ.
Data and Statistical Analysis.
Results are presented as the mean ± SEM. Statistical differences were determined by a Student’s two-tailed t test with unequal variance or a one- or two-way analysis of variance with Tukey’s post hoc test, as needed. Significance was considered as P < 0.05.
Supplementary Material
Acknowledgments
We thank Dr. Joseph Miano for insightful comments and Jose Cabrera for graphics. E.M.S. was supported by a Scientist Development Grant from the American Heart Association, a joint University of Rochester/Moulder Center for Drug Discovery Pilot Award, and startup funds from the Aab Cardiovascular Research Institute at the University of Rochester School of Medicine and Dentistry. K.E.K. was supported by the National Institutes of Health (NIH) (HL-110869) and the Moss Heart Fund. F.G. was supported by NIH Grant GM-031321. J.W.S. was supported by grants from the NIH (U01-HL-100401), AHA-Jon Holden DeHaan Foundation, and the Cancer Prevention and Research Institute of Texas. E.N.O. was supported by NIH Grants HL-077439, HL-111665, HL-093039, and U01-HL-100401; the American Heart Association–Jon Holden DeHaan Foundation (0970518N); Foundation Leducq Networks of Excellence; Cancer Prevention and Research Institute of Texas; and the Robert A. Welch Foundation (Grant 1-0025).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1316764110/-/DCSupplemental.
References
- 1.Eyden B. The myofibroblast: Phenotypic characterization as a prerequisite to understanding its functions in translational medicine. J Cell Mol Med. 2008;12(1):22–37. doi: 10.1111/j.1582-4934.2007.00213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127(3):526–537. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 3.Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell. 2001;12(9):2730–2741. doi: 10.1091/mbc.12.9.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3(5):349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 5.Tomasek JJ, McRae J, Owens GK, Haaksma CJ. Regulation of alpha-smooth muscle actin expression in granulation tissue myofibroblasts is dependent on the intronic CArG element and the transforming growth factor-beta1 control element. Am J Pathol. 2005;166(5):1343–1351. doi: 10.1016/s0002-9440(10)62353-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453(7193):314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- 7.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214(2):199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pipes GC, Creemers EE, Olson EN. The myocardin family of transcriptional coactivators: Versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 2006;20(12):1545–1556. doi: 10.1101/gad.1428006. [DOI] [PubMed] [Google Scholar]
- 9.Du KL, et al. Myocardin is a critical serum response factor cofactor in the transcriptional program regulating smooth muscle cell differentiation. Mol Cell Biol. 2003;23(7):2425–2437. doi: 10.1128/MCB.23.7.2425-2437.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li S, Wang DZ, Wang Z, Richardson JA, Olson EN. The serum response factor coactivator myocardin is required for vascular smooth muscle development. Proc Natl Acad Sci USA. 2003;100(16):9366–9370. doi: 10.1073/pnas.1233635100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Small EM, et al. Myocardin is sufficient and necessary for cardiac gene expression in Xenopus. Development. 2005;132(5):987–997. doi: 10.1242/dev.01684. [DOI] [PubMed] [Google Scholar]
- 12.Wang D, et al. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell. 2001;105(7):851–862. doi: 10.1016/s0092-8674(01)00404-4. [DOI] [PubMed] [Google Scholar]
- 13.Hoofnagle MH, et al. Myocardin is differentially required for the development of smooth muscle cells and cardiomyocytes. Am J Physiol Heart Circ Physiol. 2011;300(5):H1707–H1721. doi: 10.1152/ajpheart.01192.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang J, et al. Myocardin regulates BMP10 expression and is required for heart development. J Clin Invest. 2012;122(10):3678–3691. doi: 10.1172/JCI63635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaw PE, Schröter H, Nordheim A. The ability of a ternary complex to form over the serum response element correlates with serum inducibility of the human c-fos promoter. Cell. 1989;56(4):563–572. doi: 10.1016/0092-8674(89)90579-5. [DOI] [PubMed] [Google Scholar]
- 16.Cen B, Selvaraj A, Prywes R. Myocardin/MKL family of SRF coactivators: Key regulators of immediate early and muscle specific gene expression. J Cell Biochem. 2004;93(1):74–82. doi: 10.1002/jcb.20199. [DOI] [PubMed] [Google Scholar]
- 17.Wang DZ, et al. Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc Natl Acad Sci USA. 2002;99(23):14855–14860. doi: 10.1073/pnas.222561499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olson EN, Nordheim A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol. 2010;11(5):353–365. doi: 10.1038/nrm2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elberg G, et al. MKL1 mediates TGF-beta1-induced alpha-smooth muscle actin expression in human renal epithelial cells. Am J Physiol Renal Physiol. 2008;294(5):F1116–F1128. doi: 10.1152/ajprenal.00142.2007. [DOI] [PubMed] [Google Scholar]
- 20.Fan L, et al. Cell contact-dependent regulation of epithelial-myofibroblast transition via the rho-rho kinase-phospho-myosin pathway. Mol Biol Cell. 2007;18(3):1083–1097. doi: 10.1091/mbc.E06-07-0602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao XH, et al. Force activates smooth muscle alpha-actin promoter activity through the Rho signaling pathway. J Cell Sci. 2007;120(Pt 10):1801–1809. doi: 10.1242/jcs.001586. [DOI] [PubMed] [Google Scholar]
- 22.Guettler S, Vartiainen MK, Miralles F, Larijani B, Treisman R. RPEL motifs link the serum response factor cofactor MAL but not myocardin to Rho signaling via actin binding. Mol Cell Biol. 2008;28(2):732–742. doi: 10.1128/MCB.01623-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113(3):329–342. doi: 10.1016/s0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- 24.Kuwahara K, Barrientos T, Pipes GC, Li S, Olson EN. Muscle-specific signaling mechanism that links actin dynamics to serum response factor. Mol Cell Biol. 2005;25(8):3173–3181. doi: 10.1128/MCB.25.8.3173-3181.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun Q, et al. Defining the mammalian CArGome. Genome Res. 2006;16(2):197–207. doi: 10.1101/gr.4108706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Small EM, et al. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res. 2010;107(2):294–304. doi: 10.1161/CIRCRESAHA.110.223172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Russell JL, Goetsch SC, Aguilar HR, Frantz DE, Schneider JW. Targeting native adult heart progenitors with cardiogenic small molecules. ACS Chem Biol. 2012;7(6):1067–1076. doi: 10.1021/cb200525q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grinnell F, Petroll WM. Cell motility and mechanics in three-dimensional collagen matrices. Annu Rev Cell Dev Biol. 2010;26:335–361. doi: 10.1146/annurev.cellbio.042308.113318. [DOI] [PubMed] [Google Scholar]
- 29.Schneider JW, et al. Small-molecule activation of neuronal cell fate. Nat Chem Biol. 2008;4(7):408–410. doi: 10.1038/nchembio.95. [DOI] [PubMed] [Google Scholar]
- 30.Small EM. The actin-MRTF-SRF gene regulatory axis and myofibroblast differentiation. J Cardiovasc Transl Res. 2012;5(6):794–804. doi: 10.1007/s12265-012-9397-0. [DOI] [PubMed] [Google Scholar]
- 31.Crider BJ, Risinger GM, Jr, Haaksma CJ, Howard EW, Tomasek JJ. Myocardin-related transcription factors A and B are key regulators of TGF-β1-induced fibroblast to myofibroblast differentiation. J Invest Dermatol. 2011;131(12):2378–2385. doi: 10.1038/jid.2011.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cogan JG, Subramanian SV, Polikandriotis JA, Kelm RJ, Jr, Strauch AR. Vascular smooth muscle alpha-actin gene transcription during myofibroblast differentiation requires Sp1/3 protein binding proximal to the MCAT enhancer. J Biol Chem. 2002;277(39):36433–36442. doi: 10.1074/jbc.M203232200. [DOI] [PubMed] [Google Scholar]
- 33.Subramanian SV, et al. Induction of vascular smooth muscle alpha-actin gene transcription in transforming growth factor beta1-activated myofibroblasts mediated by dynamic interplay between the Pur repressor proteins and Sp1/Smad coactivators. Mol Biol Cell. 2004;15(10):4532–4543. doi: 10.1091/mbc.E04-04-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hautmann MB, Madsen CS, Owens GK. A transforming growth factor beta (TGFbeta) control element drives TGFbeta-induced stimulation of smooth muscle alpha-actin gene expression in concert with two CArG elements. J Biol Chem. 1997;272(16):10948–10956. doi: 10.1074/jbc.272.16.10948. [DOI] [PubMed] [Google Scholar]
- 35. Davis J, Burr AR, Davis GF, Birnbaumer L, Molkentin JD (2012) A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev Cell 23(4):705–715. [DOI] [PMC free article] [PubMed]
- 36.Russell JL, et al. Regulated expression of pH sensing G protein-coupled receptor-68 identified through chemical biology defines a new drug target for ischemic heart disease. ACS Chem Biol. 2012;7(6):1077–1083. doi: 10.1021/cb300001m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z, et al. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature. 2004;428(6979):185–189. doi: 10.1038/nature02382. [DOI] [PubMed] [Google Scholar]
- 38.Muehlich S, et al. Serum-induced phosphorylation of the serum response factor coactivator MKL1 by the extracellular signal-regulated kinase 1/2 pathway inhibits its nuclear localization. Mol Cell Biol. 2008;28(20):6302–6313. doi: 10.1128/MCB.00427-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dioum EM, et al. A small molecule differentiation inducer increases insulin production by pancreatic β cells. Proc Natl Acad Sci USA. 2011;108(51):20713–20718. doi: 10.1073/pnas.1118526109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cao D, et al. Modulation of smooth muscle gene expression by association of histone acetyltransferases and deacetylases with myocardin. Mol Cell Biol. 2005;25(1):364–376. doi: 10.1128/MCB.25.1.364-376.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koegel H, et al. Loss of serum response factor in keratinocytes results in hyperproliferative skin disease in mice. J Clin Invest. 2009;119(4):899–910. doi: 10.1172/JCI37771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Desmoulière A, et al. Alpha-smooth muscle actin is expressed in a subpopulation of cultured and cloned fibroblasts and is modulated by gamma-interferon. Exp Cell Res. 1992;201(1):64–73. doi: 10.1016/0014-4827(92)90348-c. [DOI] [PubMed] [Google Scholar]
- 43.Liu X, Kelm RJ, Jr, Strauch AR. Transforming growth factor beta1-mediated activation of the smooth muscle alpha-actin gene in human pulmonary myofibroblasts is inhibited by tumor necrosis factor-alpha via mitogen-activated protein kinase kinase 1-dependent induction of the Egr-1 transcriptional repressor. Mol Biol Cell. 2009;20(8):2174–2185. doi: 10.1091/mbc.E08-10-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Z, et al. Increased transcriptional response to mechanical strain in keloid fibroblasts due to increased focal adhesion complex formation. J Cell Physiol. 2006;206(2):510–517. doi: 10.1002/jcp.20486. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.