

Significance
Heme, traditionally viewed as a stable protein cofactor such as in hemoglobin, also serves as an acute signaling molecule and is cytotoxic at high concentrations. Here, we show that free intracellular heme potently enhances A-type potassium channel function. Such channels determine action potential frequency in excitable cells, and their dysfunction often contributes to pathological hyperexcitability, such as in pain and epilepsy. Binding of free heme at nanomolar concentrations to the “ball-and-chain” N terminus of A-type potassium channels, which typically closes the channels, introduces a stable structure in the otherwise disordered region and allows for a greater efflux of potassium ions, thus reducing cellular excitability. Heme therefore could be a powerful negative-feedback regulator in brain and muscle function.
Keywords: intrinsically disordered domain, NMR, cysteine, A-type channel, N-type inactivation
Abstract
Fine-tuned regulation of K+ channel inactivation enables excitable cells to adjust action potential firing. Fast inactivation present in some K+ channels is mediated by the distal N-terminal structure (ball) occluding the ion permeation pathway. Here we show that Kv1.4 K+ channels are potently regulated by intracellular free heme; heme binds to the N-terminal inactivation domain and thereby impairs the inactivation process, thus enhancing the K+ current with an apparent EC50 value of ∼20 nM. Functional studies on channel mutants and structural investigations on recombinant inactivation ball domain peptides encompassing the first 61 residues of Kv1.4 revealed a heme-responsive binding motif involving Cys13:His16 and a secondary histidine at position 35. Heme binding to the N-terminal inactivation domain induces a conformational constraint that prevents it from reaching its receptor site at the vestibule of the channel pore.
A-type K+ channels, a family of voltage-gated K+ (Kv) channels, play a vital role in the control of neuronal excitability, regulation of presynaptic spike duration, Ca2+ entry, and neurotransmitter release (1). One of the prominent features of A-type K+ channels is their inactivation, which is mediated by two structurally distinct processes (2, 3). The fast inactivation is initiated by the N-terminal protein structure, thereby termed N-type inactivation, whereas the slow C-type inactivation is related to the pore structure (2, 3). N-type inactivation proceeds according to a “ball-and-chain” mechanism; the positive charges of the N-terminal ball domains bring the structures to the pore domain of the channel and the distal segment of one of the four intrinsically disordered N-terminal ball domains enters the hydrophobic central cavity/vestibule of the inner pore of the channel thus obstructing the flow of K+ (2–5).
Acute enzymatic or mutational removal of the distal N terminus eliminates N-type inactivation, and in such inactivation-removed channels, intracellular application of peptides corresponding to the N-terminal sequence restores inactivation (4, 6, 7). Structural analysis suggests that the N-terminal inactivation structure needs to be flexible or even intrinsically disordered to reach the receptor in the channel’s cavity (8, 9).
“Tuning” of rapid N-type inactivation is an effective way of adapting cells to specific needs. For example, molecular processes affecting the speed and degree of N-type inactivation in Kv1.4 (KCNA4) channels include redox regulation of a cysteine residue in the N-terminal ball structure (C13) (10), protonation of histidine at position 16 (11), interaction with membrane lipids (12), and Ca2+-dependent phosphorylation (13). Furthermore, low-molecular-weight compounds affecting N-type inactivation (N-type disinactivators) have been discussed as potential drugs regulating cellular excitability (14).
Heme [Fe(II) protoporphyrin-IX] is well known as a protein cofactor, often conferring gas sensitivity as exemplified in hemoglobin, cytochromes, myoglobin, and soluble guanylyl cyclase. In many heme proteins including soluble guanylyl cyclase, heme is bound or coordinated in part by an amino acid sequence typically containing a histidine or cysteine residue, which acts as an axial fifth ligand (in addition to the four bonds provided by the nitrogen atoms of the protoporphyrin-IX ring to the iron center) to the redox-sensitive iron center, and water or a bound gas molecule acts as the sixth ligand (15). However, recent advances revealed a novel role of heme as a nongenomic modulator of ion channel functions, first exemplified for the large-conductance voltage- and Ca2+-dependent K+ channel (Slo1 BK) (16) and later for the epithelial Na+ channel (17). Detailed analysis of the biophysical action of heme [ferrous iron (Fe2+)] or hemin [ferric iron (Fe3+)] on the Slo1 BK channel demonstrated that hemin is a potent modulator of the allosteric gating mechanism of the channel (18), and mutagenesis studies have indicated the sequence CKACH located in the cytoplasmic C terminus of the channel plays a critical role (16, 19). However, neither for Slo1 BK channels nor for epithelial Na+ channels, the interaction of heme with the ion channel protein is structurally resolved. In this study, we found that the fast N-type inactivation of Kv1.4 A-type K+ channels is potently modulated by heme/hemin. Furthermore, we provide structural insight into heme interaction with a channel explaining how heme prevents A-type channels from entering an inactivated state.
Results
Intracellular Heme Slows Down Inactivation of Kv1.4 Channels.
Kv1.4 channels were expressed in Xenopus oocytes, and channel function was assayed in the inside-out patch-clamp configuration. Because fast inactivation of Kv1.4 is known to depend on the redox potential (10), the intracellular solution contained 1 mM reduced glutathione (GSH) in all cases. Application of hemin (200 nM) slowed down the time course of inactivation considerably (Fig. 1A); estimated with a single-exponential function, the inactivation time constant increased from 48.2 ± 2.8 to 245 ± 21 ms (n = 11, P < 0.001; Fig. 1B). Recovery from inactivation at –80 mV, however, was not altered by hemin (n = 5, P = 0.08; Fig. 1B). The degree of channel inactivation was estimated with the ratio of current measured at 200 ms after depolarization onset to the maximal current (I200 ms/Imax); it increased on hemin application in a single-exponential fashion, whereas washout of hemin resulted in slow and partial recovery (Fig. 1C). The onset of the hemin effect was steeply concentration dependent (Fig. 1D). The concentration dependence of the effect on inactivation (Fig. 1E) was described with a Hill function yielding an apparent EC50 value of 22.9 ± 3.5 nM and a Hill coefficient of 2.0 ± 0.5; the maximal effect on I200 ms/Imax was 0.484 ± 0.014, indicative of residual C-type inactivation on complete removal of N-type inactivation by hemin (see below).
Fig. 1.
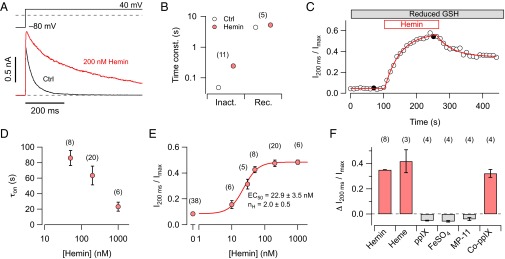
Hemin eliminates N-type inactivation of Kv1.4 channels. (A) Superposition of current traces recorded from inside-out patches taken from Xenopus oocytes expressing Kv1.4 K+ channels before (black) and 200 s after application of 200 nM hemin (red). Pulse protocol is shown above. (B) Time constants of inactivation at 40 mV before and after application of 200 nM hemin (Left) and time constants of recovery from inactivation at –80 mV (Right). (C) Degree of channel inactivation, expressed as current after 200 ms relative to peak current, as a function of time in the presence of reduced glutathione (GSH, 1 mM). Patches were exposed to 200 nM hemin followed by washing with hemin-free solutions. The filled symbols mark data from the traces shown in A. Superimposed curves are single-exponential fits. (D) Time constants of the onset of heme-induced loss of inactivation, determined as in C, as a function of hemin concentration. (E) Steady-state loss of inactivation as a function of hemin concentration. The continuous curve is the result of a Hill fit. (F) Loss of inactivation induced by hemin (50 nM), heme (50 nM, 1 mM Na2S2O4), protoporphyrin-IX (ppIX, 50 nM), FeSO4 (1 µM), MP-11 (2 µM), and Cobalt-protoporphyrin-IX (Co-ppIX, 50 nM). The number of independent experiments is provided in parentheses in B and D–F.
To reveal which structural aspect of hemin is required for its interaction with the N-type inactivation of Kv1.4 channels, we first compared the effect on N-type inactivation of the nearly saturating concentration of 50 nM hemin (Fe3+) with 50 nM heme (Fe2+), i.e., hemin reduced with 1 mM sodium dithionite, and found no significant difference (Fig. 1F). However, 50 nM protoporphyrin-IX, 1 µM FeSO4, and 2 µM MP-11, i.e., hemin bound to a peptide of microperoxidase, failed to affect the time course of inactivation. Cobalt-protoporphyrin-IX (Co-ppIX, 50 nM) was almost as effective as hemin and heme (Fig. 1F). These results indicate that only metal-ligated protoporphyrin in a free form affects N-type inactivation of Kv1.4 channels.
Analysis of the N-Terminal Inactivation Domain.
Given the specific effect of heme/hemin on the N-type inactivation of Kv1.4 channels, it is expected that the channel protein complex harbors one or more binding sites for heme with an affinity in the nanomolar range. To rule out effects of heme on mechanisms other than those on N-type inactivation, we assayed Kv1.4 Δ2–110, i.e., a mutant with a substantially shortened N terminus and only exhibiting residual C-type inactivation (20) (Fig. 2 A and B, Second row). As shown in Fig. 2B (Second row), 200 nM hemin neither affected the time course of inactivation nor altered the size of the current signal showing that Kv1.4 channels without N-type inactivation are largely insensitive to hemin at this concentration. In addition, the similarity of the inactivation time course of N-terminally truncated channels (Fig. 2B, second row) and WT Kv1.4 channels in the presence of hemin (Fig. 1A) suggests that hemin effectively eliminates N-type inactivation.
Fig. 2.

Mutagenesis of N-terminal inactivation domain. (A) Schematic presentation of the Kv1.4 protein comprising the cytosolic N and C termini, as well as the membrane-delimited 6TM domain (S1–S6). Residue numbers, based on rat Kv1.4, are indicated and the amino acid sequence corresponding to the red bar is shown below, highlighting C13, H16, and H35. (B) Inside-out patch-clamp recordings on depolarization to 40 mV for the indicated channel constructs before (black) and about 200 s after application of 200 nM hemin (red). (C) Current after 200 ms of the depolarization onset relative to the peak current for the indicated channel constructs before (white bars) and after application of hemin (red bars) from experiments as in B. Blue bars indicate the loss of inactivation induced by hemin. The numbers of independent measurements are given in parentheses. Statistical indicators reflect a two-sided paired t test, followed by Bonferroni correction, testing for a change of inactivation induced by hemin: ns, not significant; P > 0.05; **P < 0.01; ***P < 0.001.
In many heme-binding proteins, Cys and His residues are the most likely amino acids to serve as axial ligands for the iron moiety (Table S1). Inspection of the Kv1.4 primary structure did not reveal classic heme binding motifs such as that found in cytochrome-c or Slo1 BK channels (CxxCH), but the N-terminal ball structure (Fig. 2A) possesses a histidine residue (His16) close to Cys13, the latter formerly identified to be important for the channel’s redox sensitivity (10), forming a putative heme-responsive motif CxxH. We thus mutated these residues to amino acids not expected to take part in the axial ligation of heme iron (C13S and H16A) individually and in combination. Although C13S slightly affected the hemin sensitivity of inactivation, H16A had no effect. However, combination of both mutations (C13S:H16A) made the inactivation resistant to hemin (Fig. 2 B and C, Second to last row). The Kv1.4 N terminus contains another histidine at position 35 (H35; Fig. 2A). Mutation H35A significantly reduced hemin sensitivity; the same was true for the combination H16A:H35A, whereas combination with C13S (C13S:H35A) completely removed hemin sensitivity of the N-type inactivation (Fig. 2 B and C). These data suggest a complex mode of heme interaction with the ball domain involving a heme-regulatory motif and a histidine residue at position 35.
To study the biochemical properties of hemin interacting with the N-terminal ball domain of Kv1.4 channels, we produced a recombinant peptide encoding the first 61 residues of Kv1.4 (Pep61; Fig. 3A) and mutants thereof as His-tagged maltose binding protein (MBP) fusions. When produced in Escherichia coli cells using culture medium supplemented with 1 mM hemin, the WT His-tagged MBP-fusion protein appeared brown, whereas the triple mutant (C13S:H16A:H35A) yielded a clear protein solution (Fig. 3B, Left), illustrating that the His-tag alone does not bind heme in a significant manner and that the WT Pep61 must have some heme-binding capacity. UV-vis spectra of the WT protein showed a clear Soret peak indicative of hemin binding (Fig. 3B, Right). This result shows that Pep61 has a binding capacity for hemin and that hemin binding already occurs under physiological conditions within the bacteria. On cleavage of the His-tag-MBP portion, Pep61 and mutants thereof were HPLC purified for further investigations.
Fig. 3.

Hemin antagonizes channel block by free inactivation peptides. (A) Sequence of the N-terminal ball peptide, Pep61, with critical residues in red. (B) Purified His-tagged MBP-fusion proteins with WT Pep61 (CHH), mutant C13S:H16A:H35A (SAA), and MBP alone, each at about 5 mg/mL; M9 E. coli culture medium was supplemented with 1 mM hemin. (Right) UV-vis spectra indicating a Soret peak for the indicated peptides at a concentration of 4 mg/mL. (C) Currents mediated by Kv1.1 channels were measured in inside-out patches on depolarization to 40 mV (black traces). Subsequently, the indicated recombinant Kv1.4 inactivation peptides were applied (50 µM, blue) and the peptides together with hemin (50 µM each, red). Intracellular solution always contained 1 mM reduced GSH. (D) Statistics for the degree of inactivation for the peptides shown in C: white bars, control; blue bars, peptide only; red bars, peptide in presence of hemin. n values are indicated in parentheses. Statistical indicators reflect two-sided paired t tests testing for an effect of hemin on the peptide-induced degree of inactivation: ns, not significant; **P < 0.01.
Because small N-terminal inactivation peptides were shown to block ion conduction when applied as free peptide via the intracellular solution, we also investigated this function of Pep61. Noninactivating Kv1.1 channels were expressed in Xenopus oocytes, and Pep61 was applied to the cytosolic face of an inside-out patch resulting in channel inactivation. As illustrated in Fig. 3 C and D, Pep61 efficiently induced inactivation of Kv1.1 channels, and equimolar hemin obliterated that effect. Concomitant mutagenesis of the critical Cys and at least one of the His residues in this peptide eliminated its hemin sensitivity (Fig. 3 C and D). The inactivation induced by the triple-mutant peptide (C13S:H16A:H35A) in the presence of hemin demonstrates that hemin directly interacts with the inactivation domain and is otherwise independent of the channel complex; it does not interfere with the function of the receptor for the inactivation domain or the cavity within the channel pore (5).
Heme Physically Binds to Kv1.4-IP and Alters Its Structure.
The data presented thus far suggest that hemin physically binds to the inactivation ball domain of Kv1.4 channels. To quantify this interaction, we performed UV-vis absorption spectroscopy and electron paramagnetic resonance (EPR) studies with Pep61 and mutants thereof. UV-vis absorption spectroscopic measurements of hemin and hemin together with variants of Pep61 were performed in the range of 320–500 nm. With increasing hemin concentrations, difference spectra for Pep61 showed a prominent Soret absorption peak at 418 nm indicative of hemin ligation to the peptide (Fig. 4A). By measuring the absorbance at 418 nm with 10 µM Pep61 and increasing hemin concentration, binding of hemin to Pep61 was estimated with a Hill equation (Fig. 4B) yielding a half-maximal binding at a hemin:peptide ratio of 1.08 and a Hill coefficient of about 2 for the WT. In contrast, absorbance only increased slightly for Pep61-C13S:H16A:H35A. The binding capacity of all mutants was estimated for equal concentrations of peptide and hemin (10 µM) (Fig. 4C), clearly showing that all individual, double, and triple mutations resulted in impaired binding.
Fig. 4.

Hemin binding to Pep61. (A) UV-vis difference spectra using 10 µM Pep61 and increasing concentration of hemin. The black trace is for a 1:1 ratio. Experiments were performed in the presence of 2 mM GSH. (B) Titration curve for Pep61 (open circles) at 418 nm yielding a half-maximal binding at a hemin:peptide ratio of 1.08 and a Hill coefficient of 2. For mutant Pep-C13S:H16A:H35A (SAA, filled symbols) only a weak increase in absorbance was observed. (C) Absorbance change at 418 nm for 10 µM of the indicated peptides and 10 µM hemin. CHH refers to the WT signifying C13, H16, and H35. (D) Electron paramagnetic resonance spectra for 210 µM hemin (pink in all panels) and hemin plus 280 µM of the indicated peptides. Arrows mark the g factors at 2.26, which are part of rhombic signals typical for axial ligation of the low-spin hemin iron. From the full spectra, the following sets of g factors were determined: Pep61, [2.46, 2.26, 1.91]; Pep61-H35A, [2.47, 2.27, 1.90].
The coordination of hemin in a protein environment can be characterized by the EPR signals of the low-spin ferric iron. Measurement of X-band EPR spectra at 5 K revealed a clear rhombic signal when Pep61 was present (Fig. 4D, Upper Left). Such an indication of hemin ligation was absent for all mutants containing C13S. Mutants H16A and H16A:H35A only showed a very weak feature in the spectra that could be indicative of very loose binding. Interestingly, the EPR spectra of the single-point mutant H35A also showed a clear binding signal (Fig. 4D, Lower Left). From the spectral features (g factors), the rhombicity and tetragonality of the ligand field parameters and their relation were derived and compared with those from the literature describing cysteine thiolate ligation of heme (Table S1). Based on such a classification (Fig. S1), EPR spectra of hemin bound to Pep61 and mutant H35A are compatible with a penta-coordination of the hemin iron via a cysteine residue with the sixth ligand being water or histidine. Therefore, hemin is primarily bound by C13 and H16, but H35 may interfere with this configuration.
The functional consequence of hemin binding, as shown above, is a loss of fast channel inactivation. A feasible explanation could be that hemin induces a conformational change and thus reduces the peptide flexibility required to reach the receptor site inside the channel cavity. We therefore performed various assays with respect to potential conformational changes of Pep61 on hemin binding.
As a first approach, we introduced a tryptophan instead of alanine at position 23 (A23W). This tryptophan is then the only Trp of Pep61-A23W, and it is situated between the CxxH motif and the secondary His35. Although control Pep61 did not yield any fluorescence in the range of 320–380 nm when excited with 295-nm light, Pep61-A23W showed strong Trp fluorescence (Fig. 5A). Increasing concentrations of hemin quenched this fluorescence (Fig. 5B), and the concentration dependence (Fig. 5C) suggests an apparent binding constant of 195 ± 17 nM hemin. Almost no such quenching was observed for the mutant peptide Pep61-C13S:H16A:A23W:H35A (SAA), l-tryptophan, or N-acetyl-tryptophanamide (NATA) as a mimic of peptide-bound tryptophan (Fig. 5C).
Fig. 5.

Hemin changes the conformation of Pep61. (A) Fluorescence spectra on excitation at 295 nm for the buffer (green), Pep61 (blue), and Pep61-A23W (black). (B) Tryptophan fluorescence spectra for Pep61-A23W (0.5 µM) without (black) and with increasing concentrations of hemin (up to 720 nM). (C) Normalized tryptophan fluorescence at 349 nm for increasing hemin concentrations for Pep61 (gray circles) with superimposed Hill fit. Also shown are data for mutant Pep61-C13S:H16A:H35A (SAA, open circles), l-tryptophan (blue triangles), and NATA (pink triangles), all connected by straight lines. (D) Circular dichroism of Pep61 (Upper) and Pep61-C13S:H16A:H35A (Lower) (12.5 µM) without (black) and with hemin (50 µM, red) at 37 °C. The superimposed curves are secondary structural element predictions according to Reed and Reed (21). (E) Predicted structural element contents of the indicated peptides before (light) and after hemin application (dark). The bottom graph presents the hemin-induced changes of the indicated structural element contents.
Pep61 and mutant Pep61-C13S:H16A:H35A were subjected to circular dichroism (CD) spectroscopy at various temperatures without and with addition of hemin. Although at low temperatures (5 °C) hemin only had a minor impact on the shape of the CD spectra, at higher temperatures, it appeared to induce additional order. In contrast, for Pep61-C13S:H16A:H35A, hemin failed to change the structure appreciably even at high temperatures (Fig. S2). Examples of CD spectra at 37 °C are shown in Fig. 5D. Using an algorithm of Reed and Reed (21), the contents of the structural elements α-helix, β-sheet, and random coil were estimated. For the WT peptide, addition of hemin resulted in a marked increase in α-helicity, whereas the content of the β-sheet substantially decreased. In contrast, for mutant Pep61-C13S:H16A:H35A, hemin did not change the structure. Inspection of all mutants revealed that the structural changes observed for the WT are largely preserved when the mutants either harbored C13 or the combination of H16 and H35 (Fig. 5E). The same mutants also retained some hemin sensitivity when tested in the physiological assay (Fig. 2), strongly suggesting a correlation of hemin-induced change in peptide structure and functional impact as inactivating ball domain.
NMR spectroscopy was used to obtain further structural features of Pep61. The approach applied consists of the following steps: (i) production of stable isotope labeled NMR samples; (ii) establishment of the resonance assignments at 10 °C; (iii) chemical shift–based structure generation for the free Pep61 to be used as starting structure; and (iv) in silico docking of Pep61 with heme.
By means of heteronuclear 3D experiments, 91% of the protons and 97% of the heteroatoms could be assigned. The initial chemical shift index–based structure (Fig. S3, Upper) comparison indicated potential differences to a structure of an elongated construct reported earlier [1KN7 (22)]. We therefore decided to obtain an independent 3D starting structure by in silico structure predictions with CS-Rosetta and CHESHIRE to be used for the docking experiments with the heme moiety. These programs use an extended set of chemical shift assignments, i.e., the 1Hα, 1HN, 13Cα, 13Cβ, 13C', and 15N' resonances, the latter obtained from [1H,15N]-heteronuclear single quantum coherence (HSQC) spectra (Fig. S4A). Considering the structural knowledge about protein fragments with similar sequences in the Protein Data Bank (PDB) structure database, structures were generated following a Monte Carlo or molecular dynamics approach. The resulting structures were cross-checked for compliance with the experimentally acquired structure and dynamics data (Fig. S4B), and, for example, models in which residues in the N- and C-terminal regions were in close proximity were excluded on the basis of missing cross-peaks in nuclear Overhauser enhancement spectroscopy (NOESY)-type spectra. Finally, a CS-Rosetta–generated model showing an extended conformation encompassing the predicted central α-helix between residues 22 and 47 (Fig. 6) was selected as starting structure for the heme docking. The selected structure was consistent with the results of heteronuclear 15N{1H}-nuclear Overhauser enhancement effect (NOE) measurements, which revealed a low mobility of the Pep61 backbone in this helical region and a high flexibility of the N- and C-terminal ends (Fig. S3, Lower). The flexibility of the structural element around the CxxH motif could not be assessed experimentally due to spectral overlap and missing assignments. At 37 °C and in the Ga-ppIX complex, the NMR signals are broadened beyond detection, indicating a different motional regime for the Pep61 backbone.
Fig. 6.

Docking of heme to an NMR-derived model of Pep61. (Left) Full-length Pep61 is depicted with secondary structure elements without the cloning overhangs; heme, the residues C13 (yellow), as well as H16 and H35 (magenta) are presented using sticks. The N terminus of the peptide (M1) is shown in pink; residue A23 used for the introduction of a tryptophan for fluorescence quenching experiments shown in Fig. 5 A–C is shown in gray. (Right) Magnified section of the peptide focusing on the heme-binding site. Residues that are presumably taking part in heme ligation are shown in thick lines. (A) Mutant H35A with penta-coordination via C13. (B) WT Pep61 with penta-coordination via H35. (C) WT with hexa-coordination via H16 and H35.
The docking studies of hemin to the Pep61 structure included the following scenarios: the WT, all single mutants, and all double mutants were docked using ambiguous restraints, i.e., the distances of the heme iron to the potentially coordinating atoms of all respective candidate amino acids, with a fully flexible amino acid chain. Representative models were then selected according to lowest High Ambiguity Driven Protein-Protein Docking (HADDOCK) score values (Table S2), which are calculated on the basis of van der Waals repulsions, electrostatic interactions, ambiguous interaction restraint energy, desolvation energy, and buried surface area according to de Vries et al. (23). In general, the best/lowest scores were observed for a C13 coordination of the heme moiety. Although resulting in moderately higher score values, there was a clear tendency for a H35-coordinated heme when this residue was available in the docking scenario.
Docking the NMR-based starting structure with a heme moiety thus indicates the following. (i) For the WT and mutants C13S, H16A, and C13S:H16A, H35 is preferred over C13/H16 for penta-coordinated binding of the heme (Fig. 6B). Such a conformation can be stabilized by side-chain interactions of R28, E31, and R32 with the heme propionates. Compared with the starting structure in most of the models, the helix shortens by approximately one turn (residues 22–38). (ii) For mutants H35A and H16A:H35A, a penta-coordination via C13 is observed with lower score values (Fig. 6A). (iii) Hexa-coordination of the heme ring involving the distal and proximal sides at the same time by a His/His sandwich was preferred over a Cys/His sandwich only when an unambiguous restraint set was used (Fig. 6C). (iv) The docking data also suggest that heme coordination at the C13xxH16 motif can only be realized by a penta-coordinated heme with its ring system oriented parallel to the Pep61 surface.
Discussion
In this study, we showed that heme is a potent regulator of Kv1.4 channels. Independently of the redox properties of heme, it specifically binds to the N-terminal inactivation ball-and-chain domain and impairs N-type inactivation. Half-maximal removal of inactivation was observed at around 20 nM. This concentration is greater than binding constants reported for heme-binding proteins such as heme oxygenase (24), HasA (25), or hemopexin (26), but it is comparable to those of cytochrome b562 (27) and much below constants reported for albumin (28). In particular, this concentration is in the range—or even lower—of reported binding constants of heme-regulated transcription factors (29), indicating that heme-dependent regulation of N-type inactivation is expected to occur under physiological conditions. Given this high affinity and because the effect of heme on Kv1.4 channels is a gain-of-function phenomenon, even very small concentrations of heme (nanomolar levels) will be functionally effective. Under pathophysiological conditions, such as increased heme levels related to a local hemorrhage (30), the impact of channel function may even become greater.
Several studies have shown that the fast N-type inactivation is associated with states of hyperexcitability such as those occurring in pain and epilepsy (31, 32). Kv1.4 channels are present in nerve terminals where they regulate spike duration, Ca2+ entry, and neurotransmitter release. Because Kv1.4 channels normally inactivate, depolarization can develop to facilitate Ca2+ influx, which broadens presynaptic action potentials and potentiates neurotransmitter release (33). Loss of inactivation induced by elevated heme levels would counteract this mechanism and lead to a diminished neurotransmitter release.
Cell systems where this process may occur include nociceptive dorsal root ganglia (DRG) neurons. As suggested by Binzen et al. (31), Kv1.4 channels are possibly implicated in pain signaling. Excess heme levels would diminish the output of DRG neurons and may, therefore, have an analgesic potency.
Fast N-type inactivation of Kv1.4 and Kv1.1/Kvβ1.1 is implicated in autosomal dominant lateral temporal lobe epilepsy (ADLTE) (32). The Kv channels coassemble with the leucine-rich glioma inactivated gene 1 protein (Lgi1), and its presence normally prevents N-type inactivation. This influence is impaired by the ADLTE mutations resulting in neuronal hyperexcitability (32). Thus, heme-mediated modulation of Kv1.4 or Kvβ1.1, which also harbors a CxxH motif in the N terminus, may have potential to reduce the severity of epileptic seizures.
It has been reported that openers of Kv1.4 channels may serve as neuroprotective agents and may be target candidates for pain therapy (34, 35). Riluzole, which is already on the market and used for treating the degenerative motor neuron disease amyotrophic lateral sclerosis (ALS), increases the activity of Kv1.4 channels by slowing their N-type inactivation; this inhibits glutamate release in nerve terminals by a diminished depolarization-dependent Ca2+ influx (34). Thus, in all of the examples mentioned, the heme-mediated slowing of Kv1.4 inactivation may serve as an endogenous neuroprotective mechanism in various neuropathological conditions.
Our functional studies have shown that hemin has a specific impact on the N-type inactivation of Kv1.4 channels; neither the slow C-type inactivation nor voltage-dependent channel opening and closing or ion conductance is affected. Furthermore, hemin or heme must be free, and the individual components Fe2+ and protoporphyrin-IX are inactive. Mutagenesis and structural studies have identified C13xxH16 in the ball domain of the N terminus as a heme-responsive motif with additional influence of H35. NMR-based structural analysis at 10 °C (pH 7.4) revealed a disordered N-terminal ball domain (M1–M17) followed by an extended helix (Y19–A47), an assignment similar but not identical to a structure by Wissmann et al. (22) for the Kv1.4 inactivation domain of 75 residues at 10 °C and pH 4.4. Although C13xxH16 is located in the very flexible transition between the N-terminal domain and the helix, H35 is in the center of the helix. This observation is consistent with the study by Antz et al. (36), where it was shown that a peptide encompassing the inactivation domain of Kv1.4 possesses a highly flexible disordered N terminus and a well-defined helix. Based on CD measurements and NMR data in aqueous solution, Pep61 is even more flexible and disordered at body temperature (37 °C) compared with 10 °C, and this flexibility enables the distal end to enter the cavity from the intracellular side. Binding of hemin to the Kv1.4 ball domain might reduce the flexibility of the ball-and-chain machinery and induce a partial secondary structure, which makes it impossible for the ball peptides to reach the channel’s cavity. Using residue 23 of the inactivation peptide as a probe, tryptophan fluorescence was significantly quenched by hemin, whereas this did not happen for mutant Pep61-C13S:H16A:H35A (Fig. 5 A–C). This may also indicate that part of the peptide undergoes conformational changes when hemin is applied, but at present we cannot totally exclude the possibility that the diminished fluorescence in the presence of hemin is a result of resonant energy transfer from the excited tryptophan to the hemin moiety, when bound to the peptide. Changes in the secondary structure have also been observed in other ball domain peptides: addition of detergent to the Shaker ball domain peptide and anionic lipids to the Kv3.4 ball domain peptide promoted a partial β-structure (37, 38).
The exact binding mode of heme to the Kv1.4 ball peptide is unclear. In the absence of H35, both the EPR analysis (Table S1; Fig. S1) and heme docking to an NMR-based structure (Fig. 6A) strongly suggest a penta-coordination of the heme iron via C13, with the sixth ligand most likely being water. N14 and Y21 come close to the heme plane and could act as stabilizing factors. For the WT peptide (Pep61), the situation appears more complex because there are alternative binding scenarios, such as a penta-coordination via H35, presumably with E31 and R32 taking part in ligating the porphyrin ring structure (Fig. 6B). A mixture of such binding modes may be the cause of the shift in the EPR signals with respect to mutant H35A (Fig. S1). A hexa-coordination via H16 and H35, as depicted in Fig. 6C, however, is not compatible with the EPR measurements. Also taking into account the functional data (Figs. 2 and 3), the strongest ligation appears to originate from C13. The residual heme dependence of inactivation observed for mutant C13S may be the result of various weak binding components involving H16, H35, and possibly other residues of the N-terminal domain.
Based on the ball-and-chain mechanism of N-type inactivation, we propose the following mechanism of the heme action on Kv1.4. In the absence of heme, the N-terminal ball and chain domain located N-terminal to P18 is disordered and long enough such that the distal N terminus enters the cavity and blocks ion conduction. Ligation of heme by the side-chain of C13 induces a higher degree of conformational order near C13 probably extending to S10 and, consequently, only the first nine residues are available to approach the channel cavity. In its most extended conformation, the nine residues may be up to ∼30 Å in length. The pore domain radius at the cytoplasmic side based on the Kv1.2/2.1 structure (39) is ∼25 Å and the approximately nine-residue ball and chain is not long enough to reach up into the cavity to block ion conduction. In this manner, heme influences the functional availability of the N-terminal inactivation ball and has great potential to regulate cellular excitability.
Materials and Methods
Chemicals and Stock Solutions.
Hemin [Fe(III) protoporphyrin-IX], Co(II) protoporphyrin-IX, protoporphyrin-IX, microperoxidase (MP-11), FeSO4, and Na-dithionite were from Sigma-Aldrich. Stock solutions (1 mM) of these reagents were prepared weekly in 30 mM KOH and kept at −20 °C in aliquots. Working solutions were prepared immediately before use, stored at 4 °C, and used within 20 min.
Channel Constructs and Mutagenesis.
In this study, we used expression clones of rat Kv1.1 (rKv1.1 and KCNA1, accession no. X12589) and rat Kv1.4 (rKv1.4 and KCNA4, accession no. X16002). Overlap-extension mutagenesis as described previously (40) was performed to generate the following constructs: Kv1.4Δ2–110, C13S, H16A, H35A, and combinations thereof.
Channel Expression in Xenopus Oocytes.
Capped mRNA was synthesized in vitro using the mMessage mMachine kit (Ambion). Oocytes were surgically removed from the ovarian tissue of Xenopus laevis that had been anesthetized by immersion in ice water/tricaine according to an institutionally approved protocol. The oocytes were defolliculated, and healthy stage V and VI oocytes were isolated and microinjected with 50 nL of a solution containing channel WT or mutant mRNA. Inside-out patch-clamp recordings were performed 2–4 d after mRNA injection.
Electrophysiological Measurements.
Ionic currents were recorded in the inside-out configuration at room temperature using an EPC-9 patch-clamp amplifier operated with PatchMaster software (both HEKA Elektronik). Macroscopic currents were measured using aluminum silicate glass pipettes with resistances of about 1 MΩ. The intracellular solutions contained (in mM) 100 K-aspartate, 15 KCl, 1 GSH (reduced glutathione), 10 EGTA, and 10 Hepes (pH 8.0 with KOH). The extracellular solution contained (in mM) 103.6 Na-aspartate, 11.4 KCl, 1.8 CaCl2, and 10 Hepes (pH 7.2 with NaOH). Solution changes in patch-clamp experiments were performed using a multichannel perfusion system in which the patch was placed directly in the center of streaming solution.
Purification of Recombinant Kv1.4 Inactivation Peptide.
The gene encoding 61 amino acids of the ball domain of Kv1.4 channels and mutants thereof (C13S, H16A, H35A, A23W, and combinations) were inserted into NcoI/NotI sites of the pETM-41 expression vector (EMBL), providing N-terminal His6-MBP tags and tobacco etch virus (TEV) cleavage sites. All proteins were expressed in BL-21 (DE3-codon) E. coli cells by using M9 medium supplemented either with unlabeled or labeled 15NH4Cl and 13C-glucose and purified by affinity chromatography.
Overnight primary cultures (25 mL) of the desired expression construct were inoculated into 500 mL of M9 medium, grown at 37 °C until an OD600 nm of ∼ 0.6. Bacteria were induced with 0.5 mM isopropyl beta-D-1-thiogalactopyranoside (IPTG) for 16 h at 22 °C. Proteins with 15N and 13C labeled amino acids but without labeled alanine were produced by adding external alanine (22 mM) to the M9 medium 30 min before induction and then induced with 0.5 mM IPTG for 5 h at 37 °C. For the in vivo heme binding assay, hemin (1 mM) was added to M9 medium before the IPTG induction, and cells were grown for 16 h at 22 °C.
After growth and induction, the cells were lysed with sonication and then centrifuged at 15,000 × g. The clear cell lysates were applied to Ni-nitrilotriacetic acid (NTA) agarose resin (Qiagen) and incubated 2–3 h with constant mixing at 4 °C. Subsequently, the resin was thoroughly rinsed, and the purified protein was eluted with 0.15 M imidazole. The His-MBP tag was removed by adding 70 µg of recombinant His-TEV to 700 µg of purified protein and incubated overnight at 4 °C, leaving two additional amino acids (Gly-Ala) attached at the N terminus after cleavage. Numbering is based on Met at position 1. The overnight TEV-digested protein was passed through a 30-kDa Amicon filter, and the flow-through consisting of purified peptide samples was collected. The cleaved peptide was concentrated by using a 3-kDa Amicon filter and then further purified by using a MonoQ HR5/5 anionic exchange column. The amount of protein collected in the eluted fraction was measured by using the Bradford assay, and the collected samples were stored at −80 °C until further testing.
UV-vis Spectroscopy.
UV-vis spectra were recorded on a Gene Quant 1300 spectrophotometer (GE Healthcare) using quartz cells of 0.2- and 1.0-cm path lengths. Concentrations of WT and mutant peptides were 10 μM, and the experiments were carried out in the presence of 2 mM reduced GSH. Hemin incorporation was achieved by directly mixing hemin and peptide in 100 mM K-aspartate, 10 mM EGTA, 15 mM KCl, and 10 mM Hepes, pH 8.0 (with KOH). Aliquots of hemin (0.2–20 μM) were added to the sample cuvette at 25 °C, and spectra were recorded immediately after the addition of hemin. Difference spectra were obtained by subtracting hemin signals from those of the hemin–peptide samples.
CD.
CD experiments were performed on a JASCO 710 CD spectropolarimeter with 1-mm quartz cuvettes, operated by the Spectra Manager Software (both Jasco International). CD spectra were recorded in the range from 190–260 nm with a data pitch and bandwidth of 1 nm and a scan speed of 50 nm/min. The spectra shown are averages of seven readings, and the secondary structure parameters were calculated with the JASCO software according to Reed and Reed (21). For thermal denaturation, the temperature was controlled with a Peltier thermoelectric element.
Tryptophan Fluorescence Quenching.
Fluorescence measurements were performed at 20 °C with a JASCO-FP6500 fluorescence spectrometer. The intrinsic fluorescence of Pep61, Pep61-A23W, Pep61-C13S:H16A:A23W:H35A, l-tryptophan, and NATA (Sigma) was observed by exciting at 295 nm and taking emission spectra in the range of 320–380 nm. Fluorescence quenching with increasing concentrations of hemin was performed in 3 mL of 20 mM phosphate buffer with 0.1 µM GSH (pH 7.6) and 0.5 µM of the respective peptide; hemin, at final concentrations of 120–720 nM, was added from concentrated stock solutions such that the change in peptide concentration was negligible (≤0.2%).
EPR Analysis.
Samples of the recombinant Pep61 and the respective mutants were prepared in 20 mM Tris, pH 7.5, buffer with 40–50% (vol/vol) ethylene glycol at 25 °C. The protein solutions were transferred to 4-mm EPR quartz tubes (Wilmad) and quickly frozen in liquid nitrogen. EPR spectra were recorded on a Bruker Elexsys spectrometer operating in the continuous wave mode with a modulation amplitude of 0.9 mT and a frequency of 100 kHz. An Oxford 900 cryostat was used to control the temperature (5 K). The magnetic field and the microwave frequency were determined with an NMR gauss meter and a frequency counter (Hewlett Packard), respectively. Spectra were accumulated to yield a good signal-to-noise ratio. The background signals were eliminated by subtracting control spectra from buffer samples (without protein and hemin) measured under identical conditions. The apparent g factors were determined using the WINEPR or XeprView programs (Bruker). The ligand field parameters were calculated from the experimental g values using a method previously described (41–43). The obtained values are summarized in Table S1 together with representative values of relevant cysteine thiolate ligation of heme from the literature. The analysis of the axial ligation pattern at the low-spin ferric heme was performed with the derived ligand field parameters using a plot relating the rhombicity (|V/Δ|) to the tetragonal splitting (|Δ/λ|) (Fig. S1) (44, 45).
NMR Spectroscopy and Structure Modeling.
Uniformly [U-13C,15N]-labeled Pep61 for NMR spectroscopy was obtained by bacterial expression on minimal medium supplemented with 13C-glucose and 15NH4Cl using the protocol described above. Additionally, an alanine unlabeling strategy was used for fast identification of the HN resonances arising from the 19 alanines (constituting ∼30% of the total peptide sequence).
The NMR experiments were acquired at 283 K and a concentration of ∼150 µM, pH 7.4, 150 mM NaCl, 25 mM sodium phosphate buffer with 2 mM DTT on Bruker Avance III spectrometers (Bruker Biospin) with proton frequencies of 600 and 750 MHz equipped with cryogenic probes. The assignment of the 1H, 13C, and 15N resonances was performed as described previously (46, 47) using CcpNmr (48). Based on the [1H,15N]-HSQC spectrum (Fig. S4A), HNCACB and CC(CO)NH experiments were used for the identification of the amino acid type and to link the residues. By comparison of the [1H,13C]-HSQC data with HCCH-correlated spectroscopy (COSY) and HCCH-total correlated spectroscopy (TOCSY) spectra, a nearly complete resonance assignment was obtained. Figures were produced using University of California San Francisco (UCSF) Chimera (49). The chemical shift index was calculated according to Wishart and Sykes (50). The programs CS-Rosetta (51) and CHESHIRE (52) were used to screen for initial structural models based on the chemical shift assignments for the 1Hα, 1HN, 13Cα, 13Cβ, 13C', and 15N' atoms. The docking of the Pep61 model with heme was performed with the HADDOCK installation at the WeNMR grid server (23). The selected starting structure was used for all of the eight different docking scenarios. Final models (Fig. 6) were selected according to their HADDOCK scores (Table S2) and analyzed for their binding/coordination geometries.
Data Analysis.
Data were analyzed with FitMaster (HEKA Elektronik) and IgorPro (WaveMetrics). Averaged data are presented as means ± SEM (n = number of independent measurements) unless specified otherwise. Averaged data were compared with a two-sided Student t test, followed by Bonferroni-Holm correction as applicable. The resulting P values are specified.
Supplementary Material
Acknowledgments
We thank E. Leipold for help with initial CD measurements and K.-H. Gührs for mass spectrometry recordings. This work was funded by the Deutsche Forschungsgemeinschaft FOR 1738 (to T.H., S.H.H., and O.O.) and the National Institutes of Health (T.H.). The WeNMR project (European FP7 e-Infrastructure grant, Contract 261572, www.wenmr.eu), supported by the national GRID Initiatives of Belgium, France, Italy, Germany, Netherlands (via the Dutch BiG GRID project), Portugal, Spain, United Kingdom, South Africa, Taiwan, and the Latin America GRID infrastructure via the Gisela project is acknowledged for the use of web portals, computing, and storage facilities.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The chemical shift assignments have been deposited in the Biological Magnetic Resonance Data Bank (BMRB), www.bmrb.wisc.edu (accession no. 19426).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1313247110/-/DCSupplemental.
References
- 1.Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330(9):613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- 2.Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250(4980):533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- 3.Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+ channels: Effects of alterations in the carboxy-terminal region. Neuron. 1991;7(4):547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- 4.Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 1990;250(4980):568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]
- 5.Zhou M, Morais-Cabral JH, Mann S, MacKinnon R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 2001;411(6838):657–661. doi: 10.1038/35079500. [DOI] [PubMed] [Google Scholar]
- 6.Antz C, Fakler B. Fast inactivation of voltage-gated K+ channels: From cartoon to structure. News Physiol Sci. 1998;13:177–182. doi: 10.1152/physiologyonline.1998.13.4.177. [DOI] [PubMed] [Google Scholar]
- 7.Antz C, et al. Control of K+ channel gating by protein phosphorylation: Structural switches of the inactivation gate. Nat Struct Biol. 1999;6(2):146–150. doi: 10.1038/5833. [DOI] [PubMed] [Google Scholar]
- 8.Baker KA, et al. NMR-derived dynamic aspects of N-type inactivation of a Kv channel suggest a transient interaction with the T1 domain. Biochemistry. 2006;45(6):1663–1672. doi: 10.1021/bi0516430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prince-Carter A, Pfaffinger PJ. Multiple intermediate states precede pore block during N-type inactivation of a voltage-gated potassium channel. J Gen Physiol. 2009;134(1):15–34. doi: 10.1085/jgp.200910219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ruppersberg JP, et al. Regulation of fast inactivation of cloned mammalian IK(A) channels by cysteine oxidation. Nature. 1991;352(6337):711–714. doi: 10.1038/352711a0. [DOI] [PubMed] [Google Scholar]
- 11.Padanilam BJ, et al. Molecular determinants of intracellular pH modulation of human Kv1.4 N-type inactivation. Mol Pharmacol. 2002;62(1):127–134. doi: 10.1124/mol.62.1.127. [DOI] [PubMed] [Google Scholar]
- 12.Oliver D, et al. Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science. 2004;304(5668):265–270. doi: 10.1126/science.1094113. [DOI] [PubMed] [Google Scholar]
- 13.Roeper J, Lorra C, Pongs O. Frequency-dependent inactivation of mammalian A-type K+ channel KV1.4 regulated by Ca2+/calmodulin-dependent protein kinase. J Neurosci. 1997;17(10):3379–3391. doi: 10.1523/JNEUROSCI.17-10-03379.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu Q, et al. Disruption of Kv1.1 N-type inactivation by novel small molecule inhibitors (disinactivators) Bioorg Med Chem. 2008;16(6):3067–3075. doi: 10.1016/j.bmc.2007.12.031. [DOI] [PubMed] [Google Scholar]
- 15.Tsiftsoglou AS, Tsamadou AI, Papadopoulou LC. Heme as key regulator of major mammalian cellular functions: Molecular, cellular, and pharmacological aspects. Pharmacol Ther. 2006;111(2):327–345. doi: 10.1016/j.pharmthera.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 16.Tang XD, et al. Haem can bind to and inhibit mammalian calcium-dependent Slo1 BK channels. Nature. 2003;425(6957):531–535. doi: 10.1038/nature02003. [DOI] [PubMed] [Google Scholar]
- 17.Wang S, Publicover S, Gu Y. An oxygen-sensitive mechanism in regulation of epithelial sodium channel. Proc Natl Acad Sci USA. 2009;106(8):2957–2962. doi: 10.1073/pnas.0809100106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horrigan FT, Heinemann SH, Hoshi T. Heme regulates allosteric activation of the Slo1 BK channel. J Gen Physiol. 2005;126(1):7–21. doi: 10.1085/jgp.200509262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaggar JH, et al. Heme is a carbon monoxide receptor for large-conductance Ca2+-activated K+ channels. Circ Res. 2005;97(8):805–812. doi: 10.1161/01.RES.0000186180.47148.7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heinemann SH, Rettig J, Graack HR, Pongs O. Functional characterization of Kv channel β-subunits from rat brain. J Physiol. 1996;493(Pt 3):625–633. doi: 10.1113/jphysiol.1996.sp021409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reed J, Reed TA. A set of constructed type spectra for the practical estimation of peptide secondary structure from circular dichroism. Anal Biochem. 1997;254(1):36–40. doi: 10.1006/abio.1997.2355. [DOI] [PubMed] [Google Scholar]
- 22.Wissmann R, et al. Solution structure and function of the “tandem inactivation domain” of the neuronal A-type potassium channel Kv1.4. J Biol Chem. 2003;278(18):16142–16150. doi: 10.1074/jbc.M210191200. [DOI] [PubMed] [Google Scholar]
- 23.de Vries SJ, van Dijk M, Bonvin AMJJ. The HADDOCK web server for data-driven biomolecular docking. Nat Protoc. 2010;5(5):883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- 24.Trakshel GM, Kutty RK, Maines MD. Purification and characterization of the major constitutive form of testicular heme oxygenase. The noninducible isoform. J Biol Chem. 1986;261(24):11131–11137. [PubMed] [Google Scholar]
- 25.Deniau C, et al. Thermodynamics of heme binding to the HasA(SM) hemophore: Effect of mutations at three key residues for heme uptake. Biochemistry. 2003;42(36):10627–10633. doi: 10.1021/bi030015k. [DOI] [PubMed] [Google Scholar]
- 26.Miller YI, Shaklai N. Kinetics of hemin distribution in plasma reveals its role in lipoprotein oxidation. Biochim Biophys Acta. 1999;1454(2):153–164. doi: 10.1016/s0925-4439(99)00027-7. [DOI] [PubMed] [Google Scholar]
- 27.Robinson CR, Liu Y, Thomson JA, Sturtevant JM, Sligar SG. Energetics of heme binding to native and denatured states of cytochrome b562. Biochemistry. 1997;36(51):16141–16146. doi: 10.1021/bi971470h. [DOI] [PubMed] [Google Scholar]
- 28.Gattoni M, Boffi A, Sarti P, Chiancone E. Stability of the heme-globin linkage in alphabeta dimers and isolated chains of human hemoglobin. A study of the heme transfer reaction from the immobilized proteins to albumin. J Biol Chem. 1996;271(17):10130–10136. doi: 10.1074/jbc.271.17.10130. [DOI] [PubMed] [Google Scholar]
- 29.Ogawa K, et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 2001;20(11):2835–2843. doi: 10.1093/emboj/20.11.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wagner KR, Dwyer BE. Hematoma removal, heme, and heme oxygenase following hemorrhagic stroke. Ann N Y Acad Sci. 2004;1012:237–251. doi: 10.1196/annals.1306.020. [DOI] [PubMed] [Google Scholar]
- 31.Binzen U, et al. Co-expression of the voltage-gated potassium channel Kv1.4 with transient receptor potential channels (TRPV1 and TRPV2) and the cannabinoid receptor CB1 in rat dorsal root ganglion neurons. Neuroscience. 2006;142(2):527–539. doi: 10.1016/j.neuroscience.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 32.Schulte U, et al. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron. 2006;49(5):697–706. doi: 10.1016/j.neuron.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 33.Geiger JRP, Jonas P. Dynamic control of presynaptic Ca(2+) inflow by fast-inactivating K(+) channels in hippocampal mossy fiber boutons. Neuron. 2000;28(3):927–939. doi: 10.1016/s0896-6273(00)00164-1. [DOI] [PubMed] [Google Scholar]
- 34.Xu L, Enyeart JA, Enyeart JJ. Neuroprotective agent riluzole dramatically slows inactivation of Kv1.4 potassium channels by a voltage-dependent oxidative mechanism. J Pharmacol Exp Ther. 2001;299(1):227–237. [PubMed] [Google Scholar]
- 35.Takeda M, et al. Potassium channels as a potential therapeutic target for trigeminal neuropathic and inflammatory pain. Mol Pain. 2011;7:5. doi: 10.1186/1744-8069-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Antz C, et al. NMR structure of inactivation gates from mammalian voltage-dependent potassium channels. Nature. 1997;385(6613):272–275. doi: 10.1038/385272a0. [DOI] [PubMed] [Google Scholar]
- 37.Fernandez-Ballester G, et al. Adoption of β structure by the inactivating “ball” peptide of the Shaker B potassium channel. Biophys J. 1995;68(3):858–865. doi: 10.1016/S0006-3495(95)80262-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abbott GW, Mercer EA, Miller RT, Ramesh B, Srai SK. Conformational changes in a mammalian voltage-dependent potassium channel inactivation peptide. Biochemistry. 1998;37(6):1640–1645. doi: 10.1021/bi972350c. [DOI] [PubMed] [Google Scholar]
- 39.Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309(5736):897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 40.Schönherr R, Löber K, Heinemann SH. Inhibition of human ether à go-go potassium channels by Ca(2+)/calmodulin. EMBO J. 2000;19(13):3263–3271. doi: 10.1093/emboj/19.13.3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taylor CPS. The EPR of low spin heme complexes. Relation of the t2g hole model to the directional properties of the g tensor, and a new method for calculating the ligand field parameters. Biochim Biophys Acta. 1977;491(1):137–148. doi: 10.1016/0005-2795(77)90049-6. [DOI] [PubMed] [Google Scholar]
- 42.Bohan TL. Analysis of low-spin ESR spectra of ferric heme proteins: A reexamination. J Magn Reson. 1977;26(1):109–118. [Google Scholar]
- 43. Palmer G (2000) Physical Methods in Bioinorganic Chemistry: Spectroscopy and Magnetism, ed Que, Jr L (University Science Books, Sausalito, CA), pp 121–185.
- 44.Blumberg WE, Peisach J. Low-spin compounds of heme proteins. Adv Chem Ser. 1971;100:271–291. [Google Scholar]
- 45.Lever ABP, Gray HB. Iron Porphryins, Part Two. Reading, MA: Addison-Wesley Publishing; 1983. pp. 55–62. [Google Scholar]
- 46.Schlott B, et al. Interaction of Kazal-type inhibitor domains with serine proteinases: Biochemical and structural studies. J Mol Biol. 2002;318(2):533–546. doi: 10.1016/S0022-2836(02)00014-1. [DOI] [PubMed] [Google Scholar]
- 47.Ohlenschläger O, et al. NMR secondary structure of the plasminogen activator protein staphylokinase. J Biomol NMR. 1997;9(3):273–286. doi: 10.1023/a:1018678925512. [DOI] [PubMed] [Google Scholar]
- 48.Vranken WF, et al. The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins. 2005;59(4):687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 49.Pettersen EF, et al. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 50.Wishart DS, Sykes BD. The 13C chemical-shift index: A simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR. 1994;4(2):171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 51.Shen Y, et al. Consistent blind protein structure generation from NMR chemical shift data. Proc Natl Acad Sci USA. 2008;105(12):4685–4690. doi: 10.1073/pnas.0800256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cavalli A, Salvatella X, Dobson CM, Vendruscolo M. Protein structure determination from NMR chemical shifts. Proc Natl Acad Sci USA. 2007;104(23):9615–9620. doi: 10.1073/pnas.0610313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.