

Significance
This study reports the cloning of a tomato gene, SlKLUH, that controls fruit mass by increased cell layers and delayed fruit ripening. In addition, we identified a potential regulatory SNP in the promoter of SlKLUH that is significantly associated with the fruit mass. Altogether, our study encompasses several genetic analyses, as well as association mapping, plant transformation experiments, and phenotypic evaluations to offer insights into the molecular basis of the regulation of tomato fruit mass, a critical trait in the domestication of fruit and vegetable crops.
Abstract
Domestication of crop plants had effects on human lifestyle and agriculture. However, little is known about the underlying molecular mechanisms accompanying the changes in fruit appearance as a consequence of selection by early farmers. We report the fine mapping and cloning of a tomato (Solanum lycopersicum) fruit mass gene encoding the ortholog of KLUH, SlKLUH, a P450 enzyme of the CYP78A subfamily. The increase in fruit mass is predominantly the result of enlarged pericarp and septum tissues caused by increased cell number in the large fruited lines. SlKLUH also modulates plant architecture by regulating number and length of the side shoots, and ripening time, and these effects are particularly strong in plants that transgenically down-regulate SlKLUH expression carrying fruits of a dramatically reduced mass. Association mapping followed by segregation analyses revealed that a single nucleotide polymorphism in the promoter of the gene is highly associated with fruit mass. This single polymorphism may potentially underlie a regulatory mutation resulting in increased SlKLUH expression concomitant with increased fruit mass. Our findings suggest that the allele giving rise to large fruit arose in the early domesticates of tomato and becoming progressively more abundant upon further selections. We also detected association of fruit weight with CaKLUH in chile pepper (Capsicum annuum) suggesting that selection of the orthologous gene may have occurred independently in a separate domestication event. Altogether, our findings shed light on the molecular basis of fruit mass, a key domestication trait in tomato and other fruit and vegetable crops.
Plant domestication and artificial selection led to improved agricultural production resulting from dramatic increases in fruit and seed weight (1). At the start of the Neolithic era ∼10,000 y ago, domestication of animals and plants accompanied the change in lifestyle from hunter-gatherer to a farming routine (2, 3). Driven by the selection of alleles from wild relatives and those that arose after the initial domestication events, characters associated with the domestication syndrome such as larger fruit and seed are typically differentiating wild from cultivated forms (4). Genome-wide genetic diversity analyses support the notion that the initial domestication of tomato was situated in Northern Peru and Ecuador (5). Selections from the red-fruited wild relative Solanum pimpinellifolium L. evolved into the semidomesticated Solanum lycopersicum L. var cerasiforme (hereafter referred to as S. l. cerasiforme) bearing fruit of small to medium weight. S. l. cerasiforme was further domesticated in Mexico giving rise to the large fruited tomato Solanum lycopersicum var lycopersicum (hereafter referred to as S. l. lycopersicum), which to date is cultivated throughout the world (5).
Tomato is an agriculturally important vegetable crop and is used as model for fruit development including ripening and morphological studies (6–8). Genetic studies have identified several quantitative trait loci (QTLs) associated with fruit mass in tomato, of which six loci [fruit weight1.1 (fw1.1), fw2.2, fw2.3, fw3.1/fw3.2, fw4.1, and fw9.1] are postulated to be major QTLs (9). The only cloned fruit mass gene from vegetable or fruit crops is FW2.2/Cell Number Regulator (CNR) (10). The gene encodes a negative regulator of cell division and controls tomato fruit mass as well as organ size in other species, e.g., maize (10, 11) and nitrogen-fixing nodule number (12). fw3.2 is the second major tomato fruit mass QTL, which explained 19% of the phenotypic variance in a F2 population derived from a cross between cultivated tomato accession “Yellow Stuffer” and wild tomato accession “LA1589” (13, 14). Despite the importance of fruit mass in the evolution of fruit and vegetable plants, and the numerous genetic loci that underlie the trait (7, 9, 15–19), cloning of domestication genes of fruit and vegetable crops has lagged behind that of the cereal crops. Therefore, insights into the molecular mechanisms that led to the transition of the fruit from small to large remain largely unknown.
Our current study focuses on fine mapping of a tomato fruit mass locus fw3.2 and cloning of the underlying gene. Association mapping, segregation analysis, and transgenic studies led us to identify the putative molecular basis of fruit weight at this locus and a likely regulatory SNP in the promoter of the gene that is highly associated with fruit mass. Phenotypic evaluations demonstrated the cellular basis of increased fruit mass and pleiotropic effects associated with the locus. We also investigated the likely origin of the derived allele and the potential role of this gene in the regulation of fruit mass in other crop species.
Results
fw3.2 Regulates Fruit Mass, Delays Ripening, and Modulates Plant Architecture.
To elucidate whether fruit mass differences were determined during flower or fruit development, we evaluated the ovary size at anthesis. Evaluations of the nearly isogenic lines (NILs) that differ for the allele at fw3.2 showed that the ovary perimeter at anthesis was similar, whereas the mature fruit perimeter was significantly different. Further analyses revealed that in particular the pericarp and septum areas were significantly larger in the large fruited NIL fw3.2(ys) (Table 1 and Fig. S1 A and B). The pericarp of the mature fruit carrying the large fruit allele fw3.2(ys) showed an increase in cell number, whereas cell size remained the same (Table 1 and Fig. S1 C and D). The time from anthesis to ripe fruit was prolonged in the fw3.2(ys) NIL (Table 1). In addition to fruit, fw3.2(ys) increased seed mass, although seed number was not significantly different (Table S1). Number of fruits were significantly higher in the lines carrying the small fruited fw3.2(wt) allele, whereas yield per plant was the same (Table 2). Additionally, fw3.2(wt) NIL contained more inflorescences and side shoots, including increased side shoot length (Table 2 and Table S1). The number of flowers per inflorescence remained the same. Retaining only 12 fruits per plant led to increased fruit weight in both NILs (Table S1) demonstrating that the increase in fruit mass controlled by fw3.2 was not due to changes in source–sink relationships. Thus, the results demonstrate that increases in fruit mass coincide with a reduction of number of fruit per plant caused by a reduction in side shoot number and length yielding fewer inflorescences.
Table 1.
Effect of fw3.2 on fruit attributes and ripening
| Measurements | fw3.2(ys) | fw3.2(wt) | P value | |
| Perimeter, cm | ||||
| Ovary | 0.61 ± 0.05 | 0.61 ± 0.06 | 0.75900 | |
| Mature fruit | 19.56 ± 0.51 | 16.68 ± 0.30 | 9.6E-08 | |
| Area of mature fruit parts, cm2 | ||||
| Columella and Placenta | 3.05 ± 0.35 | 2.65 ± 0.19 | 0.01475 | |
| Septum | 2.62 ± 0.34 | 1.40 ± 0.23 | 1.5E-05 | |
| Pericarp | 11.48 ± 0.47 | 8.36 ± 0.36 | 1.6E-07 | |
| Pericarp cell size, µm | ||||
| Ovary | 7.23 ± 0.62 | 7.64 ± 0.93 | 0.32222 | |
| Mature fruit | 616.67 ± 68.12 | 624.63 ± 54.65 | 0.74632 | |
| Pericarp cell number | ||||
| Ovary | 9.04 ± 0.52 | 9.26 ± 0.49 | 0.40223 | |
| Mature fruit | 19.20 ± 1.75 | 17.07 ± 0.93 | 0.00458 | |
| Fruit ripening, days | ||||
| Anthesis to orange | 48.87 ± 1.06 | 43.14 ± 0.38 | 8.1E-08 | |
| Anthesis to red | 53.44 ± 0.97 | 47.27 ± 1.11 | 3.2E-08 | |
Data were taken from 6 to 10 plants per genotype and are given as mean ± SD.
Table 2.
Effect of fw3.2 on yield parameters
| Yield parameters | fw3.2(ys) | fw3.2(wt) | P value | |
| Plant height, cm | ||||
| At 35 das | 6.00 ± 1.94 | 5.50 ± 1.65 | 0.55705 | |
| At 56 das | 59.00 ± 10.25 | 54.20 ± 8.79 | 0.12792 | |
| No. of nodes | ||||
| At 35 das | 7.10 ± 1.20 | 7.90 ± 0.74 | 0.15266 | |
| At 56 das | 17.90 ± 1.79 | 19.40 ± 1.07 | 0.07138 | |
| No. of side shoots | ||||
| At 56 das | 7.11 ± 1.36 | 10.80 ± 1.23 | 1.3E-05 | |
| At 70 das | 9.67 ± 1.66 | 12.90 ± 0.99 | 0.00022 | |
| Total side shoot length, cm | ||||
| At 56 das | 116.25 ± 42.47 | 177.85 ± 41.93 | 0.01761 | |
| At 70 das | 283.60 + 65.24 | 405.95 ± 50.14 | 0.00346 | |
| No. of fruits | ||||
| Red fruits | 175.30 ± 42.28 | 257.90 ± 77.70 | 0.01582 | |
| Green fruits | 163.90 ± 40.06 | 209.40 ± 57.46 | 0.01036 | |
| Total fruits | 339.20 ± 71.19 | 467.30 ± 126.60 | 0.00600 | |
| Total fruit weight per plant, kg | ||||
| Red fruits | 10.17 ± 3.10 | 10.44 ± 3.87 | 0.85921 | |
| Green fruits | 5.66 ± 1.88 | 5.00 ± 1.69 | 0.20173 | |
| Total yield | 15.83 ± 4.56 | 15.44 ± 5.37 | 0.83032 | |
Data were collected from 10 plants per genotype and are given as mean ± SD das, days after sowing.
Fine Mapping Delimited the fw3.2 Locus to a 24.4-kb Region.
Previously, the fw3.2 locus was fine mapped to a 51.4-kb region comprised of seven candidate genes (14). An additional recombinant screen delimited the locus to a 24.4-kb region comprised of three candidate proteins: a cytochrome P450 (ORF6) belonging to CYP78A subfamily, an ABC transporter (ORF7) distantly related to PGP transporters, and a Kelch domain-containing protein of unknown function (ORF8). The fine-mapping experiment excluded most of the ORF8 gene to only include the promoter, first exon, and first intron until the NDF9 marker (Fig. 1 and Table S2). Gene action analysis revealed the additive nature of the fw3.2 alleles, suggesting that neither allele is a null (Table S2).
Fig. 1.
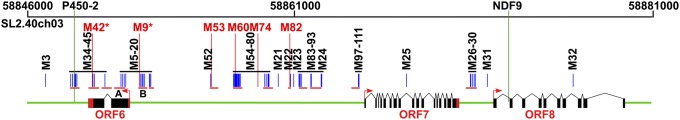
Genome structure of the fw3.2 locus and association mapping of the polymorphisms. UTRs, exons, introns, and intergenic regions are represented with red boxes, black boxes, black lines, and green lines, respectively. The direction of transcription is denoted with red arrows. Blue vertical lines represent polymorphisms not associated with fruit mass; red vertical lines represent polymorphisms significantly associated with fruit mass. Two highly significant SNPs are represented with an asterisk. Red horizontal lines depict the sequenced regions. Two markers, P450-2 and NDF9, which delimited the fw3.2 locus to 24.4-kb region, are shown with green vertical lines.
Association Mapping and Genetic Segregation Analysis Identified a Potential Regulatory SNP in the Promoter of ORF6.
To further investigate which of the candidate genes may underlie the fw3.2 locus, we sought to identify the underlying nucleotide polymorphisms that were correlated with larger fruit. We conducted an association mapping study using a core collection of tomato genotypes demonstrated to have broad genetic diversity within red-fruited tomatoes and comprised of domesticated S. l. lycopersicum, the wild relative S. pimpinellifolium and semidomesticated S. l. cerasiforme accessions (20). We sequenced ∼7 kb of the fw3.2 locus and genotyped 115 markers inside and outside the fw3.2 region spanning 48.5 kb (Fig. 1 and Dataset S1). Accounting for kinship and population structure, we identified six SNPs that were significantly associated with fruit mass (Fig. 1 and Table S3). With the exception of one SNP, all were located in the upstream region of ORF6 and ORF7. The two most significant SNPs were found in or near ORF6, 512 bp upstream of the start of transcription (M9) and 72 bp from the stop codon in the 3′UTR (M42) (Fig. 1). Successful association mapping typically requires rapid linkage disequilibrium (LD) decay. However, low rates of LD decay were found around the fw3.2 locus, suggesting few natural recombination events in this region that would potentially compromise the interpretation of the association mapping results (21). Therefore, to determine the validity of the association of these SNPs to fruit mass, we performed intraspecific segregation analyses using populations derived from parents that showed few polymorphisms including M9 and three to five additional SNPs (Dataset S1 and Table S4). Genetic analyses using the F2 and BC1F2 populations showed segregation for fruit mass with the allele of M9 in all families in either one or both populations (Table S4). Importantly, family 12S74 segregated for M9 but not for M42 and showed that fruit mass segregated with the allele of M9 (Dataset S1 and Table S4). This finding demonstrated that the M9 SNP was more critical in regulating fruit mass than M42, lending support for the notion that the ORF6 promoter SNP may regulate fruit mass differences at fw3.2.
Down-Regulation of ORF6 Leads to Reduced Fruit Mass.
To further investigate the role of the candidate genes in regulating fruit mass, we down-regulated transcript accumulation of ORF6 and ORF7. Because expression of ORF6 and ORF7 was lower in the lines that carried small fruit (Fig. 2A), the constructs were transformed into the fw3.2(ys) NILs typically yielding large fruit. Down-regulation of ORF6 transcript levels dramatically reduced fruit and seed mass (Fig. 2 B–E and Table S5). Similar to the NILs, down-regulation of ORF6 led to a higher number of side shoots and faster ripening (Table S5). The transgenic lines showed additional defects compared with the NILs (e.g., reduced plant height, smaller leaves and leaflets, and severely reduced seed number), indicating that extensive down-regulation of ORF6 led to phenotypes affecting the entire plant (Table S5). The RNAi-2 lines targeting the coding region led to less severe phenotypes, including slightly larger fruit with viable seeds. Even though overall seed number was significantly reduced, fruit with 50–100 seeds showed reduced fruit weight compared with the control with the same number of seed (Fig. S1H). Thus, when accounting for seed number, fruit mass is still reduced in lines that down-regulate ORF6. Contrary to ORF6, down-regulation of ORF7 using four amiRNA constructs did not show a correlation between reduction in fruit mass and transcript level (see Table S7 and Fig. S1I), and the plants were indistinguishable from the fw3.2(ys) NIL. Thus, it is unlikely that ORF7 underlies the fw3.2 locus. In light of these findings, we assumed that ORF6 underlies the fw3.2 locus and renamed it SlKLUH after one of the founding members of this subfamily of P540s (22). Expression of SlKLUH was the highest in vegetative meristems and in the young flower buds (Fig. 2F). Compared with SlKLUH, the expression of SlFW2.2/CNR was much lower in most tissues examined and differed in tissue specificity. Also, expression of SlKLUH was very high in developing seed and not in the growing pericarp, contrary to SlFW2.2/CNR (Fig. 2G).
Fig. 2.

Transcript accumulation and effect of SlKLUH knock down on fruit and seed mass. (A) Expression of ORF6 (SlKLUH) and ORF7 in fw3.2(ys) and fw3.2(wt) NILs. (B) Regions of SlKLUH targeted by two RNAi constructs. (C and D) Effect of SlKLUH knock down on fruit and seed mass, respectively. (E) Transcript accumulation of SlKLUH in fw3.2(ys) and fw3.2(wt) NILs and SlKLUH knock down lines. (F) Average transcript accumulation of SlKLUH and SlFW2.2/CNR in different tissues of S. pimpinellifolium LA1589. (G) Average transcript accumulation of SlKLUH and SlFW2.2/CNR in S. l. lycopersicum pericarp and developing seeds at different fruit development stages. dpa, days postanthesis; P, pericarp; S, seed; Veg. meristem, vegetative meristem.
Molecular Diversity and Phylogenetic Analysis of SlKLUH.
To examine the evolutionary history of the fw3.2 locus, DNA sequence variation of the segments spanning part of the coding region (fragment A) and promoter (fragment B) of SlKLUH were investigated. The SlKLUH region showed reduced nucleotide diversity (π) in S. lycopersicum (0 and 0.000195 for fragment A and B, respectively) compared with S. pimpinellifolium (0.002874 and 0.004962) and S. l. cerasiforme (0.003203 and 0.004364) (Fig. 3A). The ratio of nucleotide diversity of S. l. lycopersicum to S. pimpinellifolium at SlKLUH was significantly lower compared with the average diversity for chromosome 3 [percentage of πS. lycopersicum/πS.pimpinellifolium for fragment A, B, and chromosome 3 are 0%, 3.9%, and 56.9%, respectively]. Tajima’s D analysis of all accessions combined showed significant values for parts of the two sequenced fragments, ranging from −2.0851 to −1.5187 and from −2.1922 to −1.9117 for fragment A and B, respectively, compared with the average Tajima’s D for chromosome 3 (−1.3247) (Fig. 3A). The findings of the molecular diversity analysis were also supported by the results from the phylogenetic analysis using the SlKLUH gene sequences. One cluster consisted of all S. pimpinellifolium and several S. l. cerasiforme accessions and displayed relatively high sequence diversity. The other cluster is comprised of the S. l. lycopersicum and the remaining S. l. cerasiforme accessions and showed limited genetic diversity (Fig. 3B). The derived allele designated by the M9 SNP was only found in a single subclade with some S. l. cerasiforme and all of the cultivated tomato accessions, suggesting the mutation arose in the S. l. cerasiforme background.
Fig. 3.

Molecular diversity and phylogenetic analysis of SlKLUH, and origin of M9 mutation. (A) Nucleotide diversity (π) and Tajima’s D were calculated in fragment A and B spanning part of the coding region and promoter of SlKLUH (Fig.1). The Tajima’s D is calculated from the SlKLUH sequences of S. pimpinellifolium (S. p), S. l. cerasiforme (S. l. c), and S . lycopersicum lycopersicum (S. l. l) combined. (B) Phylogenetic analysis of SlKLUH. S. l. l, S. l. c, S. p, and Solanum pennellii are depicted in red, black, green, and blue, respectively. Large (>60 g), medium (>10 g and <60 g), and small (<10 g) fruit-bearing lines are represented as large, medium, and no dots after their name, respectively. Accessions carrying the M9 mutation are depicted with an asterisk (*). (C) Frequency of derived and ancestral M9 SNP allele in tomato subpopulations. Imp, improved accessions; Ecu, Ecuador; Mes, Mesoamerica; N.Per, Northern Peru; Per, Peru.
Possible Origin of the M9 Mutation and Identification of Fruit Mass QTL Overlapping with fw3.2 in Chile Pepper.
To further explore where the derived allele arose, we genotyped the M9 SNP in a tomato collection that covers the proposed trajectory of tomato domestication (5). The M9 SNP associated with increased fruit mass was already present at ∼30% frequency in the Ecuadorian and Northern Peruvian S. l. cerasiforme that were closely related to the wild progenitor S. pimpinellifolium. The allele frequency increased slightly in the Mesoamerican S. l. lycopersicum accessions, and increased further in the S. l. lycopersicum landraces from Europe and elsewhere to become practically fixed in modern accessions (Fig. 3C and Table S6). This finding suggested that even though the derived allele arose early, selections for larger fruit weight may not have been as critical or this allele was not selected early and only became relevant in recent times.
Based on coinciding QTLs, it has been proposed that orthologous genes may have been selected independently in different crops (7, 23). To evaluate whether chile pepper (Capsicum annuum) may harbor a fruit mass QTL at a region that is syntenic to tomato fw3.2, we determined whether the pepper ortholog CaKLUH was associated with fruit mass in this species. A population of 106 F9 recombinant inbred lines (RILs) derived from a cross between a large-fruited pepper and a small-fruited wild relative Capsicum frutescens showed fruit mass segregated significantly with the large fruited allele of KLUH from the C. annuum parent [dry fruit weight (mean ± SD, g) for groups carrying the C. frutescens and the C. annuum allele of fw3.2 are 3.21 ± 0.36 and 4.99 ± 0.34, respectively; P value < 0.0005]. These findings demonstrate that fw3.2 QTL is found in both tomato and chile pepper, and that KLUH may regulate fruit mass in two independently domesticated species.
Discussion
SlKLUH Is Most Likely Underlying fw3.2, and a Critical SNP in Its Promoter Is Proposed to Regulate Fruit Mass.
ORF6 encodes a protein with the highest similarity to the Arabidopsis KLUH/CYP78A5 protein controlling vegetative and reproductive organ size, and also seed size (22, 24). Other members of this subfamily of cytochrome P450 also control organ and plant size, e.g., CYP78A9, CYP78A6 in Arabidopsis, and CYP78A11 in rice (25–27). Two additional members of this subfamily, CYP78A27 and CYP78A28, control colony size in moss (28). This highly conserved function of CYP78A subfamily in regulating organ size led us to postulate that ORF6 was the most likely candidate gene for the fw3.2 QTL.
The first experiment to test this hypothesis was to identify SNPs that were significantly associated with fruit mass. Our expectations were that these SNPs would be found near the most likely candidate gene. The most significant SNPs identified from the association mapping were the M9 SNP located 512 bp upstream of the transcription start site and the M42 SNP located in the 3′ UTR at 72 bp downstream from the stop codon of ORF6. However, LD decay rate was low (21), which was likely due to the lack of outcrossing and recombination events during domestication of tomato. However, when the M9 polymorphism arose, its effect on fruit mass could have been of functional significance. This pattern of few SNPs in large LD blocks has also been found in human (29–32) and may be common in plants as well. The low LD yet significant association of the SNP with fruit mass led us to conduct segregation analyses; these analyses excluded the importance of M42, leaving M9 as the most significantly associated SNP of potential functional relevance (i.e., the most likely to regulate fruit mass). Interestingly, the M9 SNP was located within the third repeat of four 30-bp tandem repeats, with each repeat sharing 83–100% homology with its consensus sequence. The tandem repeat was located 440 bp to 559 bp upstream from the SlKLUH transcription start site. Moreover, M9 mutation is located within a putative cis element, related to the organ-specific element (OSE) found in nodulin and leghemoglobin genes in soybean and Vicia faba (33–35). In the future, it will be interesting to assess the role of this putative cis-element in gene expression and to explore whether the tandem repeat has any bearing on the modulation of SlKLUH expression. Even though the M9 SNP appears most promising to regulate fruit mass, we cannot exclude the roles of three other SNPs (M60, M74, and M82) in the regulation of fruit mass, and additional SNPs that may be found in the remaining regions that were not sequenced cannot be excluded either.
The second experiment was to transgenically down-regulate the expression of the candidate genes, ORF6 and ORF7. Our expectation was that expression levels positively correlate with fruit mass since the fw3.2(wt) NIL exhibit smaller fruit and lower transcript levels. Reduced expression of ORF6, and not of ORF7, had pronounced effects on fruit mass, seed mass, side shoot number, and fruit ripening, which were traits that were also significantly altered in the NILs. The severity of these phenotypes followed a gradient from fw3.2(ys), fw3.2(wt) to the RNAi line. Thus, fine mapping, association mapping, and functional segregation analyses of a highly significant SNP in the promoter of ORF6 combined with the plant transformation results and phenotypic evaluations strongly implied that ORF6 underlies the fw3.2 locus. The expression of ORF6, which we renamed SlKLUH, was particularly high in the developing seeds. However, the effect of increased fruit mass was found in the pericarp of the maternal tissues. Thus, it is plausible that a seed-derived signal might play a role in regulation of fruit mass by SlKLUH. A similar hypothesis has been proposed to explain the action of Arabidopsis KLUH (22).
The Role of SlKLUH in Regulating Fruit Mass, Ripening, and Plant Architecture.
Fruit development starts with the fertilization of the ovules in the ovary at the time of anthesis (flower opening) leading to the initiation of seed development. Immediately following fertilization, the fruit undergoes a short period of cell division, which is followed by a longer period of cell expansion until the final dimensions are reached (36–39). The increase in fruit mass was due to growth processes taking place after fertilization, particularly of the pericarp and septum tissues. These findings showed that the extra cell divisions led to enlarged fruit and a concomitant delay in ripening. Therefore, the delay in ripening was likely the result of an extension of the cell division stage resulting from increased expression. SlFW2.2/CNR also controls fruit mass by extending the cell division period (40). Thus, the increases in fruit mass resulting from domestication may operate on similar processes during fruit development yet through different mechanisms based on putative gene function. The lengths of other plant organs were not affected in the NILs, suggesting a specific role of the natural alleles of SlKLUH in the regulation of fruit and seed mass. Our results also indicated that plant architecture was affected, implying a pleiotropic effect of SlKLUH on plant growth and development.
Diversity of SlKLUH and Origin of M9 Mutation.
Our findings indicate reduced nucleotide diversity in S. l. lycopersicum in the SlKLUH region compared with S. pimpinellifolium, and an overall reduced diversity compared with the entire chromosome. Parts of two sequenced regions around SlKLUH showed significant Tajima’s D values, which may suggest an excess of low frequency SNPs in certain regions of the gene in tomato, which is potentially the result of a population expansion after a bottleneck or a selective sweep. Taken together, these results support a selective pressure around the fw3.2 region, but this selection is likely to have predated domestication because reduction is found in both S. pimpinellifolium and S. l. lycopersicum. Exploration of possible origin of M9 mutation using a wide array of tomato accessions showed that the increase in the derived allele frequency followed the same path as the proposed domestication of tomato and was consistent with the selection for a larger fruit along this process (5). However, the allele did not become fixed early during domestication of tomato, further supporting the notion that this region may not have been under strong selection during domestication. Even though the derived M9 allele was detected in low frequency in the Peruvian S. pimpinellifolium, without further study we cannot ascertain the validity of this finding and whether the allele may have originated in a wild relative.
Conclusion
To date, only one gene underlying a fruit mass QTL is known (10). Our study identified SlKLUH as the second major gene controlling fruit mass in a vegetable or fruit crop, shedding light on the molecular regulation of this trait in plants. Additionally, previous studies had shown an overlap of the fw2.2/cnr QTL in chile pepper, albeit that the effect of the pepper QTL was minor (7, 41), whereas the existence of a pepper fw3.2 QTL was less certain. The findings from this study raise the possibility that KLUH may regulate fruit mass in chile pepper and potentially other crops.
Materials and Methods
Plant Materials and Phenotypic Evaluation.
Plants were grown under field and greenhouse condition at Ohio Agricultural Research and Development Center (Wooster, OH) in 2010–2012, and a subset in Gainesville, FL, in 2011. Most experiments were conducted with 10 plants for fw3.2(ys) and fw3.2(wt) NILs and multiple independent lines for the ORF6_RNAi and ORF7_amiRNA constructs. Fruit and seed mass analyses in fw3.2 NILs, ORF6_RNAi and ORF7_amiRNA lines were performed with 20 ripe fruits per plant, unless fruit set was impaired. Side shoot number and lengths were measured at 56 and 70 d after sowing, whereas plant height and number of nodes were recorded at 35 and 56 d after sowing. Leaf attributes were measured on 12th and 13th leaves. Floral organ measurements were performed with five flowers per plant. For cell measurement of the ovaries at anthesis, three ovaries per plant and three sections per ovary were analyzed. For cell measurements at mature fruit stage, two fruits per plant and two sections per fruit were analyzed. To test source–sink relationship, a total of 12 fruits per plant were kept and fruit weight from these plants was compared with control plants with no fruit removal. All phenotypic evaluations were performed independently at least twice, except the fruit removal experiment, which was done once.
Whole Fruit, Pericarp, and Cell Measurements.
Mature fruit perimeter, area of pericarp, septum, columella, and placenta were analyzed using Tomato Analyzer (version 2.3) (42). For the cell measurements in the pericarp, thick sections were made with a razor blade, stained with 0.5% Toluidine Blue in 0.1% Sodium Carbonate solution (SPI, Electron Microscopy Supplies), and photographed with a Leica MZFLIII dissecting microscope coupled to a digital camera (SPOT RT KE, Diagnostic Instruments), followed by analysis with ImageJ software. For cell measurements of the ovary at anthesis, the tissue was fixed in 0.1 M phosphate buffer (pH 7.2) containing 3% (wt/vol) glutaraldehyde and 2% (wt/vol) paraformaldehyde. Samples were dehydrated in 25–90% ethanol series, followed by infiltration in 50–100% (vol/vol) LR White resin (Electron Microscopy Supplies) gradient at room temperature. Samples were embedded in 100% LR White for 24 h at 55 °C. For each ovary, 5-µm transverse sections were stained and photographed and cell measurements were similar as described for the pericarp cell measurements.
Fine Mapping, Gene Action, and Genetic Segregation Analysis.
To identify additional recombinants, 178 seedlings of 09S69-68 line were screened, resulting in recombinant line 10S187-82. The progeny testing of the other line was from a previous study (14). For the genetic segregation analysis of the M9 SNP, we developed five F2 and BC1F2 populations each that were derived from parents from the core collection showing few polymorphisms in addition to M9. Alleles for the other known fruit weight/shape genes that were segregating in the populations were fixed such that only fw3.2 was segregating. For the progeny testing and segregation analysis, 10–13 plants per genotype [genotypes carrying homozygous fw3.2(ys) and homozygous fw3.2(wt) alleles] were grown and 20 ripe fruits representing the average per plant were weighed. The average fruit mass of each plant was contrasted between the genotypes and the significance of fruit mass segregation in each family was determined using Student t test. For gene action analysis, 10 plants each for fw3.2(ys), fw3.2(wt), and heterozygous for fw3.2 were analyzed. Gene action was represented as D/A, where D = Aa − (AA + aa)/2 and A = (AA − aa)/2. Gene action experiments were repeated with the same NIL family grown in two separate fields.
Gene Expression Analysis.
Total RNA was extracted using TRIzol (Invitrogen) as recommended by the manufacturer. Northern blots were performed as described (43). For analyzing ORF7 transcript accumulation, total RNA was isolated from mature leaves, genomic DNA was removed using TURBO DNA-free kit (Invitrogen), followed by first-strand cDNA synthesis and quantitative PCR (qPCR) analysis using iQSYBR Green Supermix (Bio-Rad). For Northern blot and qPCR analyses, the expression of eIF4α and CAC were used as internal control (for primers, see Table S7). For RNA seq analysis, strand-specific libraries of ∼250-bp fragments were prepared using 10 µg of total RNA. Four replicates were generated for each tissue type and time point. Single end sequences of 51 bp were generated on an Illumina HiSeq2000 at Weill Medical College (44). Datasets for the digital gene expression and transcriptome analysis of developing fruit using RNA seq have been published previously, in addition to those published herein (45). Analyzed data can be accessed at the Tomato Functional Genomics Database (http://ted.bti.cornell.edu/cgi-bin/TFGD/digital/experiment.cgi?ID=D006 and http://ted.bti.cornell.edu/cgi-bin/TFGD/digital/experiment.cgi?ID=D008).
Association of CaKLUH with Fruit Weight in Chile Pepper.
To identify nucleotide polymorphisms, the 5′ upstream, intron, 3′ downstream regions of the CaKLUH ortholog in pepper were sequenced in C. annuum cv. “NuMex RNaky” and the C. frutescens accession BG 2814-6 (46). Genomic DNA was extracted from 106 individuals from a F9 RIL population derived from a cross between these parents. The intron SNP was developed into a CAPS marker using the primer pair fw3.2_pepper_F and fw3.2_pepper_R and restriction enzyme digestion with HaeIII. Ten fruit were dried and weighed from each line.
Development of Transgenic RNA Interference Lines for ORF6 and ORF7.
Two RNAi hairpin constructs for ORF6, pMC2 corresponding to RNAi-3 transgenic lines and pMC3 corresponding to RNAi-2 transgenic lines, were generated. RNAi-2 targets the coding sequence of SlKLUH, whereas RNAi-3 targets mainly the 3′UTR (Fig. 2B). Sense and antisense arms were cloned in the HindIII-XhoI and SacI-XbaI sites of a modified pKYLX80 vector, where XhoI and SacI sites are separated by a 150-bp ω-3 fatty acid desaturase intron. Expression cassettes between HindIII-XbaI were subcloned into binary vector pKYLX71 under a 35S promoter with duplicated enhancer (47). Four artificial miRNAs specific for ORF7 were designed using WMD3- Web MicroRNA Designer (48) and were cloned into pKYLX71 binary vector. Primers for the RNAi and sequences for amiRNAs are shown in Table S7. All constructs were introduced in the fw3.2(ys) NIL 08S591-10 carrying an introgression of ∼130 kb. Transformations were carried out at the Plant Transformation Core Research Facility at the University of Nebraska-Lincoln.
Association Mapping.
For the association mapping, ∼7 kb of the fw3.2 locus was sequenced and additional markers were used to genotype the region in 86 accessions of the core collection (20). Genomic positions and scores of markers used in association analysis are presented in Table S3. Association analysis was done using MLM model of TASSEL2.1 software (49). Q and K matrix were generated with STRUCTURE 2.2 (50) and SPAGeDi (51), respectively. To generate Q and K matrix, 20 EST-simple sequence repeat markers distributed throughout the genome were used and the data for these markers in the core collection was obtained from a previous study (20).
Molecular Diversity and Phylogenetic Analysis.
For the molecular diversity analysis, fragment A (637 bp) and fragment B (507 bp) were sequenced in 157 and 172 lines, respectively (20). Fragment A and B span genomic positions 58850848–58851481 and 58852273–58852778, respectively (positions are based on the Tomato WGS Chromosomes SL2.40). The molecular diversity analysis π and Tajima’s D were computed using DnaSP5.0 (52) with a sliding window length 100 and step size 25. Nucleotide diversity across chromosome 3 was calculated using 10 accessions each of S. l. lycopersicum and S. pimpinellifolium.
A contig of 2,528 bp comprised of five fragments spanning the 4,382-bp region (corresponding to 58848391–58852772 in Tomato WGS chromosome SL2.40ch03) around SlKLUH was obtained from the 86 accessions of the association mapping collection (Dataset S1) and used for the phylogenetic analysis. All sequences were aligned using ClustalX2.1 with default multiple sequence alignment parameters and DNA weight matrix ClustalW (1.6). The alignment file was imported to MEGA5.05 (53) and converted into mega (.meg) file format. The phylogeny was reconstructed using the Neighbor-Joining statistical method with 1,000 bootstrap replications and Maximum Composite Likelihood model. For computing phylogenetic distance, a cut off value of 50% bootstrap value was used.
Supplementary Material
Acknowledgments
We thank Dr. D. Choi (Seoul National University) for providing DNA sequence of chile pepper CaKLUH region; and Molecular and Cellular Imaging Center, The Ohio State University, for assistance with microscopy. This work was supported by National Science Foundation Grant IOS-0922661 (to E.v.d.K.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. J.J.G. is a guest editor invited by the Editorial Board.
Data deposition: The sequences reported in this paper have been deposited in the National Center for Biotechnology Information Sequence Read Archive (SRA submission nos. SRA068200 and SRA091611) and in the Genomic Survey Sequences (GSS accession nos. KG700052–KG701213).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1307313110/-/DCSupplemental.
References
- 1.Darwin C. The Variation of Animals and Plants Under Domestication. London: John Murray; 1868. [Google Scholar]
- 2.Diamond J. Guns, Germs, and Steel. New York: WW Norton & Co; 1997. [Google Scholar]
- 3.Smith BD. Seed plant domestication in eastern North America. Last Hunters-First Farmers. 1995;354:193–213. [Google Scholar]
- 4.Pickersgill B. Domestication of plants in the Americas: Insights from Mendelian and molecular genetics. Ann Bot (Lond) 2007;100(5):925–940. doi: 10.1093/aob/mcm193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blanca J, et al. Variation revealed by SNP genotyping and morphology provides insight into the origin of the tomato. PLoS ONE. 2012;7(10):e48198. doi: 10.1371/journal.pone.0048198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klee HJ, Giovannoni JJ. Genetics and control of tomato fruit ripening and quality attributes. Annu Rev Genet. 2011;45:41–59. doi: 10.1146/annurev-genet-110410-132507. [DOI] [PubMed] [Google Scholar]
- 7.Paran I, van der Knaap E. Genetic and molecular regulation of fruit and plant domestication traits in tomato and pepper. J Exp Bot. 2007;58(14):3841–3852. doi: 10.1093/jxb/erm257. [DOI] [PubMed] [Google Scholar]
- 8.Tanksley SD. The genetic, developmental, and molecular bases of fruit size and shape variation in tomato. Plant Cell. 2004;16(Suppl):S181–S189. doi: 10.1105/tpc.018119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grandillo S, Ku HM, Tanksley SD. Identifying loci responsible for natural variation in fruit size and shape in tomato. Theor Appl Genet. 1999;99(6):978–987. [Google Scholar]
- 10.Frary A, et al. fw2.2: A quantitative trait locus key to the evolution of tomato fruit size. Science. 2000;289(5476):85–88. doi: 10.1126/science.289.5476.85. [DOI] [PubMed] [Google Scholar]
- 11.Guo M, et al. Cell Number Regulator1 affects plant and organ size in maize: Implications for crop yield enhancement and heterosis. Plant Cell. 2010;22(4):1057–1073. doi: 10.1105/tpc.109.073676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Libault M, et al. A member of the highly conserved FWL (tomato FW2.2-like) gene family is essential for soybean nodule organogenesis. Plant J. 2010;62(5):852–864. doi: 10.1111/j.1365-313X.2010.04201.x. [DOI] [PubMed] [Google Scholar]
- 13.van der Knaap E, Tanksley SD. The making of a bell pepper-shaped tomato fruit: Identification of loci controlling fruit morphology in Yellow Stuffer tomato. Theor Appl Genet. 2003;107(1):139–147. doi: 10.1007/s00122-003-1224-1. [DOI] [PubMed] [Google Scholar]
- 14.Zhang N, Brewer MT, van der Knaap E. Fine mapping of fw3.2 controlling fruit weight in tomato. Theor Appl Genet. 2012;125(2):273–284. doi: 10.1007/s00122-012-1832-8. [DOI] [PubMed] [Google Scholar]
- 15.Blas AL, et al. Genetic mapping of quantitative trait loci controlling fruit size and shape in papaya. Mol Breed. 2012;29(2):457–466. [Google Scholar]
- 16.Costantini L, Battilana J, Lamaj F, Fanizza G, Grando MS. Berry and phenology-related traits in grapevine (Vitis vinifera L.): From quantitative trait loci to underlying genes. BMC Plant Biol. 2008;8:38. doi: 10.1186/1471-2229-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doganlar S, Frary A, Daunay MC, Lester RN, Tanksley SD. Conservation of gene function in the solanaceae as revealed by comparative mapping of domestication traits in eggplant. Genetics. 2002;161(4):1713–1726. doi: 10.1093/genetics/161.4.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eduardo I, et al. QTL analysis of fruit quality traits in two peach intraspecific populations and importance of maturity date pleiotropic effect. Tree Genet Genomes. 2011;7(2):323–335. [Google Scholar]
- 19.Zhang GR, et al. Fruit size QTL analysis of an F-1 population derived from a cross between a domesticated sweet cherry cultivar and a wild forest sweet cherry. Tree Genet Genomes. 2010;6(1):25–36. [Google Scholar]
- 20.Ranc N, Muños S, Santoni S, Causse M. A clarified position for Solanum lycopersicum var. cerasiforme in the evolutionary history of tomatoes (solanaceae) BMC Plant Biol. 2008;8:130. doi: 10.1186/1471-2229-8-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang N (2012) Fine mapping and characterization of fw3.2, one of the major QTL controlling fruit size in tomato. PhD dissertation (The Ohio State Univ, Columbus, OH)
- 22.Anastasiou E, et al. Control of plant organ size by KLUH/CYP78A5-dependent intercellular signaling. Dev Cell. 2007;13(6):843–856. doi: 10.1016/j.devcel.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 23.Paterson AH, et al. Convergent domestication of cereal crops by independent mutations at corresponding genetic Loci. Science. 1995;269(5231):1714–1718. doi: 10.1126/science.269.5231.1714. [DOI] [PubMed] [Google Scholar]
- 24.Adamski NM, Anastasiou E, Eriksson S, O’Neill CM, Lenhard M. Local maternal control of seed size by KLUH/CYP78A5-dependent growth signaling. Proc Natl Acad Sci USA. 2009;106(47):20115–20120. doi: 10.1073/pnas.0907024106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang W, Wang Z, Cui R, Li J, Li Y. Maternal control of seed size by EOD3/CYP78A6 in Arabidopsis thaliana. Plant J. 2012;70(6):929–939. doi: 10.1111/j.1365-313X.2012.04907.x. [DOI] [PubMed] [Google Scholar]
- 26.Ito T, Meyerowitz EM. Overexpression of a gene encoding a cytochrome P450, CYP78A9, induces large and seedless fruit in arabidopsis. Plant Cell. 2000;12(9):1541–1550. doi: 10.1105/tpc.12.9.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyoshi K, et al. PLASTOCHRON1, a timekeeper of leaf initiation in rice, encodes cytochrome P450. Proc Natl Acad Sci USA. 2004;101(3):875–880. doi: 10.1073/pnas.2636936100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katsumata T, et al. Involvement of the CYP78A subfamily of cytochrome P450 monooxygenases in protonema growth and gametophore formation in the moss Physcomitrella patens. Biosci Biotechnol Biochem. 2011;75(2):331–336. doi: 10.1271/bbb.100759. [DOI] [PubMed] [Google Scholar]
- 29.Meyer KB, et al. A functional variant at a prostate cancer predisposition locus at 8q24 is associated with PVT1 expression. PLoS Genet. 2011;7(7):e1002165. doi: 10.1371/journal.pgen.1002165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Näkki A, et al. Allelic variants of IL1R1 gene associate with severe hand osteoarthritis. BMC Med Genet. 2010;11:50. doi: 10.1186/1471-2350-11-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nischwitz S, et al. More CLEC16A gene variants associated with multiple sclerosis. Acta Neurol Scand. 2011;123(6):400–406. doi: 10.1111/j.1600-0404.2010.01421.x. [DOI] [PubMed] [Google Scholar]
- 32.Uno S, et al. A genome-wide association study identifies genetic variants in the CDKN2BAS locus associated with endometriosis in Japanese. Nat Genet. 2010;42(8):707–710. doi: 10.1038/ng.612. [DOI] [PubMed] [Google Scholar]
- 33.Sandal NN, Bojsen K, Marcker KA. A small family of nodule specific genes from soybean. Nucleic Acids Res. 1987;15(4):1507–1519. doi: 10.1093/nar/15.4.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stougaard J, Jørgensen JE, Christensen T, Kühle A, Marcker KA. Interdependence and nodule specificity of cis-acting regulatory elements in the soybean leghemoglobin lbc3 and N23 gene promoters. Mol Gen Genet. 1990;220(3):353–360. doi: 10.1007/BF00391738. [DOI] [PubMed] [Google Scholar]
- 35.Vieweg MF, et al. The promoter of the Vicia faba L. leghemoglobin gene VfLb29 is specifically activated in the infected cells of root nodules and in the arbuscule-containing cells of mycorrhizal roots from different legume and nonlegume plants. Mol Plant Microbe Interact. 2004;17(1):62–69. doi: 10.1094/MPMI.2004.17.1.62. [DOI] [PubMed] [Google Scholar]
- 36.Gillaspy G, Ben-David H, Gruissem W. Fruits: A developmental perspective. Plant Cell. 1993;5(10):1439–1451. doi: 10.1105/tpc.5.10.1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iglesias DJ, et al. Physiology of citrus fruiting. Braz J Plant Physiol. 2007;19(4):333–362. [Google Scholar]
- 38.Marcelis L, Hofman-Eijer L. Cell division and expansion in the cucumber fruit. J Hortic Sci. 1993;68(5):665–671. [Google Scholar]
- 39.Xiao H, et al. Integration of tomato reproductive developmental landmarks and expression profiles, and the effect of SUN on fruit shape. BMC Plant Biol. 2009;9:49. doi: 10.1186/1471-2229-9-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cong B, Liu J, Tanksley SD. Natural alleles at a tomato fruit size quantitative trait locus differ by heterochronic regulatory mutations. Proc Natl Acad Sci USA. 2002;99(21):13606–13611. doi: 10.1073/pnas.172520999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rao GU, Ben Chaim A, Borovsky Y, Paran I. Mapping of yield-related QTLs in pepper in an interspecific cross of Capsicum annuum and C. frutescens. Theor Appl Genet. 2003;106(8):1457–1466. doi: 10.1007/s00122-003-1204-5. [DOI] [PubMed] [Google Scholar]
- 42.Brewer MT, et al. Development of a controlled vocabulary and software application to analyze fruit shape variation in tomato and other plant species. Plant Physiol. 2006;141(1):15–25. doi: 10.1104/pp.106.077867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao H, Jiang N, Schaffner E, Stockinger EJ, van der Knaap E. A retrotransposon-mediated gene duplication underlies morphological variation of tomato fruit. Science. 2008;319(5869):1527–1530. doi: 10.1126/science.1153040. [DOI] [PubMed] [Google Scholar]
- 44.Zhong S, et al. High-throughput illumina strand-specific RNA sequencing library preparation. Cold Spring Harb Protoc. 2011;2011(8):940–949. doi: 10.1101/pdb.prot5652. [DOI] [PubMed] [Google Scholar]
- 45.Huang ZJ, Van Houten J, Gonzalez G, Xiao H, van der Knaap E. Genome-wide identification, phylogeny and expression analysis of SUN, OFP and YABBY gene family in tomato. Mol Genet Genomics. 2013;288(3-4):111–129. doi: 10.1007/s00438-013-0733-0. [DOI] [PubMed] [Google Scholar]
- 46.Ben-Chaim A, et al. QTL analysis for capsaicinoid content in Capsicum. Theor Appl Genet. 2006;113(8):1481–1490. doi: 10.1007/s00122-006-0395-y. [DOI] [PubMed] [Google Scholar]
- 47.Schardl CL, et al. Design and construction of a versatile system for the expression of foreign genes in plants. Gene. 1987;61(1):1–11. doi: 10.1016/0378-1119(87)90359-3. [DOI] [PubMed] [Google Scholar]
- 48.Ossowski S, Schwab R, Weigel D. Gene silencing in plants using artificial microRNAs and other small RNAs. Plant J. 2008;53(4):674–690. doi: 10.1111/j.1365-313X.2007.03328.x. [DOI] [PubMed] [Google Scholar]
- 49.Bradbury PJ, et al. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23(19):2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- 50.Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol Ecol Notes. 2007;7(4):574–578. doi: 10.1111/j.1471-8286.2007.01758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hardy OJ, Vekemans X. SPAGeDi: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes. 2002;2(4):618–620. [Google Scholar]
- 52.Librado P, Rozas J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 53.Tamura K, et al. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.