

Significance
The clinical outcomes of anticancer drugs are compromised by their poor physicochemical properties, low tumor targeting, insufficient bioavailability, and side effects. Matrix metalloproteinase 2 (MMP2) has been found overexpressed in most cancers and responsible for tumor cell proliferation and metastasis. In this study, a self-assembling MMP2-sensitive paclitaxel-containing micellar nanopreparation was developed. Several major drug delivery strategies, including self-assembly, PEGylation, the enhanced permeability and retention effect, stimulus sensitivity, a cell-penetrating moiety, and the concept of prodrug, were used in a collaborative fashion to design this nanoparticle. The nanopreparation showed superior tumor targeting, cell internalization, and antitumor efficacy over its nonsensitive counterpart, free paclitaxel and conventional micelles. This uniquely engineered nanoparticle has potential for effective intracellular delivery of drug into cancer cells.
Keywords: nanomedicine, polymer-drug conjugate, polymeric micelles, multifunctional, non-small cell lung cancer
Abstract
In response to the challenges of cancer chemotherapeutics, including poor physicochemical properties, low tumor targeting, insufficient tumor cell internalization/bioavailability, and side effects, we developed a unique tumor-targeted micellar drug-delivery platform. Using paclitaxel as a model therapeutic, a nanopreparation composed of a matrix metalloproteinase 2 (MMP2)-sensitive self-assembly PEG 2000-paclitaxel conjugate (as a prodrug and MMP 2-sensitive moiety), transactivating transcriptional activator peptide-PEG1000-phosphoethanolamine (PE) (a cell-penetrating enhancer), and PEG1000-PE (a nanocarrier building block) was prepared. Several major drug delivery strategies, including self-assembly, PEGylation, the enhanced permeability and retention effect, stimulus sensitivity, a cell-penetrating moiety, and the concept of prodrug, were used in design of this nanoparticle in a collaborative manner. The nanopreparation allowed superior cell internalization, cytotoxicity, tumor targeting, and antitumor efficacy in vitro and in vivo over its nonsensitive counterpart, free paclitaxel and conventional micelles. This uniquely engineered nanoparticle has potential for effective intracellular delivery of drug into cancer cells.
Drug-loaded nanocarriers such as liposomes, micelles, polymeric and inorganic nanoparticles, and drug conjugates have demonstrated various advantages over free therapeutic molecules. These nanopreparations can be further engineered with functional moieties to improve their performance in terms of circulation longevity, targetability, cellular penetration, and stimulus sensitivity. The idea of a stimulus-sensitive drug delivery system is based on the abnormalities in the tumor microenvironment, such as acidic pH (1), altered redox potential (2), and up-regulated proteins (3). These internal conditions and external stimuli such as hyperthermia (4), magnetic field (4), and ultrasound (5) can be used to change the behavior of nanocarriers, resulting in an enhanced tumor targeting and antitumor effects.
Matrix metalloproteinases (MMPs), especially MMP2, are known to be involved and overexpressed in many stages of human cancers (3, 6). Various MMP-sensitive substrates have been designed and showed stimulus responsiveness when used in drug delivery and imaging systems (3, 6). In our previous study, a synthetic octapeptide (GPLGIAGQ) was used as the MMP2-sensitive linker in a PEGylated liposomal nanocarrier that could trigger PEG deshielding and the resultant enhanced cell internalization (3).
Although many targeted delivery strategies have shown drug disposition in the tumor, low cellular bioavailability of chemotherapeutics due to insufficient cellular internalization could represent another barrier. To enhance the target cell internalization, cell-penetrating proteins/peptides (CPPs) such as transactivating transcriptional activator peptide (TATp) have been used to modify the nanocarriers/drugs (7).
Paclitaxel (PTX) is one of the most effective antineoplastic agents. It inhibits cell proliferation by stabilization of microtubules and tubulin polymerization, resulting in cell apoptosis (8). However, its clinical application is complicated by its low water solubility, off-target toxicity, and acquired drug resistance. Among many attempts to deal with these issues, “core-shell” polymeric micelles have led to successes in delivery of PTX (9). However, the low drug loading (10), risk of premature drug release (11), and insufficient targetability (9) remain major problems.
Because many drugs with high hydrophobicity result in poor solubility and bioavailability (9), the conjugation of a hydrophilic moiety (e.g., PEG) to a hydrophobic drug molecule (e.g., PTX) improves drug solubility as well as imparts amphiphilicity to the formed conjugates, so that the resultant amphiphilic molecules assemble into a core-shell structure. Although PEGylation provides many advantages (12), various studies have shown that the stable PEG corona is not always beneficial for drug delivery. Ideally, the protective PEG should be removed before cell internalization and subsequent intracellular events (1, 3).
Here, we synthesized a self-assembling drug-polymer conjugate/prodrug, PEG2000-peptide-PTX, which contains the same MMP2-cleavable octapeptide between PEG and PTX. We hypothesized that the MMP2-sensitive and amphiphilic PEG2000-peptide-PTX would serve not only as a tumor environment-sensitive water-soluble PTX prodrug, but also as an MMP2-sensitive building block for the design of a PTX-containing nanopreparation. With this idea in mind, we prepared a unique MMP2-sensitive micellar nanopreparation composed of the PTX prodrug and two other easy-to-make polymers, TATp-PEG1000-phosphoethanolamine (PE) (a cell-penetrating enhancer) and PEG1000-PE (a nanocarrier building block) via their self-assembly in an aqueous environment (Fig. 1A). In this nanopreparation, PTX is located in the hydrophobic “core” covered by a hydrophilic PEG “shell.” Upon administration, the nanopreparation accumulates in the tumor via the enhanced permeability and retention (EPR) effect (13). In the tumor microenvironment, the peptide linker is cleaved by the up-regulated extracellular MMP2, allowing the liberation of the active drug and exposure of the previously hidden TATp for cell internalization. Compared with conventional micelles, this system ensures the following: (i) a high drug loading, (ii) a low risk of premature drug release/leakage, (iii) an enhanced tumor targeting, and (iv) an enhanced tumor cell-selective drug internalization.
Fig. 1.

Drug delivery strategy and characterization of the MMP2-sensitive nanopreparation. (A) Drug delivery strategy. (B) 1H-NMR of PEG2000-peptide-PTX. Both CDCl3 (blue) and D2O (red) were used to determine the chemical structure and nanostructure of the PTX conjugate. (C) TEM. The particle size and morphology of the nanopreparations were analyzed by TEM using negative staining with 1% phosphotungstic acid (PTA). (D) Enzymatic cleavage. To determine the digestion of PEG2000-peptide-PTX (Left) and its nanopreparation (Right), the samples were treated with 5 ng/µL MMP2 followed by TLC and Dragendorff’s reagent staining.
Herein, we described the preparation and characterization of the PTX prodrug and the suggested MMP2-sensitive nanopreparation. The cell internalization, tumor tissue penetration, and cytotoxicity of the nanopreparation were tested in monolayer cancer cells and 3D cancer cell spheroids. Furthermore, after systemic administration, the tumor targeting, antitumor efficacy, and side toxicity of the nanopreparation were examined in a non-small cell lung cancer (NSCLC) xenograft mouse model.
Results and Discussion
Synthesis and Characterization of PEG2000-peptide-PTX and TATp-PEG1000-PE.
To prepare the suggested MMP2-sensitive nanopreparation, two functional conjugates were synthesized. PEG2000-peptide-PTX was expected to have three functions by itself including solubilization of PTX, self-assembly into nanoparticles, and MMP2 sensitivity. TATp-PEG1000-PE was used as the moiety for intracellular delivery of PTX to tumor cells.
In our previous work, we successfully conjugated PEG3400 with the MMP2-cleavable peptide. The same method was used to link PEG2000 with the peptide. To link PEG2000-peptide with PTX, the coupling reagents (DCC/DMAP) were used to activate the carboxyl group of the peptide and accelerate the reaction (Scheme S1A). In thin layer chromatography (TLC) (Fig. S1A), a unique spot was visualized by both UV and Dragendorff’s reagent staining with a significantly lowered retardation factor (Rf) value than that of PTX due to the increased hydrophilicity.
Fig. 1B shows the 1H-NMR spectra of PEG2000-peptide-PTX in both CDCl3 (blue curve) and D2O (red curve). The characteristic peaks of the PTX conjugate were clearly displayed when CDCl3 was used as solvent. PTX is characterized with aromatic (7.35–7.55 ppm), NH (7.0 ppm), acetyl (2.17 and 2.35 ppm), and methyl (1.6–1.7 and 1.25 ppm) protons (14). PEG is characterized by -CH2CH2O- protons (3.65 ppm). The peaks of CH3, CH2, and CH protons in the octapeptide can be found at 1.55–1.75, 1.25, and 0.8 ppm. However, most of PTX peaks disappeared when D2O was used as solvent. The disappearance of PTX peaks in water could be due to the formation of core-shell structure in which the hydrophobic PTX is entrapped in its core and isolated by the hydrophilic PEG shell, whereas the conjugate would be fully dissolved in chloroform. The integration of the characteristic peaks showed that the molar ratio between PEG (-CH2CH2O-) and PTX (aromatic protons) was approximately 1:1. After reaction, the content of PTX per conjugate was approximately 24 wt % based on its molecular mass.
For the synthesis of TATp-PEG1000-PE (Scheme S1B), NHS-PEG1000-MAL was first linked to DOPE (PE) via an ester bond. Then, PE-PEG1000-MAL was conjugated with the cysteine of Cys-TATp. After conjugation, because of the increased hydrophilicity, TATp-PEG1000-PE remained near the starting point on the TLC plates and could be visualized by three staining methods (Dragendorff’s reagent staining for PEG, Ninhydrin reagent staining for peptides, and Molybdenum Blue reagent staining for phospholipids) (Fig. S1C).
Characterization of the Micelle Formation.
To study physicochemical properties of PEG2000-peptide-PTX, the particle size, morphology, and critical micelle concentration (CMC) were analyzed (Fig. 1C and Fig. S2). The CMC of PEG2000-peptide-PTX was approximately 3.2 × 10−5 M, which is in the range of the CMC of the micelles formed by PEG2000-PE (10), indicating the formation of a core-shell structure. The transmission electron microscopy (TEM) showed that PEG2000-peptide-PTX formed nonspherical particles with a size of 61.3 ± 15 nm. These data suggest that the hydrophobic interaction/force among PTX molecules is not strong enough to hold them together tightly, resulting in a large and loose core-shell structure. In contrast, PEG1000-PE containing a strong hydrophobic lipid moiety formed uniform micelles with a spherical shape, small size (11.9 ± 2.0 nm), and low CMC (1 × 10−6 M). Mixing PEG1000-PE with PEG2000-peptide-PTX faciliates the micelle formation as evidenced by a decreased CMC (3.9 × 10−6 M), near-spherical shape, and intermediate size (22.5 ± 2.7 nm). The measured CMC of PEG2000-peptide-PTX/PEG1000-PE micelles was similar to the theoretical CMC (1.9 × 10−6 M) using the equation: 1/CMC = X1/CMC1 + X2/CMC2, suggesting the formation of a mixed micelle. Compared with the CMC obtained in the serum-free medium, the CMC of PEG2000-peptide-PTX/PEG1000-PE in the presence of serum was even lower (4 × 10−7 M), probably due to the high ionic strength of the serum. The increased ionic strength usually decreases the CMC of the amphiphilic polymers, such as lipids and lipid derivatives, which could be well explained by the “binding model” theory (15). The low CMC of the nanopreparation ensures the in vitro and in vivo stability of these core-shell/micellar structures (11).
Stability of the MMP2-Sensitive Nanopreparation.
The particle size of the nanopreparations was measured by the dynamic light scattering as a measure of stability of the micellar structures (11). After incubation with Hank’s balanced salt solution (HBSS) at 37 °C for 4 h, there was no significant change in the size of the nanopreparation. After long-term storage (3 wk) at 4 °C, a slight aggregation (3.6%) was observed (Fig. S3A). These data indicated that the formed micelles were quite stable in the aqueous buffer. In normal mouse sera, the small number of larger aggregates (>500 nm) caused by the interaction of nanopreparations and blood proteins was slightly increased from 0.5% (serum only) to 1.1% after 4-h incubation at 37 °C (Fig. S3B), indicating little in vivo protein adsorption/interaction/opsonization due to the high density of PEG and appropriate PEG length on the surface of the nanopreparation (12, 16). The MMP2-sensitive nanopreparation with the minimized protein adsorption and small size are more likely to “escape” capture by immune cells (16).
Cleavage of PEG2000-peptide-PTX by MMP2.
The cleavability of PEG2000-peptide-PTX was determined by enzymatic digestion followed by TLC (3). After incubation with 5 ng/µL human MMP2, the spot of the PTX conjugate disappeared, whereas two new spots were seen in the TLC plate (Fig. 1D). These data indicated that the MMP2 completely cleaved the peptide linker resulting in two digestion fragments (IAGQ-PTX and PEG-GPLG), in agreement with our previous data (3). Furthermore, incubation of PEG2000-peptide-PTX/PEG1000-PE mixed micelles with MMP2 showed similar results to PEG2000-peptide-PTX alone (Fig. 1D), indicating sufficient accessibility of MMP2 to the peptide even in this “compact” micellar structure. In contrast, incubation of PEG2000-peptide-PTX with mouse plasma could not cleave the linker (Fig. S1B). After MMP2-mediated cleavage, the release of IAGQ-PTX from the micelles was analyzed (Fig. S4). After dialysis for 24 h, approximately 48% of the free PTX was released from the dialysis tube (Fig. S4A, blue curve), whereas TATp-PEG1000-PE/PEG2000-peptide-PTX micelles with MMP2 pretreatment didn’t have any free PTX outside the tube (Fig. S4B, blue curve), similar to the one without MMP2 pretreatment (Fig. S4B, black curve). However, the peak of PTX appears after trypsinization of the sample inside the dialysis tube (Fig. S4B, pink curve). Trypsin is a potent protease that can digest peptides, and trypsinization removes the peptide residue or at least part of it from IAGQ-PTX. The released PTX showed the same retention time as the “naked” PTX. Although the method is not the most sensitive to quantitate the remaining PTX in the micellar core, the data indicated that, after cleavage, IAGQ-PTX was still incorporated into TATp-PEG1000-PE/PEG1000-PE micelles because of its high hydrophobicity (Fig. 1D). This negligible drug leakage guaranteed the enhanced cell internalization of IAGQ-PTX, which is mediated by the micelle surface-attached TATp (Fig. 2 C and E). After internalization, the peptide residue of PTX fragments would be digested/removed by the intracellular enzymes (such as endosomal proteases) and not be the obstacle of the pharmacological activity of the liberated PTX (Fig. 2 D and F).
Fig. 2.
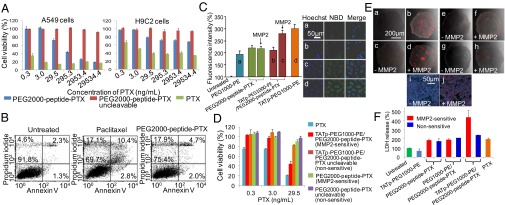
In vitro evaluation of paclitaxel conjugate and its nanopreparation. (A) Cytotoxicity of PEG2000-peptide-PTX in A549 and H9C2 cells. The cytotoxicity of monolayer cells was determined by CellTiter-Blue Cell Viability Assay after 72-h treatments. (B) Apoptosis analysis. The apoptosis of A549 cells was determined by FACS using annexin V/propidium iodide double staining after 72-h treatments. (C) Cellular uptake in A549 cell monolayer. Cells were treated with NBD-PE–labeled formulations (green) for 2 h before measurement. For FACS (Left), cells were trypsinized and collected. For confocal microscopy (Right), cells were fixed and stained with Hoechst 33342. (D) Cytotoxicity of the nanopreparations in A549 cell monolayer. Cells were treated with moderate to low doses of PTX formulations for 72 h before CellTiter-Blue Cell Viability Assay. (E) Penetration of the nanopreparations in A549 spheroids. The spheroids were treated with rhodamine-PE-labeled formulations for 2 h before confocal microscopy (a–h). The sections from c and d were stained by Hoechst 33342 (i and j). (a), PEG1000-PE. (b) TATp-PEG1000-PE. (c and d) TATp-PEG1000-PE/PEG2000-peptide-PTX. (e and f) PEG1000-PE/PEG2000-peptide-PTX. (g and h) PEG2000-peptide-PTX. (i and j) TATp-PEG1000-PE/PEG2000-peptide-PTX. (F) Cytotoxicity of the nanopreparations in A549 spheroids. The spheroids were treated with PTX formulations at the dose of 29.5 ng/mL every other day for 6 d, and the cytotoxicity was estimated by the LDH release.
MMP2-Triggered Tumor Cell-Specific Cytotoxicity of PEG2000-peptide-PTX.
PEG2000-peptide-PTX and its uncleavable counterpart were tested in A549 tumor cells and H9C2 normal cardiomyocytes (Fig. 2A). The decreased cytotoxicity of PTX was observed in both cell lines after PEGylation. In tumor cells, PTX and PEG2000-peptide-PTX showed comparable strong toxicity (approximately 20% cell viability) at high doses, whereas PEG2000-peptide-PTX was much safer in normal cells. Higher cytotoxicity of PTX in A549 cells than H9C2 cells is understandable because tumor cells have higher proliferation rates than normal cells, resulting in the different response to the same treatment (17).
The cytotoxicity of PTX and its conjugate was dose dependent. The data indicated that the released IAGQ-PTX was still cytotoxic after the MMP2-mediated cleavage, which is in agreement with the previous reports that esterification at either C-2′ or C-7′ did not significantly influence PTX’s activity (18). In a recent study, the PTX-peptide conjugate was reported to have equivalent or even higher pharmacological activity compared with free PTX in cancer cells because of the increased solubility despite the C-2′ position of PTX was occupied (19). However, the cytotoxicity of PEG2000-peptide-PTX was lower than that of free PTX, because PEG inhibits the cellular uptake of the conjugate (Fig. 2C), and only released PTX fragments can be efficiently taken up by cells. The drug release process (cleavage) delayed PTX’s action and lowered its activity.
The apoptosis-inducing ability of PEG2000-peptide-PTX was analyzed by fluorescence-activated cell sorting (FACS) (Fig. 2B). The percentage of viable cells of PTX (69.7%) and its conjugate (75.4%) treated groups was much lower than that of untreated cells (>90%). The similar percentage of early apoptotic cells (Annexin positive only) was detected in PTX (2.8%) and PEG2000-peptide-PTX (2.0%) groups. As expected, the percentage of late apoptotic cells (both Annexin and PI positive) in PEG2000-peptide-PTX was lower than that of PTX (4.7% vs. 10.4%), but it is still much higher than that of untreated cells (2.3%). Both treatments significantly increased the number of necrotic/dead cells (PI positive only) with 17.1% for PTX and 17.9% for its conjugate compared with only 4.6% in untreated cells. To further clarify the mechanism of the PTX conjugates, the treated cells were stained by the monoclonal anti–β-tubulin antibody. PEG2000-peptide-PTX induced a significant tubulin polymerization, as evidenced by the visualized green fluorescent filaments around cell nuclei, compared with the uncleavable PTX conjugate and untreated cells. However, the fluorescence intensity was somewhat lower than that of free PTX-treated cells (Fig. S5). In our design, this decreased activity of PEG2000-peptide-PTX caused by PEGylation was used to minimize the nonspecific cytotoxicity of the PTX conjugate to normal cells (Fig. 2A). To exert effective anticancer effects, the decreased activity can be easily compensated by a higher dose (Fig. 2A), appropriate nanocarriers (Fig. 2D), or a longer treatment time (Fig. 2F). By contrast, the uncleavable PTX conjugate did not show cytotoxicity with all doses in either cell line (Fig. 2A).
These data are consistent with the extracellular MMP2 levels (Fig. S6 A–C). Both cell types secreted proteins with gelatinase activity and a molecular mass close to active MMP2 (66.5 KDa; EMD Biosciences). The MMP2 level in A549 cell media was much higher than that from H9C2 cells, and efficiently cleaved the peptide linker, allowing PTX liberation from its nontoxic prodrug. The normal cardiomyocyte was selected as the control because of its low extracellular MMP2 level. The cytotoxicity of PEG2000-peptide-PTX in normal cells is probably due to the basal MMP2, which is required to maintain a cell’s normal activity (20). We also cannot rule out an induction/activation of MMP2 in normal cells by the toxic chemotherapeutics like PTX (21) and doxorubicin (22). However, compared with tumoral MMP2, its influence on the PTX conjugate is limited. These data suggested that PEG2000-peptide-PTX has MMP2-triggered tumor cell-specific cytotoxicity.
Cellular Uptake and Cytotoxicity of the MMP2-Sensitive Nanopreparation.
The self-assembled MMP2-sensitive nanopreparation composed of PEG2000-peptide-PTX, PEG1000-PE, and TATp-PEG1000-PE (Fig. 1A) had a relatively high drug loading (15 wt %) and stable structure, which is superior to most conventional PTX polymeric micelles with low drug loading (usually, less than 5 wt %) (10) and a higher risk of drug leakage (11).
The cellular uptake of NBD-PE–labeled nanopreparations was evaluated by FACS and confocal microscopy (Fig. 2C). PEG2000 in the MMP2-sensitive nanopreparation prevented the TATp-mediated cell internalization (b), which was restored to the level (c) similar to that obtained with TATp-PEG1000-PE micelles (d) after MMP2-induced cleavage (Fig. 2C). Because TATp’s role in nanocarriers has been systematically evaluated in our previous studies (7), and this work might be considered as one of its applications, the competition effect of the free TATp was examined to confirm the function of TATp in this nanopreparation. The data showed that adding free TATp to cell media decreased the TATp-mediated cellular uptake of rhodamine-PE–labeled nanopreparations. The competition effect was dose dependent (Fig. S7). As a result of the enhanced cellular uptake, the MMP2-sensitive nanopreparation killed more tumor cells (44% cell viability, at 29.5 ng/mL) compared with its nonsensitive counterpart, PTX conjugate and uncleavable conjugate (Fig. 2D).
However, the 2D cell culture cannot fully represent in vivo tumors because they are different in terms of cellular heterogeneity, nutrient and oxygen gradients, cell–cell interactions, matrix deposition, and gene expression profiles, resulting in different drug responses and poor in vitro-in vivo correlation (23). To better mimic the real tumor conditions in vitro, A549 multicellular spheroids were established (24) to study the penetration and cell internalization of rhodamine-PE–labeled nanopreparations (Fig. 2E). The presence of the long-chain PEG in the nanopreparation lowered its cell association. However, MMP2 pretreatment significantly increased nanopreparations’ penetration into spheroids (d, f, and h vs. c, e, and g). Strong red fluorescence around cell nuclei was clearly shown upon pretreatment of TATp-PEG1000-PE/PEG2000-peptide-PTX with MMP2 (j vs. i), indicating that MMP2-triggered PEG deshielding allows the exposure of the previously hidden TATp and enhanced cell internalization.
The cytotoxicity of the nanopreparations was also evaluated by using this in vitro model (Fig. 2F). After three treatments at 29.5 ng/mL, all PTX formulations except TATp-PEG1000-PE/PEG2000-peptide-PTX showed similar cytotoxicity with approximately twofold increase of the lactate dehydrogenase (LDH) release compared with untreated spheroids, whereas the empty nanocarrier (TATp-PEG1000-PE) showed no cytotoxicity. The limited cytotoxicity of the nonsensitive nanopreparations was probably a result of the nonspecific cell internalization and cumulative effect of the treatments, which would not be reproduced in the in vivo dynamic conditions. It was also notable that free PTX did not cause the highest cytotoxicity, probably because of its poor penetration of the spheroids (25). By contrast, TATp-PEG1000-PE/PEG2000-peptide-PTX showed the highest cytotoxicity with a more than fourfold LDH release.
In Vivo Tumor Targeting and Antitumor Efficacy.
The tumor targetability and antitumor efficacy of the MMP2-sensitive nanopreparation were evaluated in a NSCLC xenograft mouse model. The in vivo cell internalization of rhodamine-PE–labeled nanopreparations was analyzed by flow cytometry after cell dissociation at 2 h after i.v. injection (Fig. 3A). No significant fluorescence in heart, spleen, lung, and kidney cells was observed after the administration, indicating negligible accumulation of the nanopreparations there. In contrast, the cellular uptake in the liver and tumor was significantly higher, because these tissues contain a large amount of the MMP2 (Fig. S6D). The high MMP2 level might be related to the high cell internalization of the nanopreparation in the liver, whereas the high tumor accumulation of the nanopreparation was the result of the combination effect of the EPR effect and the up-regulated MMP2 in the tumor. The data were confirmed by the enhanced red fluorescence around cell nuclei (blue) in confocal micrographs (Fig. 3B). Furthermore, to see the PTX tissue distribution, the PTX concentration in the tumor, organs, and blood was measured by reverse phase (RP)-HPLC (Fig. S8). No significant difference in the PTX concentration was observed in the major organs and blood between the MMP2-sensitive nanopreparation and nonsensitive one. In contrast, the MMP2-sensitive nanopreparation resulted in a more than 2.5-fold higher PTX concentration in the tumor tissue compared with its nonsensitive counterpart, which is consistent with the in vivo cellular uptake data (Fig. 3 A and B). The liver didn’t show the highest drug accumulation. Instead, the lung and spleen showed the high drug accumulation similar to the tumor. The difference between the two methods is understandable. The tissue accumulation of PTX (HPLC data) showed the overall PTX concentration including both intracellular and extracellular drugs, whereas the in vivo cell internalization data showed only the intracellular nanoparticles/drug. Because the TATp was expected to mediate the enhanced cellular uptake after the MMP2-mediated cleavage, the real in vivo cellular uptake data might be more informative (26). The tissue accumulation data cannot differentiate the intracellular nanoparticles/drug from the extracellular ones (27), therefore, might be not enough to fully describe the MMP2-sensitive nanopreparation’s in vivo behavior. Although the data obtained from the two methods had different meanings, they were actually consistent and delivered the same information that the MMP2-sensitive nanopreparation had the excellent tumor targetability. A small size and PEG corona minimized the distribution/cell internalization of nanoparticles in nontarget tissues, whereas the combined use of the MMP2-sensitive moiety, a cell-penetrating enhancer in the nanoparticles, and the tumoral EPR effect enhanced their tumor cell-selective internalization.
Fig. 3.

In vivo tumor targeting and antitumor efficacy. (A) In vivo cell internalization. HBSS, the rhodamine-labeled nanopreparation, and its nonsensitive counterpart were injected i.v. in tumor-bearing mice at 5 mg/kg PTX, respectively. At 2 h after injection, tumor and major organs were collected. The cells were dissociated from fresh tissue and analyzed immediately by FACS. (B) Intratumor localization. Tumor sections were stained by Hoechst 33342 and detected by confocal microscopy. (C) Tumor growth inhibition (% of the starting tumor volume). Tumor size was measured every 3 d and calculated as V = lw2/2. *P < 0.05 compared with other groups. (D) Tumor cell apoptosis. Tumor sections were stained by Hoechst 33342, and apoptosis was analyzed by TUNEL assay under confocal microscopy.
Like with most chemotherapeutics, we cannot rule out the internalization of the nanopreparation in the tumor by noncancer cells (including immune cells, such as myeloid cells). However, the population of these cells is much lower than that of tumor cells, and myeloid cells themselves also contribute to the tumor invasion in the aggressive tumors (28), which makes the MMP2-sensitive nanopreparation efficacious to inhibit the tumor growth. As a secreted soluble protein, the proteolytically active MMP2 is not only residing in the extracellular matrix and circulating in the blood but also attached on the surface of invasive cells, such as cancer cells, by interaction with integrin αvβ3 (29). The cancer cell-surface integrin receptor regulates both cell migration and matrix degradation, facilitating cancer cell’s invasion (30). In addition to the soluble form of MMP2 in the tumor’s extracellular matrix, the cell surface-bound MMP2 may also contribute to the MMP2-sensitive tumor-targeted drug delivery.
To test the therapeutic activity of the MMP2-sensitive nanopreparation, tumor-bearing mice were injected with PTX formulations twice a week for 4 wk at the dose of 5 mg/kg PTX. Our in vitro data clearly showed that PEG1000-PE/PEG2000-peptide-PTX micelles and the formulations with no TATp or with the blocked TATp do not improve the cellular uptake and antitumor activity, and only the exposed TATp could efficiently enhance the internalization of the nanocarrier by target cells (Fig. 2 C–F). This conclusion has been repeatedly proved in different nanocarriers including the similar PEG-PE micelles by our (1, 3, 7) and other groups (31). Therefore, the only absolutely required experimental groups were tested and compared in vivo to decrease the number of used animals. The formulation with the blocked TATp (nonsensitive nanopreparation) and the formulation with the exposed TATp (MMP2-sensitive nanopreparation) were compared to show the difference between the hidden TATp and the exposed one. Besides, the micelle group PEG2000-PE/PTX, which has no TATp modification, could be considered as another negative control for the TATp modification. The tumor growth of the MMP2-sensitive nanopreparation group (e) was significantly inhibited compared with HBSS (a), the nonsensitive nanopreparation (b), PTX conventional micelles (c), and free PTX (d) (Fig. 3C). It was also notable that the tumor growth inhibition was well correlated with significant apoptosis seen in tumor tissues (green fluorescent dots in the TUNEL assay) (Fig. 3D).
No significant changes were observed after treatment of mice with the MMP2-sensitive nanopreparation in terms of mouse body weights, activities of alanine transaminase (ALT) and aspartate transaminase (AST), and white blood cell counts (Fig. 4 A–C). The free PTX-treated mice showed significantly lower white blood cell counts (approximately 30% of the HBSS group), in agreement with the reported neutropenia or leucopenia (32). By contrast, the nanocarrier improved PTX’s pharmacokinetic profile and biodistribution, resulting in low side toxicity. The hematoxylin and eosin (H&E) staining lacked histological signs of toxicity in major organs with the MMP2-sensitive nanopreparation. However, necrotic areas were clearly present in the MMP2-sensitive nanopreparation-treated tumors (Fig. 4D), in agreement with antitumor effects observed in Fig. 3 C and D. The high therapeutic index of the MMP2-sensitive nanopreparation is most likely a result of the collaborative functions including the “stealth” character of PEGylation (12), the EPR effect (13), the MMP2 sensitivity (3, 6), the TATp-mediated intracellular drug delivery (7), and the enhanced penetration/diffusion (33).
Fig. 4.

In vivo side toxicity assessment. (A) Mouse body weight (% of starting body weight). (B) White blood cell counts. At the end of the experiment, white blood cells were counted by a hemocytometer. (C) Activity of ALT and AST. (D) H&E staining.
Most recently, micellar nanocarriers with high drug loading have been reported (34). However, compared with these micelles, which usually load the drug via physical forces between drug and polymer hydrophobic fragments (11), the risk of drug leakage of the MMP2-sensitive nanopreparation is minimized by the covalent bond between PTX and the polymer (Fig. S4). The most important is that: (i) the PEG2000 and MMP2-sensitive linker in the PTX conjugate allowed the tumor cell-specific cytotoxicity (Fig. 2); (ii) the small size and PEG corona of the MMP2-sensitive nanopreparaion decreased the nonspecific tissue distribution/cell internalization (Fig. 3); and (iii) the combined use of various functions in a collaborative manner enhanced nanopreparations’ tumor cell-selective internalization, resulting in high anticancer activities and low side effects (Fig. 3 and 4).
Conclusion
This work demonstrates a unique drug delivery strategy by the combined use of many collaborative strategies in a single nanocarrier to deal with the drug delivery concerns of anticancer drug. The self-assembled MMP2-sensitive TATp-modified micellar nanopreparation with high paclitaxel loading, low risk of drug leakage, and enhanced tumor targeting and cellular penetration is superior to its nonsensitive counterpart, free paclitaxel and conventional polymeric micelles, resulting in enhanced anticancer activity both in vitro and in vivo. This strategy shows great potential of cancer cell-selective intracellular delivery of an anticancer drug for enhanced cancer chemotherapy.
Materials and Methods
A full description of materials and methods are provided in SI Materials and Methods.
Preparation of the MMP2-Sensitive Nanopreparation.
To prepare the MMP2-sensitive nanopreparation (TATp-PEG1000-PE/PEG2000-peptide-PTX), PEG2000-peptide-PTX (50 mol%), PEG1000-PE (40 mol%), and TATp-PEG1000-PE (10 mol%) were dissolved in chloroform and dried on a freeze-dryer overnight, followed by hydration with HBSS at room temperature. The nonsensitive nanopreparation (TATp-PEG1000-PE/PEG2000-peptide-PTX uncleavable), nanopreparations without TATp modification (PEG1000-PE/PEG2000-peptide-PTX and PEG1000-PE/PEG2000-peptide-PTX uncleavable), and the empty micelle (TATp-PEG1000-PE) were prepared by using the same method.
In Vitro Cellular Uptake of PEG2000-peptide-PTX and Its Nanopreparations.
The A549 cells were incubated with NBD-PE–labeled formulations for 2 h. Then, the cells were analyzed by FACS and confocal microscopy. To study the cellular uptake by spheroids, A549 spheroids were incubated with rhodamine-PE–labeled formulations for 2 h. The spheroids were imaged by confocal microscopy.
In Vitro Cytotoxicity of PEG2000-peptide-PTX and Its Nanopreparations.
Cancer (A549 cells) and normal (H9C2 cells) cells were incubated with PTX or PTX conjugates. To study the cytotoxicity of assembled nanopreparations, various formulations were incubated with A549 cells. After 72-h incubation, the cell viability was determined. To study the response of spheroids to PTX formulations, various formulations were incubated with A549 spheroids at 29.5 ng/mL PTX every other day for 6 d. The cytotoxicity was determined on day 12 by measurement of the LDH release.
Biodistribution and Intratumoral Localization.
HBSS, the rhodamine-labeled MMP2-sensitive nanopreparation, and its nonsensitive counterpart were i.v. injected in tumor-bearing mice at the dose of 5 mg/kg PTX. At 2 h after injection, the tumor and major organs were collected, followed by cell dissociation. The single-cell suspension was analyzed by FACS. The tumors were sectioned and analyzed by confocal microscopy. To determine the PTX’s tissue accumulation, the tissues and blood were homogenized and the PTX was measured by HPLC.
Antitumor Efficacy and Side Toxicity.
HBSS, PTX, PEG2000-PE/PTX, TATp-PEG1000-PE/PEG2000-peptide-PTX, and the nonsensitive nanopreparation were injected i.v. in tumor-bearing mice at the dose of 5 mg/kg PTX twice a week for 4 wk. The tumor volume and body weight were monitored. The blood was collected and white blood cells were counted. The ALT and AST were measured. Tumor sections were stained by Hoechst 33342, and the tumor cell apoptosis was analyzed. Additionally, H&E staining of organs and tumor sections was conducted for histological study of toxicity.
Statistical Analysis.
Data were presented as mean ± SD. The difference between the groups was analyzed by using a one-way ANOVA analysis by the commercial software PASW Statistics 18 (SPSS). P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Dr. Rupa Sawant, Ms. Pooja Kate, and Mr. Amey Kanitkar for their kind help during this study and Dr. William C. Hartner for helpful comments on the manuscript. This work was supported by the National Institutes of Health Grants R01CA121838 and U54CA151881 (to V.T.) and in part by Postdoctoral Fellowship PF-13-361-01-CDD from American Cancer Society-Ellison Foundation (to L.Z.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1304987110/-/DCSupplemental.
References
- 1.Sawant RM, et al. “SMART” drug delivery systems: Double-targeted pH-responsive pharmaceutical nanocarriers. Bioconjug Chem. 2006;17(4):943–949. doi: 10.1021/bc060080h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu L, Mahato RI. Targeted delivery of siRNA to hepatocytes and hepatic stellate cells by bioconjugation. Bioconjug Chem. 2010;21(11):2119–2127. doi: 10.1021/bc100346n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu L, Kate P, Torchilin VP. Matrix metalloprotease 2-responsive multifunctional liposomal nanocarrier for enhanced tumor targeting. ACS Nano. 2012;6(4):3491–3498. doi: 10.1021/nn300524f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhu L, et al. Targeted delivery of methotrexate to skeletal muscular tissue by thermosensitive magnetoliposomes. Int J Pharm. 2009;370(1-2):136–143. doi: 10.1016/j.ijpharm.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 5.Schroeder A, et al. Ultrasound triggered release of cisplatin from liposomes in murine tumors. J Control Release. 2009;137(1):63–68. doi: 10.1016/j.jconrel.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 6.Olson ES, et al. Activatable cell penetrating peptides linked to nanoparticles as dual probes for in vivo fluorescence and MR imaging of proteases. Proc Natl Acad Sci USA. 2010;107(9):4311–4316. doi: 10.1073/pnas.0910283107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Torchilin VP, Rammohan R, Weissig V, Levchenko TS. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc Natl Acad Sci USA. 2001;98(15):8786–8791. doi: 10.1073/pnas.151247498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jordan MA, Toso RJ, Thrower D, Wilson L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc Natl Acad Sci USA. 1993;90(20):9552–9556. doi: 10.1073/pnas.90.20.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Torchilin VP. Micellar nanocarriers: Pharmaceutical perspectives. Pharm Res. 2007;24(1):1–16. doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]
- 10.Lukyanov AN, Torchilin VP. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Adv Drug Deliv Rev. 2004;56(9):1273–1289. doi: 10.1016/j.addr.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 11.Owen SC, Chan DP, Shoichet MS. Polymeric micelle stability. Nano Today. 2012;7(1):53–65. [Google Scholar]
- 12.Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nat Rev Drug Discov. 2003;2(3):214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 13.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 14.Chmurny GN, et al. 1H- and 13C-nmr assignments for taxol, 7-epi-taxol, and cephalomannine. J Nat Prod. 1992;55(4):414–423. doi: 10.1021/np50082a002. [DOI] [PubMed] [Google Scholar]
- 15.Palladino P, Ragone R. Ionic strength effects on the critical micellar concentration of ionic and nonionic surfactants: The binding model. Langmuir. 2011;27(23):14065–14070. doi: 10.1021/la202897q. [DOI] [PubMed] [Google Scholar]
- 16.Walkey CD, Olsen JB, Guo H, Emili A, Chan WC. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J Am Chem Soc. 2012;134(4):2139–2147. doi: 10.1021/ja2084338. [DOI] [PubMed] [Google Scholar]
- 17.Matsuoka H, Furusawa M, Tomoda H, Seo Y. Difference in cytotoxicity of paclitaxel against neoplastic and normal cells. Anticancer Res. 1994;14(1A):163–167. [PubMed] [Google Scholar]
- 18.Deutsch HM, et al. Synthesis of congeners and prodrugs. 3. Water-soluble prodrugs of taxol with potent antitumor activity. J Med Chem. 1989;32(4):788–792. doi: 10.1021/jm00124a011. [DOI] [PubMed] [Google Scholar]
- 19.Yamada R, et al. Biological evaluation of paclitaxel-peptide conjugates as a model for MMP2-targeted drug delivery. Cancer Biol Ther. 2010;9(3):192–203. doi: 10.4161/cbt.9.3.10656. [DOI] [PubMed] [Google Scholar]
- 20.Hua H, Li M, Luo T, Yin Y, Jiang Y. Matrix metalloproteinases in tumorigenesis: an evolving paradigm. Cell Mol Life Sci. 2011;68(23):3853–3868. doi: 10.1007/s00018-011-0763-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aggarwal BB, et al. Curcumin suppresses the paclitaxel-induced nuclear factor-kappaB pathway in breast cancer cells and inhibits lung metastasis of human breast cancer in nude mice. Clin Cancer Res. 2005;11(20):7490–7498. doi: 10.1158/1078-0432.CCR-05-1192. [DOI] [PubMed] [Google Scholar]
- 22.Spallarossa P, et al. Matrix metalloproteinase-2 and -9 are induced differently by doxorubicin in H9c2 cells: The role of MAP kinases and NAD(P)H oxidase. Cardiovasc Res. 2006;69(3):736–745. doi: 10.1016/j.cardiores.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 23.Desoize B, Jardillier J. Multicellular resistance: A paradigm for clinical resistance? Crit Rev Oncol Hematol. 2000;36(2-3):193–207. doi: 10.1016/s1040-8428(00)00086-x. [DOI] [PubMed] [Google Scholar]
- 24.Hirschhaeuser F, et al. Multicellular tumor spheroids: An underestimated tool is catching up again. J Biotechnol. 2010;148(1):3–15. doi: 10.1016/j.jbiotec.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 25.Kuh HJ, Jang SH, Wientjes MG, Weaver JR, Au JL. Determinants of paclitaxel penetration and accumulation in human solid tumor. J Pharmacol Exp Ther. 1999;290(2):871–880. [PubMed] [Google Scholar]
- 26.Leuschner F, et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29(11):1005–1010. doi: 10.1038/nbt.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kirpotin DB, et al. Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 2006;66(13):6732–6740. doi: 10.1158/0008-5472.CAN-05-4199. [DOI] [PubMed] [Google Scholar]
- 28.Kitamura T, et al. SMAD4-deficient intestinal tumors recruit CCR1+ myeloid cells that promote invasion. Nat Genet. 2007;39(4):467–475. doi: 10.1038/ng1997. [DOI] [PubMed] [Google Scholar]
- 29.Brooks PC, et al. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell. 1996;85(5):683–693. doi: 10.1016/s0092-8674(00)81235-0. [DOI] [PubMed] [Google Scholar]
- 30.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 31.Sethuraman VA, Bae YH. TAT peptide-based micelle system for potential active targeting of anti-cancer agents to acidic solid tumors. J Control Release. 2007;118(2):216–224. doi: 10.1016/j.jconrel.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spencer CM, Faulds D. Paclitaxel. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in the treatment of cancer. Drugs. 1994;48(5):794–847. doi: 10.2165/00003495-199448050-00009. [DOI] [PubMed] [Google Scholar]
- 33.Cabral H, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6(12):815–823. doi: 10.1038/nnano.2011.166. [DOI] [PubMed] [Google Scholar]
- 34.Matsumura Y, Kataoka K. Preclinical and clinical studies of anticancer agent-incorporating polymer micelles. Cancer Sci. 2009;100(4):572–579. doi: 10.1111/j.1349-7006.2009.01103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.