

Significance
Protocols for NMR determination of high-resolution protein structures in solution require, among other things, an accurate method with which to assess the quality of protein structures. It is important that such a validation method provide information as to where structural flaws are and how they can be repaired. So far, no generally accepted validation method with these characteristics appears to exist for evaluation of protein structures in solution. As an approach to find a solution to this long-standing problem, we developed a method to detect flaws in protein structures at the residue level and also to provide a way to repair these flaws.
Abstract
A method that makes use of information provided by the combination of 13Cα and 13Cβ chemical shifts, computed at the density functional level of theory, enables one to (i) validate, at the residue level, conformations of proteins and detect backbone or side-chain flaws by taking into account an ensemble average of chemical shifts over all of the conformations used to represent a protein, with a sensitivity of ∼90%; and (ii) provide a set of (χ1/χ2) torsional angles that leads to optimal agreement between the observed and computed 13Cα and 13Cβ chemical shifts. The method has been incorporated into the CheShift-2 protein validation Web server. To test the reliability of the provided set of (χ1/χ2) torsional angles, the side chains of all reported conformations of five NMR-determined protein models were refined by a simple routine, without using NOE-based distance restraints. The refinement of each of these five proteins leads to optimal agreement between the observed and computed 13Cα and 13Cβ chemical shifts for ∼94% of the flaws, on average, without introducing a significantly large number of violations of the NOE-based distance restraints for a distance range ≤ 0.5 Ǻ, in which the largest number of distance violations occurs. The results of this work suggest that use of the provided set of (χ1/χ2) torsional angles together with other observables, such as NOEs, should lead to a fast and accurate refinement of the side-chain conformations of protein models.
Since the seminal observation by Kendrew (1) that “it is the spatial relations between the side-chains which determine the chemical behavior and biological specificity of the protein molecule as a whole and these relations cannot be determined, except in a fragmentary manner, by purely chemical techniques” interest has been focused on the development of accurate methods to validate and determine side-chain conformations in proteins (ref. 2 and references therein). Side-chain chemical shifts have also been used for protein structure validation because these observables are highly sensitive to protein structural packing (3). The latter interest arises because it is largely accepted that a proper protein structure determination requires validation methods in which the observable values used to validate the structures are not used in their determination (4, 5), and it will assure spectroscopists and other users that a given protein model is a good representation of the native structure in solution. However, the validation process involves two crucial steps: (i) detecting flaws in the structure at the residue level and (ii) providing details as to how these flaws can be repaired. Existing validation methods are mainly concerned with determining the quality of the whole structure but only sometimes with highlighting where the flaws are located at the residue level (3, 6–12). To the best of our knowledge, these validation methods do not provide detailed information as to how such flaws can be eliminated. Therefore, it is left to the spectroscopists to find a reliable solution for the detected structural problems. Consequently, to develop a validation method capable of detecting and repairing structural flaws at the residue level, we focused our effort on a combined use of 13Cα and 13Cβ chemical shifts, rather than use of separate 13Cα or 13Cβ chemical shifts, to provide complementary information regarding the quality of a given structural model.
It is worth noting that the observed 13Cβ chemical shifts have so far been used predominantly to determine conformational preferences of the backbones of polypeptide chains (13), although they also contain very valuable information about side-chain conformations, which would be a very important contribution to accurate validation, determination, and refinement of protein models. Despite this, the use of chemical shifts for determination and refinement of protein structures (14–17) is not the aim of this work, although a simple refinement routine is used here only as a tool to assess the reliability of a proposed methodology to repair flaws in proteins.
Evidence has been accumulated showing that the 13Cα chemical shift is determined mainly by its amino acid residue without significant influence of the nearest-neighbor residues, except for residues preceding proline (18–21). There is also evidence that not only the backbone but also the side-chain conformation influences the 13Cα chemical shift to some extent (22, 23). However, the question whether 13Cβ rather than 13Cα chemical shifts are more sensitive to χ1/χ2 side-chain torsional angles remains to be investigated; that is, to what extent are the 13Cα and 13Cβ chemical shifts of an amino acid residue in a protein affected by its side-chain orientation? This query is relevant to the fact that the three torsion angles ϕ, ψ, and χ1 are not independent of each other because they involve a common N−Cα group (24, 25). To answer this important question, we have expanded our 13Cα-based CheShift-2 Web server (12) to include the computation of 13Cβ chemical shifts. This database of 13Cβ chemical shifts contains ∼600,000 conformations and can be used (together with the database for 13Cα) for a detailed analysis of the combined dependence of the 13Cα and 13Cβ chemical shifts on the χ1 torsional angle for a given fixed χ2 for all 20 naturally occurring amino acids, not including the 13Cβ chemical shifts for Ala and Gly, which do not contain a side chain with χ1 and χ2, and Pro, for which χ1 and χ2 are fixed.
Overall, use of the updated version of the CheShift-2 Web server containing both 13Cα and 13Cβ chemical shifts will enable users to (i) assess the quality of protein structures and detect flaws at the residue level by making use of the ensemble average of the chemical shifts computed over all of the conformations used to represent a protein and not the chemical shift values for one conformation and (ii) obtain possible solutions as to how these flaws can be fixed by generating a list of side-chain χ1 and χ2 torsional angles that can be used to improve the agreement between observed and predicted 13Cα and 13Cβ chemical shifts. A series of tests of the methodology includes one to determine the sensitivity of the updated version of the CheShift-2 Web server to validate protein structures at the residue level. In addition, the reliability of the provided set of (χ1/χ2) torsional angles to repair existent flaws is assessed here by refinement of the NMR-determined structures of five proteins (26), namely, 1D3Z, 2KIF, 2LQ9, 2LU1, and 2M2J.
Results and Discussion
Comparison of the Dependence of Separate 13Cα and 13Cβ Chemical Shifts on the Variation of Backbone and Side-Chain Conformations.
To examine the relative dependence of these two chemical shifts for a given residue on its conformational changes, we selected a set of variable χ1 torsional angles in 30° intervals for each of the most frequently observed χ2 torsional angles. Then, the mean values of each of the 13Cα and 13Cβ chemical shifts over all values of χ1 for fixed ϕ, ψ, and χ2 and the corresponding standard deviations, σα and σβ, were computed. The difference between the standard deviations, Δσ = (σα − σβ), computed for the 13Cα and 13Cβ chemical shifts, provides information about the relative ability of each of these chemical shifts to sense χ1 variations for these chosen values of the fixed torsional angles. A negative value of Δσ indicates a greater variation of the 13Cβ shielding compared with the 13Cα shielding upon changes in χ1.
A large database (∼1,200,000) of DFT-computed 13Cα and 13Cβ shieldings was used to calculate the standard deviation differences, Δσ, as a function of side-chain and backbone variations for each of the five residues, listed in SI Appendix, Table S1, as an example when they are in a tripeptide. On one hand, due to the side-chain variations reported in SI Appendix, Figs. S1 and S2, the results of this analysis show a larger variation of Δσ with changes of the χ1 and χ2 torsional angles for the 13Cβ shielding than for the 13Cα shielding. Thus, for Asn, the variations of the Δσ values are within the range +0.87 > Δσ > −4.9 ppm (SI Appendix, Fig. S1).
On the other hand, the magnitudes of the variations of the 13Cβ and 13Cα shieldings, caused by variations in their backbone conformations, depend on both the residue type and the adopted values of the fixed χ1/χ2 torsional angles (SI Appendix, Table S1); for example, for residues such as Asn and Ser showing at least one Δσ > 0, the variations in the 13Cα shielding display a wider range, i.e., larger σα, of values in response to variations of their backbone compared with those of 13Cβ. The opposite (Δσ < 0) is observed for Leu and Asp, whereas for Tyr, the 13Cα and 13Cβ shieldings show similar values of Δσ but with opposite sign in response to variations of the side-chain (χ1/χ2) values.
Taken all together, these results indicate that the combined use of 13Cα and 13Cβ chemical shifts should be a better probe with which to validate backbone and side-chain conformations of protein models compared with the separate use of either the 13Cα or 13Cβ chemical shifts. Evidence supporting this conclusion is shown in the next section.
Graphical Representation of the Chemical Shift Differences for Ubiquitin.
Using the color representation described in Materials and Methods, Fig. 1 shows the validation results for ubiquitin, a protein whose structure was solved by NMR spectroscopy (1D3Z) (27) and by X-ray crystallography (1UBQ) at 1.8-Ǻ resolution (28). The CheShift-2 Web server was used to validate the structures using either the 13Cα (Fig. 1 A and B) or the 13Cβ (Fig. 1 C and D) chemical shifts. The left and right columns in Fig. 1 illustrate the graphical validation for the NMR- and the X-ray–determined structures, 1D3Z and 1UBQ, respectively.
Fig. 1.

Structures of ubiquitin in the left-hand column were determined by NMR spectroscopy; those in the right-hand column were determined by X-ray crystallography. Structures in A and B pertain to validation of proteins 1D3Z and 1UBQ, respectively, by using only 13Cα chemical shifts; structures in C and D pertain to validation of proteins 1D3Z and 1UBQ, respectively, by using only 13Cβ chemical shifts; and structures in E and F pertain to validation of proteins 1D3Z and 1UBQ, respectively, by using a combination of 13Cα and 13Cβ chemical shifts. All of the residues for which the agreement between observed and computed 13Cα or 13Cβ chemical shifts (A–D) can be improved, i.e., by varying the side-chain torsional angles to any of the solutions provided by the CheShift-2 Web server, are highlighted in blue in E and F. Those residues showing good, marginally good, and poor agreement with the observed chemical shift values are highlighted in green, yellow, and red, respectively.
By comparing the residue color distribution between Fig. 1 A and C or B and D, it can be seen that the use of either the 13Cα or 13Cβ nuclei leads to similar but not identical results. The graphical validation of these residues, obtained by using either the 13Cα or 13Cβ chemical shifts, yields different color codes for ∼29% and ∼41% of the residues in 1D3Z and in 1UBQ, respectively. This result indicates that the combined use of 13Cα and 13Cβ chemical shifts would be a better probe with which to validate protein models compared with the separate use of either the 13Cα or 13Cβ chemical shifts, as suggested in the previous section.
For the residues which display validation disagreement between 13Cα and 13Cβ chemical shifts, it should be possible to find values of side-chain torsional angles that would bring the graphical validation in terms of both nuclei to an optimal agreement, i.e., to be represented in green. This optimal set of side-chain conformations (see SI Appendix, Table S2, for Lys and Ile in boldface as an example) were generated by following the procedure described in Materials and Methods.
Overall, all residues of 1D3Z and 1UBQ, which could become green by variation of χ1 and χ2 with the CheShift-2 Web server, are highlighted in blue in Fig. 1 E and F. For the remaining residues for which no variations of χ1 and χ2 were able to change these residues to green, the backbone torsional angles should be revised.
Determining the Sensitivity and Specificity of the CheShift-2 Web Server.
The current methodology for computing shieldings (5, 15) relies on a crucial observation: after a residue conformation is established by its interactions with the rest of the protein, the 13Cα shielding of each residue depends mainly on its backbone and side-chain conformations, with no significant influence of the nature of the nearest-neighbor amino acids, except for residues immediately preceding proline (21). Evidence supporting this statement for 13Cβ shielding is presented in the first subsection of SI Appendix, Materials and Methods. Consequently, a given set of amino acid residue conformations representing the accessible conformational space for all of the 20 naturally occurring amino acids constitutes a reliable ensemble with which to determine the sensitivity and specificity of the CheShift-2 Web server. Sensitivity refers to the likelihood to detect a flaw in a given residue when it exists, and specificity refers to the likelihood that no flaw is detected when there is no flaw in a given residue. Hence, these results should be transferable to proteins of any class or size.
Based on the above observation, sensitivity and specificity of ∼90% and ∼70%, respectively (SI Appendix, Table S3), were computed as described in second subsection of SI Appendix, Materials and Methods. A sensitivity and specificity of ∼100% implies an ideal validation; that is, all flaws are detected, and all are true flaws. However, in practice, these values are not reachable. From a validation point of view, it is convenient to have high sensitivity because this will ensure that no errors are left out. Note that the sensitivity of the server to detect flaws drops from ∼90% to ∼70% (SI Appendix, Table S3) if only the 13Cα rather than both the 13Cα and 13Cβ nuclei are used, in agreement with the conclusion reached from previous sections.
Test of the Reliability of the Side-Chain Torsional Angle Predictions.
Analysis of a set of 42 NMR- and X-ray–determined structures (listed in SI Appendix, Table S4) permits an illustration that for up to ∼90% of the chosen residues, at least one of the predicted (χ1, χ2) side-chain torsional angles of a given NMR-determined structure is actually seen in the corresponding X-ray–determined structure (first subsection in SI Appendix, Results and Discussion). However, this analysis is not an accurate test of the reliability of the method because it is known that an ensemble of conformations rather than a single structure is a more accurate representation of a protein, both in the crystal and in solution (29, 30).
Because the observed chemical shift for each residue in the sequence represents the contributions from an ensemble of rapidly interconverting conformers that coexist in solution, a rigorous test of the reliability of the predicted set of χ1/χ2 side-chain torsional angles should be one that makes use of this set, rather than a single conformation, for a refinement of a protein conformational ensemble. Moreover, this test will be used to investigate whether significant NMR restraint violations appear in the already existing set as a result of the refinement. Consequently, in the next section the refinement of 10 models of the protein ubiquitin is described in detail.
Refinement of NMR-Determined Proteins.
The question of whether the set of optimal χ1/χ2 torsional angles provided by the CheShift-2 Web server is reliable for protein structure refinement is discussed here in detail for the 10 models of ubiquitin 1D3Z, solved at high accuracy.
Among many refinement options, which include use of molecular dynamics or molecular mechanics conformational searches and different types of force fields, we decided to try the simplest one consisting of the following steps: (i) all backbone torsional angles (ϕ, ψ) are kept fixed at their original values; (ii) residues for which rotation of side chains could improve the agreement between the observed and predicted 13Cα and 13Cβ chemical shifts are selected according to SI Appendix, Table S2; and (iii) for each of the selected residues, the side-chain torsional angles that are proposed by the CheShift-2 Web server and also lead to lowest atomic overlapping with the nearest neighbor atoms are chosen to replace the torsional angles of the original model. If the replacement leaves some atomic overlapping, a short torsional angle relaxation, considering only nonbonded interactions, is applied with the use of the PyMOL package (31). During the relaxation procedure, changes of torsional angles that lead to variations larger than ±15° are not considered acceptable. Overall, if none of the proposed solutions satisfies the requirement for low atomic overlapping, the original side-chain torsional angles are retained. This refinement procedure is applied to each conformation of the ensemble, although the decision whether a detected flaw is repaired rests on the evaluation of all of the conformations of the ensemble, not just a single one, as explained in detail below.
At the end of the procedure, the original and refined structures of 10 models of the protein 1D3Z were evaluated by two independent validation methods: WHAT_IF (6), to check the average number of overlaps between atoms, and the Protein Structure Validation Software Suite (10) to test the quality of the structures based on the number of violations of the NMR restraints. In addition, agreement between the observed and computed 13Cα and 13Cβ chemical shifts was evaluated only in terms of the conformational average root-mean-square deviation (21) to demonstrate the capability of the method to repair flaws in the resulting structures.
A total of 255 residues from the 10 models of the protein 1D3Z, for which a change in the χ1/χ2 torsional angles would lead to better agreement between the observed and computed 13Cα and 13Cβ chemical shifts, was selected. However, whether a given residue μ in the sequence is or is not a flaw is determined by the average value computed over all of the conformations and not by the chemical shift value for one conformation. The computed average chemical shift value of residue μ is given by 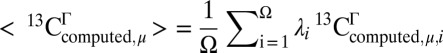 , where Ω is the total number of conformations, Γ = α or β, and λi is the Boltzmann factor for conformer i, with
, where Ω is the total number of conformations, Γ = α or β, and λi is the Boltzmann factor for conformer i, with 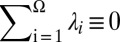 . However, as was noted previously (21), computation of the Boltzmann factors on the quantum mechanical level of theory is not possible with the present computational facilities, and hence, the approximation that each conformer contributes equally to the average chemical shift obtained by fast conformational averaging, i.e., λi = 1/Ω, is needed. In addition, because the distribution of conformations reported in an NMR bundle is not a true representation of the distribution of conformations present in solution, i.e., because they are just a collection of conformers each of which represents the best fit of the data to a single conformation, validation of residues pertaining to the bundle, or exhibiting large flexibility, should be interpreted with caution because of possible effects of the above ensemble average approximation.
. However, as was noted previously (21), computation of the Boltzmann factors on the quantum mechanical level of theory is not possible with the present computational facilities, and hence, the approximation that each conformer contributes equally to the average chemical shift obtained by fast conformational averaging, i.e., λi = 1/Ω, is needed. In addition, because the distribution of conformations reported in an NMR bundle is not a true representation of the distribution of conformations present in solution, i.e., because they are just a collection of conformers each of which represents the best fit of the data to a single conformation, validation of residues pertaining to the bundle, or exhibiting large flexibility, should be interpreted with caution because of possible effects of the above ensemble average approximation.
Overall, these 255 residues can be represented by only 39 nonidentical residues, i.e., each of them occupying different positions in the sequence among all of the conformers but not necessarily being different residues. Thus, we are able to detect 39 flaws for the ensemble of conformations representing the protein 1D3Z. The refinement method, which is carried out for each conformer of the ensemble, is able to repair the side-chain conformation for 34 out of 39 nonidentical residues. This leads to a success rate of replacement of ∼87% of the detected flaws. This is illustrated by Fig. 2, where 39 residues highlighted in blue in Fig. 2A are reduced to only 5 in Fig. 2B.
Fig. 2.

(A) Graphic validation, in terms of both the 13Cα and 13Cβ chemical shifts, for the 10 models of ubiquitin 1D3Z in which 39 residues are highlighted in blue to indicate that their validation could be improved by varying their side-chain torsional angles by using the CheShift-2 Web server solutions (SI Appendix, Table S2). (B) The refined 10 models obtained by using the CheShift-2 Web server solutions. A total of 34 residues, now colored in green, were improved, while the remaining five residues are still colored in blue because any of the solutions provided by the CheShift-2 Web server leads to an unacceptably large atomic overlapping.
The validation results for the 10 original and refined models of the protein 1D3Z in terms of the number of NOE-derived distances are shown in Fig. 3. As shown in Fig. 3A, the total numbers of NOE-based distance restraints before (black bars) and after (gray bars) the refinement are similar. The gray bars in Fig. 3B show the computed difference between the average distance violations after and before the refinement, over all of the NOE-based distance restraints, for each conformer of the ensemble. In particular, for 3 out of 10 conformers the computed differences between the average distance violations are either ∼0 or slightly negative, indicating that the refinement of ubiquitin cannot only repair flaws without introducing large additional distance violations but it can also lead to slightly better agreement with the observed NOE-based distance restraints (see results for conformations 4, 9, and 6 in Fig. 3B). In addition, Fig. 3B shows that there is no significant correlation (R2 = 0.27) between the total number of repaired flaws for each of the conformers (black squares in Fig. 3B) and the average differences of NOE distance restraint violations after and before refinement (gray bars). The values of the computed differences between the average distance violations (displayed on the left y axis of Fig. 3B) are small because the frequency of the distribution of the NOE distance violations follows a rapidly decaying dependence with the distance range of violations (Fig. 3A).
Fig. 3.

(A) Black- and gray-filled bars denote the average number, per conformer, of NOE-derived distance restraint violations obtained from the original and the refined ensembles of 10 conformations of 1D3Z, respectively. The violations are grouped within intervals of 0.5 Ǻ. At a given interval, e.g., 1.0 Ǻ, the heights of the bars represent the accumulated number of violations (X), which are in the range 0.5 < X ≤ 1.0 Ǻ. (B) The difference, as gray bars, between the total number of NOE-derived distance restraint violations after and before the refinement as a function of the conformation number for each of the 10 conformations of 1D3Z. The black-filled square for each conformation represent the total number of repaired flaws.
Among 39 nonidentical residues to be repaired, 34 were successfully repaired. All of the 34 are solvent-exposed residues, except Ile-3, Leu-43, and Ile-63. Five of the 39 residues are nonrepaired, including Ile-23 and Ile-30, which are fully buried, and Phe-4, Ile-36, and Arg-72, which are solvent-exposed. Hence, the condition for a residue to be buried does not appear as an obstacle for a successful replacement in ubiquitin.
We carried out a graphical analysis of the average side-chain heavy-atom B factors reported for the X-ray–determined structure of ubiquitin (1UBQ) (28) to find out whether the 39 nonidentical residues to be repaired are associated with side-chain B factors. For this purpose, the backbone of ubiquitin is represented as a tube with a radius proportional to the observed side-chain B factors. Then, the surface of this backbone representation was highlighted (SI Appendix, Fig. S3) using the color distribution shown in Fig. 2A. Although there is some correspondence between the locations of the 39 residues to be repaired (highlighted in blue) and the sections characterized by larger radius (higher B factors) of the tube representation of the backbone, a generalization is not straightforward (SI Appendix, Fig. S3). Residues possessing higher side-chain B factors (larger radius) tend to be repairable (highlighted in blue); that is, the agreement between observed and computed 13Cα and 13Cβ chemical shifts can be improved. On the other hand, several repairable residues do not possess high side-chain B factors; for example, see those backbone regions with a small radius section highlighted in blue in SI Appendix, Fig. S3. On the whole, this result seems to indicate that the flaws identified by the CheShift-2 Web server for the NMR-determined protein structure in solution are not necessarily mirrored by the side-chain B factors of the structure in the crystal environment.
A summary of the validation analysis carried out for ubiquitin and for another four NMR-determined protein structures (2KIF, 2LQ9, 2LU1, and 2M2J) is shown in SI Appendix, Table S5. In general, the original and the refined models of all five of these proteins show a similar number of accumulated NOE-based distance violations for a distance range ≤0.5 Ǻ, i.e., a distance range in which the largest number of distance restraint violations occurs (Fig. 3 and SI Appendix, Fig. S4). Additionally, for all five proteins, a comparable per-residue average atomic overlapping is observed between the original and the refined protein models (SI Appendix, Table S5). Considering that the NOE-based distances were not taken into account during the refinement, the obtained results represent a strong validation test (5) of the reliability of the provided set of (χ1/χ2) torsional angles.
Overall, development of an accurate refinement method, i.e., one that includes not only the 13Cα and 13Cβ chemical shifts but also the NOE-based distance restraints, is needed, although this is beyond the scope of the current work.
Conclusions
In this work, we expanded our existing database of 13Cα chemical shifts to include chemical shifts for the 13Cβ nucleus. This new expanded database was used for validation of protein structures, and the results obtained here demonstrate that the combined use of 13Cα and 13Cβ chemical shifts provides better information than the use of either alone to detect flaws in the backbones and side chains of protein conformations with a sensitivity of ∼90%.
We also proposed a simple method for generating a set of χ1/χ2 side-chain torsional angles that provide optimal agreement between the observed and computed 13Cα and 13Cβ chemical shifts for ∼94%, on average, of the residues from five NMR-determined structures. Use of this optimal set of χ1/χ2 torsional angles, taken together with other restraints, such as those from NMR spectroscopy, opens a way for accurate refinement of the side-chain conformations for protein models.
To make all these improvements available to any user interested in validation/refinement of protein structures, the CheShift-2 Web server (http://cheshift.com/) has been upgraded and is available free of charge for academic use.
During the peer review process of our manuscript, Shen and Bax (32), in a recent advance, published an artificial neural network-based hybrid system that makes use of information provided by the observed chemical shifts of several nuclei, namely, HN, Hα, 13Cα, 13Cβ, 13CO, and 15N, for an empirical prediction of the backbone (ϕ, ψ) and the χ1 torsional angles of residues in proteins. Although prediction, not validation, is the main goal of ref. 32, it is worth noting the main differences regarding the side-chain torsional angle predictions between their manuscript and ours: (i) we focused on both the χ1 and the χ2 torsional angles because the shieldings of the 13Cα and mostly the 13Cβ nuclei are sensitive to variations of these torsional angles; (ii) we developed a method to detect flaws at the residue level and also to provide a way that these flaws could be repaired, i.e., by making use of a provided set of not only χ1 but also (χ1/χ2) side-chain torsional angles; and (iii) our test of the reliability of the (χ1/χ2) side-chain predictions relies on the refinement of each conformer of the ensemble used to represent an NMR-determined protein structure rather than a single X-ray–determined structure. This assessment of the reliability of the predictions is important for two reasons: first, the observed chemical shift for each residue in the sequence represents the contributions from an ensemble of rapidly interconverting conformers that coexist in solution and second, proteins in solution and in a crystal are better represented by an ensemble of conformations than by a single structure (29, 30).
Materials and Methods
Definition of Validation and Refinement.
There are two critical words relevant to the main goal of our work, and hence, it is essential to define them. First, the CheShift-2 Web server chemical shifts are compared with the observed values to assess the quality of the reported conformation and to detect flaws (at the residue level). This is validation. Second, the values of the χ1/ χ2 torsional angles are varied to try to correct these flaws, i.e., to improve the quality of the reported structure. This is refinement.
Database of 13Cβ Chemical Shifts.
Computation of the 13Cβ database follows the same procedure used to compute the 13Cα database (11), and hence, only a brief description is provided here. The 13Cβ database is based on the generation of ∼600,000 conformations, as a function of the ϕ, ψ, ω, χ1, and χ2 torsional angles, for terminally blocked tripeptides with the sequence Ac-Gly-Xxx-Gly-NMe (Ac, acetyl; NMe, N-methyl), where Xxx is any of the 20 naturally occurring amino acids. For the generation of these ∼600,000 conformations, the following sampling procedure was used: (i) the backbone torsional angles ϕ and ψ were sampled every 10°; (ii) all ω torsional angles were assumed to be 180°, except for Pro residues for which the cis conformation (0°) was also considered; (iii) all χ1 side-chain torsional angles were sampled every 30ο; and (iv) all χ2 side-chain torsional angles were sampled according to the most frequently seen torsional values (33). For each of these conformations, the 13Cβ isotropic shielding value for the residue Xxx was calculated using an identical procedure to that used to compute the 13Cα isotropic shielding values (11).
Partitioning the Differences per Residue for the 13Cβ Chemical Shifts.
For each of the 12,935 residues belonging to the 88 X-ray–determined protein structures listed in SI Appendix, Table S6, the value of 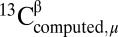 for each residue μ was obtained by using the CheShift-2 Web server, and the value of
for each residue μ was obtained by using the CheShift-2 Web server, and the value of 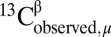 was taken from the BioMagResBank database (34) by using the corresponding accession numbers listed in SI Appendix, Table S4.
was taken from the BioMagResBank database (34) by using the corresponding accession numbers listed in SI Appendix, Table S4.
For each residue μ, the difference between observed and predicted 13Cβ chemical shifts is defined as 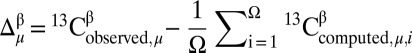 , where
, where 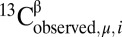 is the CheShift-2–computed chemical shift of residue μ in conformation i out of Ω conformations. The average of the predicted chemical shifts over the Ω conformations is evaluated because proteins in solution exist as an ensemble of conformations. The corresponding histogram of the frequency distribution of the differences in
is the CheShift-2–computed chemical shift of residue μ in conformation i out of Ω conformations. The average of the predicted chemical shifts over the Ω conformations is evaluated because proteins in solution exist as an ensemble of conformations. The corresponding histogram of the frequency distribution of the differences in 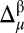 (shown in SI Appendix, Fig. S5A) can be fitted with a Gaussian or normal function with a mean value x0 (0.02 ppm), which is close to the ideal mean value (0.0), and a standard deviation σ (1.77 ppm), with σ used as a criterion to establish a three-state partition of the computed differences per residue,
(shown in SI Appendix, Fig. S5A) can be fitted with a Gaussian or normal function with a mean value x0 (0.02 ppm), which is close to the ideal mean value (0.0), and a standard deviation σ (1.77 ppm), with σ used as a criterion to establish a three-state partition of the computed differences per residue, 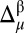 (SI Appendix, Eq. S1).
(SI Appendix, Eq. S1).
Test of the Accuracy of the 13Cβ Chemical Shift Predictions.
As a test of the accuracy of the 13Cβ chemical shift predictions, the correlation coefficient (R) between the observed and computed 13Cβ chemical shifts was computed for the 88 X-ray–determined protein structures and listed for five proteins in SI Appendix, Table S5. The result, R = 0.984, from all 88 structures, is shown in SI Appendix, Fig. S5B. A similar analysis using several other predictive methods (35) led to a higher correlation coefficient, although as noted previously in our analysis for the 13Cα chemical shifts (11), a higher correlation coefficient could mean less capability to detect subtle structural differences rather than more accurate predictions.
Visual Validation of Protein Models.
A similar procedure formulated for mapping the 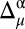 values onto a 3D protein model (12) was used for the
values onto a 3D protein model (12) was used for the 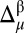 differences, and hence, it is revisited here for the convenience of the reader: first, the
differences, and hence, it is revisited here for the convenience of the reader: first, the 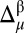 value is computed for each residue μ, and, second, the resulting value is discretized according to the rule given in SI Appendix, Eq. S1. Finally, the discretized values 1, 0, and −1 are mapped onto a 3D protein model and associated with the colors green, yellow, and red, respectively. Implicit in this color-code assignment is the assumption that average differences per residue between observed and predicted 13Cα or 13Cβ chemical shifts which are within ∼1σ (green) are considered small; within ∼2σ (yellow), they are considered medium; and beyond 2σ (red), they are considered large differences. Moreover, the color white was adopted to indicate the absence of the observed or computed 13Cα and/or 13Cβ chemical shift value. The combined use of information from the 13Cα and 13Cβ nuclei enables the CheShift-2 Web server to generate a set of χ1/χ2 side-chain torsional angles that provides optimal agreement between the observed and computed 13Cα and 13Cβ chemical shifts for any red or yellow residue; in such a case, the residues are highlighted in blue. Those residues in blue can become green if any of the proposed side-chain torsional angle solutions satisfies all of the existing restraints, such as nonatomic overlapping or NOE-derived distances. In addition, when more than one protein conformation exists, as in a reported NMR ensemble, the color representation is illustrated only on the first conformation of the ensemble, although average values of
value is computed for each residue μ, and, second, the resulting value is discretized according to the rule given in SI Appendix, Eq. S1. Finally, the discretized values 1, 0, and −1 are mapped onto a 3D protein model and associated with the colors green, yellow, and red, respectively. Implicit in this color-code assignment is the assumption that average differences per residue between observed and predicted 13Cα or 13Cβ chemical shifts which are within ∼1σ (green) are considered small; within ∼2σ (yellow), they are considered medium; and beyond 2σ (red), they are considered large differences. Moreover, the color white was adopted to indicate the absence of the observed or computed 13Cα and/or 13Cβ chemical shift value. The combined use of information from the 13Cα and 13Cβ nuclei enables the CheShift-2 Web server to generate a set of χ1/χ2 side-chain torsional angles that provides optimal agreement between the observed and computed 13Cα and 13Cβ chemical shifts for any red or yellow residue; in such a case, the residues are highlighted in blue. Those residues in blue can become green if any of the proposed side-chain torsional angle solutions satisfies all of the existing restraints, such as nonatomic overlapping or NOE-derived distances. In addition, when more than one protein conformation exists, as in a reported NMR ensemble, the color representation is illustrated only on the first conformation of the ensemble, although average values of 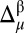 are always computed as described in SI Appendix, Eq. S1, by considering all of the Ω-deposited conformations of the ensemble.
are always computed as described in SI Appendix, Eq. S1, by considering all of the Ω-deposited conformations of the ensemble.
It should be noted that (i) residues 1 and N in the sequence are always displayed in the white representation because, by definition, CheShift-2 does not predict 13Cα or 13Cβ chemical shifts for the first and last residues in the sequence (11); (ii) the 13Cα chemical shifts, but not the 13Cβ chemical shifts, of residues preceding proline need corrections (see first subsection in SI Appendix, Materials and Methods for further details); (iii) all ionizable residues were considered neutral; and (iv) Cystine and Cysteine residues were excluded from our database of 13Cα and 13Cβ chemical shifts because large differences between observed and predicted chemical shifts were obtained.
A Protocol to Reduce Side-Chain Conformational Flaws in Protein Structures.
The combined use of the 13Cα and 13Cβ chemical shifts enables us to improve the agreement between the observed and computed 13Cα and 13Cβ chemical shifts. This is possible for a given residue if all of the following conditions are satisfied: (i) both observed and predicted 13Cα and 13Cβ chemical shifts exist; (ii) at least one of the differences 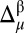 or
or 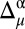 must be >σ (in other words, nuclei needing improvements cannot both be green, in terms of the graphical validation colors); (iii) a residue must pertain to a high-probability region of the Ramachandran map (as defined in third subsection of SI Appendix, Materials and Methods), or else it is not considered for further analysis; and (iv) the torsional angles (ϕ, ψ) of any residue are assumed to be fixed at the values determined by the whole protein structure. When all these conditions are satisfied, all possible χ1 and χ2 torsional angle combinations that would bring the differences
must be >σ (in other words, nuclei needing improvements cannot both be green, in terms of the graphical validation colors); (iii) a residue must pertain to a high-probability region of the Ramachandran map (as defined in third subsection of SI Appendix, Materials and Methods), or else it is not considered for further analysis; and (iv) the torsional angles (ϕ, ψ) of any residue are assumed to be fixed at the values determined by the whole protein structure. When all these conditions are satisfied, all possible χ1 and χ2 torsional angle combinations that would bring the differences 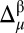 and
and 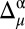 to be <σ for both the 13Cα and 13Cβ nuclei are generated and listed by the upgraded CheShift-2 server. Then, the listed solution (see SI Appendix, Table S2, as an example) can be used to improve the quality, in terms of chemical shifts, of the validated structure.
to be <σ for both the 13Cα and 13Cβ nuclei are generated and listed by the upgraded CheShift-2 server. Then, the listed solution (see SI Appendix, Table S2, as an example) can be used to improve the quality, in terms of chemical shifts, of the validated structure.
The χ1/χ2 side-chain torsional angle solutions provided by the CheShift-2 Web server are determined by assuming a fixed (ϕ, ψ) torsional angle (see condition iv above). Nevertheless, the (ϕ, ψ) torsional angles, and hence the set of χ1/χ2 side-chain torsional angle solutions, may vary among the conformations of the ensemble used to represent a protein. Therefore, the server provides solutions for each conformation in the ensemble used to represent the protein structure.
Supplementary Material
Acknowledgments
We thank James Aramini and Sai Tong from Rutgers, The State University of New Jersey for helpful assistance with the use of the Protein Structure Validation Software Suite server. We thank all spectroscopists and crystallographers who have deposited their coordinates at the Protein Data Bank and chemical shifts at the BioMagResBank; without their efforts, this work would not have been possible. The research was conducted by using the resources of Blacklight, a facility of the National Science Foundation Terascale Computing System at the Pittsburgh Supercomputer Center. This research was supported by National Institutes of Health Grant GM14312 (to H.A.S.); Grant PIP-112-2011-0100030 from Instituto de Matemática Aplicada San Luis, Consejo Nacional de Investigaciones Científicas y Técnicas de Argentina (to J.A.V.); and Project 328402 from the National University of San Luis (J.A.V.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1315525110/-/DCSupplemental.
References
- 1.Kendrew JC. The structure of globular proteins. Comp Biochem Physiol. 1962;4(2-4):249–252. doi: 10.1016/0010-406x(62)90009-9. [DOI] [PubMed] [Google Scholar]
- 2.Olson MA, Lee MS. Structure refinement of protein model decoys requires accurate side-chain placement. Proteins. 2013;81(3):469–478. doi: 10.1002/prot.24204. [DOI] [PubMed] [Google Scholar]
- 3.Sahakyan AB, Cavalli A, Vranken WF, Vendruscolo M. Protein structure validation using side-chain chemical shifts. J Phys Chem B. 2012;116:4754–4759. doi: 10.1021/jp2122054. [DOI] [PubMed] [Google Scholar]
- 4.Kleywegt GJ. On vital aid: The why, what and how of validation. Acta Crystallogr D Biol Crystallogr. 2009;65(Pt 2):134–139. doi: 10.1107/S090744490900081X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vila JA, Scheraga HA. Assessing the accuracy of protein structures by quantum mechanical computations of 13C(α) chemical shifts. Acc Chem Res. 2009;42(10):1545–1553. doi: 10.1021/ar900068s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vriend G. WHAT IF: A molecular modeling and drug design program. J Mol Graph. 1990;8(1):52–56, 29. doi: 10.1016/0263-7855(90)80070-v. [DOI] [PubMed] [Google Scholar]
- 7.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26(2):283–291. [Google Scholar]
- 8.Huang YJ, Powers R, Montelione GT. Protein NMR recall, precision, and F-measure scores (RPF scores): Structure quality assessment measures based on information retrieval statistics. J Am Chem Soc. 2005;127(6):1665–1674. doi: 10.1021/ja047109h. [DOI] [PubMed] [Google Scholar]
- 9.Nabuurs SB, Spronk CAEM, Vuister GW, Vriend G. Traditional biomolecular structure determination by NMR spectroscopy allows for major errors. PLOS Comput Biol. 2006;2(2):e9. doi: 10.1371/journal.pcbi.0020009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhattacharya A, Tejero R, Montelione GT. Evaluating protein structures determined by structural genomics consortia. Proteins. 2007;66(4):778–795. doi: 10.1002/prot.21165. [DOI] [PubMed] [Google Scholar]
- 11.Vila JA, Arnautova YA, Martin OA, Scheraga HA. Quantum-mechanics-derived 13Calpha chemical shift server (CheShift) for protein structure validation. Proc Natl Acad Sci USA. 2009;106(40):16972–16977. doi: 10.1073/pnas.0908833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin OA, Vila JA, Scheraga HA. CheShift-2: Graphic validation of protein structures. Bioinformatics. 2012;28(11):1538–1539. doi: 10.1093/bioinformatics/bts179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spera S, Bax A. Empirical correlation between protein backbone conformation and Cα and Cβ 13C nuclear magnetic resonance chemical shifts. J Am Chem Soc. 1991;113(14):5490–5492. [Google Scholar]
- 14.Kuszewski J, Qin J, Gronenborn AM, Clore GM. The impact of direct refinement against 13C α and 13C β chemical shifts on protein structure determination by NMR. J Magn Reson B. 1995;106(1):92–96. doi: 10.1006/jmrb.1995.1017. [DOI] [PubMed] [Google Scholar]
- 15.Vila JA, et al. Quantum chemical 13C(α) chemical shift calculations for protein NMR structure determination, refinement, and validation. Proc Natl Acad Sci USA. 2008;105(38):14389–14394. doi: 10.1073/pnas.0807105105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robustelli P, Kohlhoff K, Cavalli A, Vendruscolo M. Using NMR chemical shifts as structural restraints in molecular dynamics simulations of proteins. Structure. 2010;18(8):923–933. doi: 10.1016/j.str.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 17.Rosato A, et al. Blind testing of routine, fully automated determination of protein structures from NMR data. Structure. 2012;20(2):227–236. doi: 10.1016/j.str.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wishart DS, Bigam CG, Holm A, Hodges RS, Sykes BD. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J Biomol NMR. 1995;5(1):67–81. doi: 10.1007/BF00227471. [DOI] [PubMed] [Google Scholar]
- 19.Schwarzinger S, et al. Sequence-dependent correction of random coil NMR chemical shifts. J Am Chem Soc. 2001;123(13):2970–2978. doi: 10.1021/ja003760i. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Jardetzky O. Investigation of the neighboring residue effects on protein chemical shifts. J Am Chem Soc. 2002;124(47):14075–14084. doi: 10.1021/ja026811f. [DOI] [PubMed] [Google Scholar]
- 21.Vila JA, Serrano P, Wüthrich K, Scheraga HA. Sequential nearest-neighbor effects on computed 13Calpha chemical shifts. J Biomol NMR. 2010;48(1):23–30. doi: 10.1007/s10858-010-9435-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iwadate M, Asakura T, Williamson MP. C α and C β carbon-13 chemical shifts in proteins from an empirical database. J Biomol NMR. 1999;13(3):199–211. doi: 10.1023/a:1008376710086. [DOI] [PubMed] [Google Scholar]
- 23.Villegas ME, Vila JA, Scheraga HA. Effects of side-chain orientation on the 13C chemical shifts of antiparallel β-sheet model peptides. J Biomol NMR. 2007;37(2):137–146. doi: 10.1007/s10858-006-9118-6. [DOI] [PubMed] [Google Scholar]
- 24.Dunbrack RL, Karplus M. Conformational analysis of the backbone-dependent rotamer preferences of protein side chains. J Mol Biol. 1994;230:543–574. doi: 10.1006/jmbi.1993.1170. [DOI] [PubMed] [Google Scholar]
- 25.Chakrabarti P, Pal D. Main-chain conformational features at different conformations of the side-chains in proteins. Protein Eng. 1998;11(8):631–647. doi: 10.1093/protein/11.8.631. [DOI] [PubMed] [Google Scholar]
- 26.Bernstein FC, et al. The Protein Data Bank: A computer-based archival file for macromolecular structures. J Mol Biol. 1977;112(3):535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 27.Cornilescu G, Marquardt JL, Ottiger M, Bax A. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J Am Chem Soc. 1998;120(27):6836–6837. [Google Scholar]
- 28.Vijay-Kumar S, Bugg CE, Cook WJ. Structure of ubiquitin refined at 1.8 A resolution. J Mol Biol. 1987;194(3):531–544. doi: 10.1016/0022-2836(87)90679-6. [DOI] [PubMed] [Google Scholar]
- 29.Furnham N, Blundell TL, DePristo MA, Terwilliger TC. Is one solution good enough? Nat Struct Mol Biol. 2006;13(3):184–185, discussion 185. doi: 10.1038/nsmb0306-184. [DOI] [PubMed] [Google Scholar]
- 30.Arnautova YA, Vila JA, Martin OA, Scheraga HA. What can we learn by computing 13Calpha chemical shifts for X-ray protein models? Acta Crystallogr D Biol Crystallogr. 2009;65(Pt 7):697–703. doi: 10.1107/S0907444909012086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schrödinger, LLC (2013) The PyMOL Molecular Graphics System (Schrödinger, LLC, San Diego), Version 1.5.0.1.
- 32.Shen Y, Bax A. Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J Biomol NMR. 2013;56(3):227–241. doi: 10.1007/s10858-013-9741-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lovell SC, Word JM, Richardson JS, Richardson DC. The penultimate rotamer library. Proteins. 2000;40(3):389–408. [PubMed] [Google Scholar]
- 34.Ulrich EL, et al. BioMagResBank. Nucleic Acids Res. 2008;36(Database issue):D402–D408. doi: 10.1093/nar/gkm957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han B, Liu Y, Ginzinger SW, Wishart DS. SHIFTX2: Significantly improved protein chemical shift prediction. J Biomol NMR. 2011;50(1):43–57. doi: 10.1007/s10858-011-9478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.