

Abstract
The overall goal of this study was to determine the role of Rac1 in POSH/MLK/JNK signaling and delayed neuronal cell death following cerebral ischemia. Temporal studies revealed that Rac1 GTPase activation was significantly elevated in hippocampus CA1 at 10 min to 72h after cerebral ischemia reperfusion, with peak levels 30 min to 6h after reperfusion. Total Rac1 protein levels were not significantly changed following cerebral ischemia. Rac1 has been shown to interact with POSH (plenty of SH3s), a scaffold protein that binds to and regulates MLK3 and JNK activation. Co-immunoprecipitation (Co-IP) studies revealed that POSH-Rac1-MLK3 complex formation displayed a significant and prolonged elevation after reperfusion, with a correlative increase in phosphorylation/activation of MLK3 as compared to sham controls. Intracerebroventricular administration of Rac1 antisense oligonucleotides (AS-ODNs) significantly attenuated Rac1 levels and Rac1 activation at 30min after reperfusion, with a correlated significant attenuation of POSH-MLK3-Rac1 complex formation and MLK3 activation in hippocampus CA1. Infusion of Rac1 AS-ODNs also significantly attenuated post-ischemic activation of JNK, downstream of MLK3, and strongly protected the hippocampus CA1 from ischemic damage. Missense oligos had no effect on any of the parameters measured. The Rac1 AS-ODNs results were further confirmed by administration of a Rac1 inhibitor (NSC23766), which markedly attenuated activation of Rac1 and JNK, and significantly attenuated delayed neuronal cell death following cerebral ischemia. As a whole, these studies demonstrate an important role for Rac1 in activation of the prodeath MLK3-JNK kinase signaling pathway and delayed neuronal cell death following cerebral ischemia.
Keywords: Global cerebral ischemia, hippocampus, cell signaling
1. INTRODUCTION
Pyramidal neurons in hippocampus CA1 are highly sensitive to global ischemic damage, with delayed neuronal cell death typically occurring 2–4 days after the induction of global ischemia in rats and gerbils (Pulsinelli et al., 1982; Tanaka et al., 2000). The cellular decision to undergo delayed cell death is determined by the integration of multiple survival and death signals. There is increasing evidence that c-Jun N-terminal kinase (JNK) is an important pro-death signal mediating the demise of pyramidal CA1 neurons in response to cerebral ischemia (Irving and Bamford, 2002; Nozaki et al., 2001; Ozawa et al., 1999). Along these lines, JNK has been shown to be rapidly activated in hippocampus CA1 following cerebral ischemia (Gu et al., 2001; Ozawa et al., 1999; Zhang et al., 2003; Zhang et al., 2005), and inhibition of JNK activation by small peptide inhibitors or JNK antisense oligodeoxynucleotides (AS-ODNs) significantly inhibits post-ischemic neuronal death and preserves cognitive function (Carboni et al., 2008; Gu et al., 2001; Guan et al., 2006). Additionally, targeted disruption of the JNK3 gene reduced phosphorylation of the downstream effector c-Jun, and profoundly protected the mice from brain injury after cerebral ischemia–hypoxia (Kuan et al., 2003). JNK appears to induce cell apoptosis by activating caspase 3 and by inducing N-terminal phosphorylation of the inducible transcription factor, c-Jun (Gelderblom et al., 2004).
Recent work suggests that Rac1, a Rho family small GTPase, is an upstream regulator of JNK signaling (Jin et al., 2006; Kukekov et al., 2006; Teramoto et al., 1996; Xu et al., 2003). Rac1 exists in an inactive (GDP-bound) state until signals such as cellular stress, growth factors, or G-protein receptor activation convert it into an active (GTP-bound) state (Linseman and Loucks, 2008). The binding of GTP induces a conformational change in Rac1, which promotes its association with a diverse array of downstream effectors including protein kinases, lipid kinases, scaffolding proteins, and phospholipases. One recently identified Rac1-interactor protein is the multi-domain scaffold protein POSH (plenty of SH3s), which regulates JNK signaling by forming a complex with JNK and its upstream activating enzymes, mixed lineage kinases (MLK1-3), and MAP kinase kinase 4 and 7 (MKK4, MKK7) (Kukekov et al., 2006; Tapon et al., 1998; Xu et al., 2003). Recent work by our group showed that antisense oligonucleotide knockdown of POSH attenuates JNK and c-Jun activation in the hippocampus CA1 following global cerebral ischemia and protects from ischemic neuronal cell death (Zhang et al., 2005), implicating an important role for POSH in ischemic-induced JNK signaling.
Since POSH can interact with Rac1, Rac1 may have an important role in modulation of POSH complex formation following cerebral ischemia, as well as ischemia induced MLK3/JNK signaling and delayed neuronal cell death. The present study was designed to address this issue by using a variety of approaches to determine Rac1 activation changes following global cerebral ischemia, and to assess the effect of Rac1 knockdown or inhibition. The study provides novel insights by showing that Rac1 exhibits a rapid and prolonged activation in the hippocampus CA1 following global cerebral ischemia, that Rac1 activation parallels and is essential for POSH-Rac1-MLK3 complex formation, as well as MLK3 and JNK activation following cerebral ischemia, and that knockdown or inhibition of Rac1 significantly protects the hippocampus CA1 from delayed neuronal cell death following global cerebral ischemia.
2. RESULTS
Rac1 Activation in Hippocampus CA1 Following Global Cerebral Ischemia
To characterize Rac1 modulation following cerebral ischemia, we examined Rac1 levels and Rac1 GTPase activation in hippocampus CA1 at different reperfusion timepoints following 15-min global cerebral ischemia. As shown in Fig. 1A&B, Western blot analysis revealed that total Rac1 protein levels in hippocampus CA1 do not change significantly at any timepoint examined following reperfusion. In contrast, activated Rac1 (Rac1-GTP) showed a rapid and prolonged elevation from 10 min to 72h after reperfusion, as compared to sham controls. Peak Rac1 GTPase activation levels (increased approximately 3.1 fold over sham) were observed at 3 h after reperfusion, whereas by 72 h Rac1 GTPase activation had fallen, but was still 1.7 fold increased over sham controls.
Fig. 1. Time course of the activation of Rac1 in the hippocampal CA1 region after global cerebral ischemia.

(A) Homogenates from the CA1 region at various time points of reperfusion (sham, 10 min, 30 min, 3h, 6 h, 24 h and 72 h) were subjected to Rac1 activation assay. (B) Graphical depiction of results from all animals. Corresponding bands were scanned and the optical density (OD) was represented as folds versus sham control. Data are expressed as means ± SD from four independent animals (n = 4), *p < 0.05 versus sham control.
Elevation of POSH-MLK3-Rac1 Complex Formation and MLK3 Phosphorylation in Hippocampus CA1 Following Global Cerebral Ischemia
POSH is a Rac1-binding protein and scaffold protein that has been previously implicated to mediate MLK-JNK activation. Since Rac1 showed a rapid and prolonged activation after global cerebral ischemia/reperfusion, we wanted to determine if there was a correlative increase in POSH-MLK3-Rac1 complex formation and MLK3 activation in the hippocampus CA1 following global cerebral ischemia. We thus examined the biochemical ability of POSH to interact with Rac1 and MLK3 at various time points (sham, 10 min, 30 min, 6 h, 24h and 72h) of reperfusion after 15 min of ischemia. The sample proteins from the hippocampal CA1 regions were immunoprecipitated with antibody against POSH then immunoblotted with antibodies against Rac1 and MLK3, respectively. We found that global cerebral ischemia and reperfusion induced rapid and sustained increases in the interactions between POSH and Rac1, and MLK3, as shown in Fig. 2A&D, with peak increases at 30 min (2–3 fold over sham controls), indicating that Rac1 and MLK3 formed a complex with POSH after ischemic reperfusion. Total POSH expression kept unchanged. In reciprocal co-immunoprecipitation experiments, homogenates from the hippocampal CA1 regions at 30 min of reperfusion were subjected to immunoprecipitation with antibodies against Rac1 and MLK3, or nonspecific IgGs and the immunocomplexes were probed for the presence of POSH with POSH-specific antiserum. As shown in Fig. 2B, the results confirmed the interaction of POSH with Rac1 and MLK3, while non-specific IgGs as controls had negligible effects, confirming their specificity. Finally, in agreement with enhanced POSH-Rac1-MLK3 complex formation following ischemic reperfusion, we found that MLK3 phosphorylation is enhanced in the hippocampus CA1 from 10 min – 72h following reperfusion, with peak levels at 30 min (Fig. 2C&D), which paralleled the enhanced POSH-Rac1-MLK3 complex formation.
Fig. 2. Time courses of the associations of POSH with Rac1 and MLK3 and phosphorylation of MLK3 in the hippocampal CA1 region after cerebral ischemia.

(A) Homogenates from the CA1 region at various time points after reperfusion (sham, 10 min, 30 min, 6 h, 1 and 3 days) were immunoprecipitated (IP) with anti-POSH antibody, then separately blotted (WB) with anti-Rac1, MLK3 or POSH antibody. (B) In reciprocal co-immunoprecipitation experiments, homogenates were subjected to immunoprecipitation with anti-Rac1, MLK3 or non-specific IgG (control) and the immunocomplexes were probed for the presence of POSH. (C) Homogenates from the CA1 region at various time points of reperfusion were western blotted with antibody against MLK3 or p-MLK3. (D) Corresponding bands from A&C were scanned and the optical density (OD) was represented as folds versus sham control. Data are expressed as means ± SD from independent animals (n = 4–5), *p < 0.05 versus sham control.
Rac1 AS-ODNs Significantly Attenuates Rac1 Activation and POSH-Rac1-MLK3 Complex Formation in Hippocampus CA1 Following Global Cerebral Ischemia
To investigate the possible relationship between Rac1 activation and POSH-MLK3-JNK signaling activation, we next examined the alteration of Rac1 expression and activation after i.c.v. injection of the Rac1 AS-ODNs using Rac1 activation assay and Western blot analysis. The results showed that Rac1 AS-ODNs markedly decreased its protein expression when compared with vehicle or missense ODNs in the rat hippocampal CA1 region 30 min after reperfusion (Fig 3A&B). Rac1 AS-ODNs also significantly inhibited Rac1 activation when compared with vehicle or missense ODNs in the rat hippocampal CA1 region 30 min after reperfusion (Figs 3A&B).
Fig. 3. Effect of Rac1 AS-ODNs (AS) or missense oligonuleotides (MS) on Rac1 expression and activation or MLK3/Rac1/POSH complex formation in CA1 region at 30 min of reperfusion following cerebral ischemia.
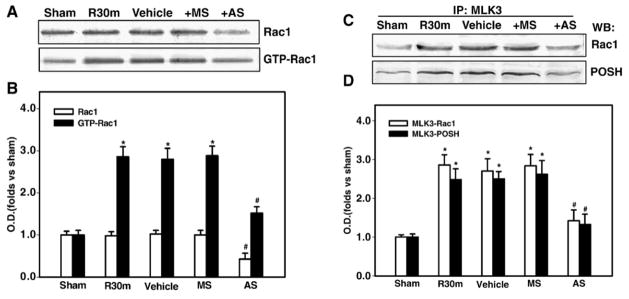
(A,B) The AS-ODNs administration significantly attenuated cerebral ischemia-induced Rac1 expression and activation at 30 min after ischemia. Missense ODNs (MS) had no significant effect on Rac1 expression or activation in CA1 as compared to Vehicle control. (C,D) Homogenates from the hippocampal CA1 region at 30 min of reperfusion were immunoprecipitated with anti-MLK3 antibody, and then separately blotted with anti-Rac1 or POSH antibody. (B,D) Bands on western blots were scanned and the intensities were expressed as folds versus sham treatment. Data are means ± SD from four independent animals (n = 4). *p < 0.05 versus sham group. # p < 0.05 versus MS group.
To further examine the contribution of Rac1 to ischemia-induced activation of POSH-MLK3-JNK signaling in the hippocampal CA1 region, rats were treated with Rac1 AS-ODNs and the samples from the hippocampal CA1 region were co-immunoprecipitated with an anti-MLK3 antibody. The immunoreactivity of MLK3-associated POSH and Rac1 were examined by western blot analysis. Figure 3C&D showed that there was a significant decrease in the expression of Rac1 and POSH precipitated by MLK3 in hippocampus CA1 at 30 min of reperfusion after AS-ODNs infusion. Missense ODNs had no effect on the expression of Rac1 or POSH precipitated by MLK3 following reperfusion, demonstrating the specificity of the Rac1 AS-ODNs effect. Additionally, reciprocal immunoprecipitation experiments confirmed the decreased interaction of MLK3 with Rac1 and POSH (data not shown).
Rac1 AS-ODNs Significantly Attenuates Cerebral Ischemia-Induced Activation of MLK3 and JNK in Hippocampus CA1
Since Rac1 was shown to be critical for ischemic regulation of POSH complex component interactions, which is known to regulate MLK3-JNK signaling, we next examined the effect of Rac1 AS-ODNs on phosphorylation of MLK3 and JNK following global cerebral ischemia. Western blot analysis using the samples from the hippocampal CA1 region showed that the phosphorylation of MLK3 and JNK were simultaneously decreased at 30 min of reperfusion by Rac1 AS-ODNs, while their activation was still maintained in rats treated with missense Rac1 (Figs 4A–D). The total proteins of MLK3 and JNK were unchanged. Taken together, these findings strongly suggest that Rac1 is a critical regulator of the POSH multiprotein complex activation following cerebral ischemia, which mediates downstream activation of the pro-death JNK signaling pathway.
Fig. 4. Effect of Rac1 AS-ODNs (AS) or missense oligonuleotides (MS) on the phosphorylation of MLK3 or JNK in CA1 region at 30 min of reperfusion following cerebral ischemia.
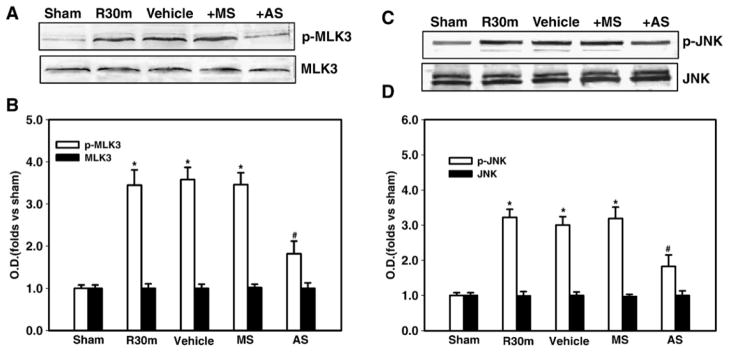
(A,C) The AS-ODNs administration significantly attenuated cerebral ischemia-induced MLK3 phosphorylation and JNK phosphorylation in hippocampus CA1 at 30 min after ischemia. Missense ODNs (MS) had no significant effect on MLK3 and JNK phosphorylation in hippocampus CA1 as compared to Vehicle control. (B,D) Bands on western blots were scanned and the intensities were expressed as folds versus sham treatment. Data are means ± SD from four to five independent animals. *p < 0.05 versus sham group. # p < 0.05 versus MS group.
Rac1 Inhibition by NSC23766 Significantly Attenuates Cerebral Ischemia-Induced Activation of Rac1 and JNK in Hippocampus CA1 Following Global Cerebral Ischemia
To further confirm the Rac1 AS-ODNs findings and the role of Rac1, we administered a Rac1 GTPase-specific small molecule inhibitor, NSC23766 via i.c.v. injection and examined the effect upon Rac1 activation and phosphorylation of JNK following cerebral ischemia. As shown in Fig. 5A&B, administration of NSC23766 15 min before cerebral ischemia significantly attenuated Rac1 GTPase activation in hippocampus CA1 at 3 h of reperfusion, demonstrating the effective inhibition of Rac1 activation by NSC23766. Furthermore, inhibition of Rac1 activation by administration of the Rac1 inhibitor was correlated with a significant inhibition of cerebral ischemia-induced phosphorylation of JNK in hippocampus CA1 at 3 h after global cerebral ischemia (Fig. 5C&D), further confirming the important role of Rac1 in JNK signaling activation following cerebral ischemia.
Fig. 5. Effect of Rac1 inhibitor (NSC23766) on Rac1 activation and JNK activation in CA1 region at 3 h of reperfusion following cerebral ischemia.
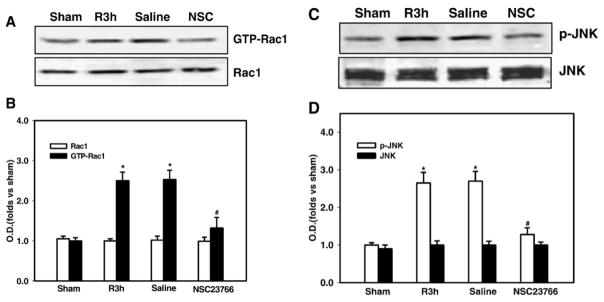
(A,C) NSC23766 injection significantly attenuated cerebral ischemia-induced Rac1 activation and phosphorylation of JNK in hippocampus CA1 at 3 h after ischemia. Saline had no significant effect on Rac1 activation and JNK activation in hippocampus CA1 as compared to ischemia reperfusion control. (B,D) Bands on western blots were scanned and the intensities were expressed as folds versus sham treatment. Data are means ± SD from three independent animals. *p < 0.05 versus sham group. # p < 0.05 versus saline group.
Administration of Rac1 AS-ODNs or the Rac1 Inhibitor (NSC23766) Significantly Attenuates Cerebral Ischemia-Induced Apoptotic Neuronal Cell Death
To establish the functional importance of Rac1 activation in cerebral ischemia events, we examined the effect of Rac1 knockdown by Rac1 AS-ODNs or Rac1 inhibition by the Rac1 inhibitor, NSC23766 upon delayed neuronal cell death 5 days after global cerebral ischemia. Cresyl violet staining was used to examine the surviving cells of CA1 pyramidal neurons. Normal cells showed round and pale stained nuclei. The shrunken cells with pyknotic nuclei after reperfusion were counted as dead cells. TUNEL staining was also used to assess the apoptotic like neurons in hippocampal CA1 region. As shown in Fig. 6, global cerebral ischemia followed by 5 days of reperfusion induced severe cell death (Figs 6A(d,I,n)) as compared to sham controls (Fig 6A (a,f,k)). However, administration of Rac1 AS-ODNs markedly attenuated apoptotic neuronal degeneration (Figs 6A(c,h,m)&B). In contrast, treatment with the non-specific ODNs did not improve the neuronal density (Figs 6A(b,g,i)&B), suggesting that antisense action was specific for Rac1. Furthermore, treatment with the Rac1 inhibitor, NSC23766 strongly attenuated neuronal cell death following global cerebral ischemia (Figs 6A(e,j,o)&B). The results demonstrate a critical role for Rac1 in delayed neuronal cell death following global cerebral ischemia.
Fig. 6. Effects of Rac1 AS-ODNs or Rac1 inhibitor (NSC23766) on delayed apoptotic neuronal death by histology analysis.

(A) Cresyl violet and TUNEL staining was performed on the sections from the hippocampi in sham (a, f, k), and rats subjected to 5 days of reperfusion after 15 min of ischemia with saline control (d, I, n), administration of the Rac1 missense ODNs (b, g, l) and Rac1 AS-ODNs (c, h, m) or Rac1 inhibitor NSC23766 (e, j, o) before ischemia. (B) The number of survival CA1 neurons per 1mm length of the medial CA1 pyramidal cell layer was counted as indicated. Note that treatment with Rac1 AS-ODNs or NSC23766 significantly increased the number of the survival neurons compared with control at 5 days of reperfusion in the CA1 region. Data were obtained from five independent animals and a typical experiment is presented. Boxed areas in left column are shown at higher magnification in right column.*p < 0.05 versus sham control. # p < 0.05 versus MS or saline group. (a–e) magnification is 5° and the scale bar = 200 μm; (f–o) magnification is 40° and the scale bar = 50 μm.
3. DISCUSSION
Previous work has shown that POSH directly binds both active Rac1 and MLK1-3, and the subsequent interaction of active Rac1 with MLK1-3 induces autophosphorylation and activation of MLK1-3 (Kukekov et al., 2006; Tapon et al., 1998; Xu et al., 2003). POSH also directly binds a second scaffold protein JIP (JNK-interacting protein 1 or 2), and JIP completes the complex by binding MKK 4/7 and JNK1-3. Activated MLK can then phosphorylate and activate MKK4/7, which in turn phosphorylates and activates JNK1-3 (Kukekov et al., 2006; Xu and Greene, 2006). Our present study demonstrates that Rac1 undergoes a rapid and prolonged activation in the hippocampus CA1 following global cerebral ischemia, that its activation parallels POSH-MLK3-Rac1 complex formation, and that MLK3/JNK phosphorylation and delayed neuronal cell death following global cerebral ischemia is mediated by Rac1 activation.
Whereas, the role of Rac1 in promoting growth cone motility, axonal migration and dendritic spine morphogenesis in the brain has been intensely studied (Linseman and Loucks, 2008), the role of Rac1 in cell death has only been recently appreciated. Work in intestinal epithelial cells has provided evidence that Rac1 has a proapoptotic role, as administration of a Rac1 inhibitor or a Rac1 dominant negative prevented TNF-α induced apoptosis (Jin et al., 2006). Furthermore, Rac1 inhibition was shown to attenuate TNF-α induced JNK1/2 activation, although the mechanism underlying the attenuation was not elucidated (Jin et al., 2006). Our work extends the pro-death role of Rac1 to the hippocampus CA1 following global cerebral ischemia, and provides mechanistic insights as to how Rac1 modulates JNK activation.
Using a Rac1 activation assay, we established that Rac1 GTPase activation in the hippocampal CA1 region undergoes a rapid and prolonged elevation following global cerebral ischemia in the rat. The mechanism underlying activation of Rac1 following cerebral ischemia is unclear. However, the Rac1 inhibitor (NSC23766) we used in our study may shed some light on this issue. NSC23766 was identified by a structure-based virtual screening of compounds that fit into a surface groove of Rac1 known to be critical for GEF specification. NSC23766 was shown to effectively inhibit Rac1 binding and activation by the Rac-specific GEF such as Trio and Tiam1 in a dose-dependent manner (Gao et al., 2004). Thus, Rac1 activation in hippocampus CA1 following cerebral ischemia is likely mediated by GEFs, based on the known specificity of NSC23766. In support of this possibility, both Trio and Tiam1 are expressed in the hippocampus CA1 (Ehler et al., 1997; Ma et al., 2005), and interestingly Tiam1 has been shown to mediate NMDA receptor activation of Rac1 in neurons (Tolias et al., 2005). This is intriguing as activation of NMDA receptors by excess glutamate is a proposed early step in the signaling cascade underlying cerebral ischemia-induced neuronal damage and cell death (Benquet et al., 2008). Interestingly, glutamate has been shown to induce activation of Rac1 in human neuroblastoma cells, an effect correlated with glutamate-induced neuronal cell death (Nikolova et al., 2005).
Our study suggests that Rac1 proapoptotic effect in cerebral ischemia involves enhancement of POSH-MLK-JNK signaling. This agrees with a growing body of evidence that POSH is an important proapototic factor in both neuronal and non-neuronal cells (Tapon et al., 1998; Xu et al., 2003; Xu and Greene, 2006). For instance, overexpression of POSH has been shown to induce apoptosis in COS-1 kidney cells, PC12 cells and cultured sympathetic neurons, an effect blocked by dominant negative forms of MLKs, MKK4/7 and c-Jun (Tapon et al., 1998; Xu et al., 2003). Additionally, POSH siRNAs or AS-ODNs suppress c-Jun phosphorylation and neuronal apoptosis induced by nerve growth factor withdrawal in differentiated PC12 cells (Tapon et al., 1998). Furthermore, our group reported that POSH antisense knockdown attenuates JNK signaling and protects the hippocampus CA1 from neuronal cell death following global cerebral ischemia (Zhang et al., 2005).
POSH appears to be a nexus for regulation by both pro-death and pro-survival signals. The present study demonstrates the critical role of the pro-death signal Rac1 in POSH complex formation and regulation of MLK3-JNK signaling activation. Recent work further demonstrates that pro-survival signals can also regulate POSH function. For instance, the pro-survival signal Akt has been shown to phosphorylate POSH at Ser304, which lies within the Rac1-binding domain (Lyons et al., 2007). Akt-induced phosphorylation of POSH at Ser304 resulted in a decreased ability of POSH to bind activated Rac1, and a decreased ability of POSH to induce apoptosis (Lyons et al., 2007). Furthermore, we recently demonstrated that enhanced Akt activation leads to phosphorylation of Rac1 and deactivation of MLK3 in hippocampus CA1 following tyrosine phosphatase inhibition in animals that had undergone global cerebral ischemia (Zhang et al., 2006). Thus, Akt negatively regulates Rac1 and POSH function by multiple mechanisms that contribute to its prosurvival effect in the brain. Since Akt activation is known to be suppressed following cerebral ischemia, the Rac1 activation following cerebral ischemia observed in our study is therefore not restrained, and activation of POSH ensues, eliciting a JNK proapoptotic signaling cascade.
After POSH-MLK1-3 complex formation and activation of MLK1-3/MKK4/7 signaling that leads to JNK activation, JNK phosphorylates Ser63 and Ser73 residues of c-Jun and increases activator protein-1 (AP-1) transcription activity (Gupta et al., 1996; Karin, 1995). Previous studies have shown that c-Jun is specifically phosphorylated in CA1 at an early stage of reperfusion and is closely associated with ischemia-induced neuronal apoptosis (Matsuoka et al., 1999). Our study further demonstrates the upstream role of Rac1 in c-Jun activation following cerebral ischemia, as evidenced by the inhibition of JNK phosphorylation following treatment with either Rac1 AS-ODNs or the Rac1 inhibitor NSC23766. In our AS-ODN studies, the specificity of the Rac1 AS-ODNs was confirmed by a lack of significant effect by missense ODNs. Additionally, use of the Rac1 inhibitor NSC23766 further confirmed the role of activated Rac1 in MLK3-JNK signaling and delayed neuronal cell death in the hippocampus CA1 following global cerebral ischemia.
Finally, it should be mentioned that active Rac1 has been shown to regulate NADPH oxidase activation and superoxide production in many cells due to its ability to bind to P67Phox and to bind and directly regulate NOX2 (Bedard and Krause, 2007; Hordijk, 2006; Kao et al., 2008). Thus, activation of NADPH oxidase/superoxide production by Rac1 may contribute to Rac1 pro-death effects in cells. To our knowledge, the role of Rac1 in NADPH oxidase and superoxide production in the hippocampus CA1 after cerebral ischemia has not been examined. However, studies in a liver ischemia/reperfusion models implicates a role for Rac1 in reactive oxygen species (ROS) production. For instance, adenoviral expression of a dominant negative Rac1 suppressed ischemia/reperfusion-induction of ROS and apoptosis in a liver ischemia/reperfusion injury model (Harada et al., 2003; Ozaki et al., 2000). Additionally, administration of the Rac1 inhibitor, NSC23766 prevented TNF-α induction of ROS production and apoptosis in epithelial cells (Jin et al., 2008). These observations, coupled with the findings in the current study, indicate that Rac1 can bind and interact with multiple protein signaling complexes in the cell to modulate cell survival. This suggests that Rac1 may be an especially attractive target for drug therapy in stroke and other neurodegenerative disorders that involve ROS and JNK activation.
In conclusion, the current study provides new mechanistic insights into the regulation and role of Rac1 in the hippocampus CA1 following cerebral ischemia. The study demonstrates that Rac1 activation is an early upstream event that is critical for POSH-MLK3 complex formation, activation of MLK3-JNK signaling, and induction of delayed neuronal cell death following global cerebral ischemia. Our finding of a significant pro-death role of Rac1 in cerebral ischemia, suggests that targeting Rac1 for inhibition may have clinical efficacy as a potential neuroprotective treatment in stroke.
4. EXPERIMENTAL PROCEDURE
Materials
Anti-Rac1, anti-phospho-MLK3 (Thr277/Ser281) antibodies were from Cell Signaling Technology (Beverly, MA, USA). Antibodies to POSH, MLK3, JNK and active JNK (recognizes active JNK1, JNK2 and JNK3) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Nitrocellulose filter was from Amersham Pharmacia (Little Chalfont, UK). BCIP (5-bromo-4-chloro-3-indolyl-phosphate) and NBT (nitro blue tetrazolium) were from Promega (Madison, WI, USA). Rac1 inhibitor NSC 23766 was from Tocris Cookson Inc. (Ballwin, MO, USA). All the other chemicals were from Sigma (St Louis, MO, USA) unless indicated otherwise.
Intracerebroventricular administration
To investigate the possible role of Rac1 in response to ischemic injury, 10 nmol of end-phosphorothioated Rac1 AS-ODNs in 10 μL TE buffer (10 mM Tris-HCl (PH 8.0), 1 mM EDTA) were administrated to the left cerebral ventricles of rats every 24 h for 3 days before cerebral ischemia by means of intracerebroventricular (i.c.v.) infusion. The same dose of missenses (MS) or vehicle (TE) was used as controls. The sequences for Rac1 AS-ODNs used in this study were 5′-ACTTGATGGCCTGCAT-3′ from the Integrated DNA Technologies, Inc. and missenses were 5′-GTATGGGACTCTACCT-3′. In other experimental conditions, 25 μg NSC23766 was administered to both of the cerebral ventricles of rats 15 min before cerebral ischemia. For i.c.v. injection, the rats were placed on ear bars of a stereotaxic instrument under anesthesia. Drug infusion was performed using a steppermotorized microsyringe (Stoelting, Wood Dale, IL, USA) at a rate of 1 μL/min through a pre-implanted cannula in the cerebral ventricle (from the bregma: anteroposterior, ±0.8 mm; lateral, 1.5 mm; depth, 3.5 mm).
Induction of forebrain ischemia
Adult male Sprague–Dawley rats weighing 250–300 g were used (all procedures were approved by the institutional animal care committee). Brain ischemia was induced by four-vessel occlusion, as described previously (Gu et al., 2001; Pulsinelli and Brierley, 1979). Briefly, under anesthesia with chloral hydrate (350 mg/kg, i.p.), vertebral arteries were electrocauterized and common carotid arteries were exposed. Rats were allowed to recover for 24 h and fasted overnight. Ischemia was induced by occluding the common arteries with aneurysm clips for 15 min. Rats which lost their righting reflex within 30 s and whose pupils were dilated and unresponsive to light during ischemia were selected for the experiments. An EEG was monitored to ensure isoelectricity within 30 s after carotid artery occlusion. Carotid artery blood flow was restored by releasing the clips. Rectal temperature was maintained at about 37°C during and 2 h after ischemia. Sham controls were performed using the same surgical exposure procedures, except that the arteries were not occluded.
Tissue preparation
For brain tissue preparation, rats were sacrificed under isoflurane anesthesia at 10 min, 30 min, 3 h, 6 h, 1 and 3 days after 15 min of global cerebral ischemia. The hippocampal CA1 region microdissected from both sides of the hippocampal fissure and immediately frozen in liquid nitrogen. Tissues were homogenized with a Teflon-glass homogenizer in ice cold homogenization medium consisting of 50 mM HEPES (pH 7.4), 150 mM NaCl, 12 mM β-glycerophosphate, 3 mM dithiotheitol (DTT), 2 mM sodium orthovanadate (Na3VO4), 1 mM EGTA, 1 mM NaF, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1% Triton X-100, and 10 μg/ml each of aprotinin, leupeptin, and pepstatin A. The homogenates were centrifuged at 15,000 ×g for 30 min at 4°C, supernatants were collected and stored at −80°C for use. The protein concentrations were determined by a Lowry protein assay kit with bovine serum albumin as standard.
Immunoprecipitation and western blotting
For immunoprecipitation, tissue homogenates (each containing 400 μg of proteins) were diluted 4-fold with HEPES buffer containing 50 mM HEPES (pH 7.4), 150 mM NaCl, 10% glycerol, 1% Triton X-100, and 1 mM each of EGTA, EDTA, PMSF and Na3VO4. Samples were pre-incubated for 1 h with 20 μL protein A/G then centrifuged to remove any protein adhered nonspecifically to the protein A/G. The supernatant was incubated with 2–5 μg proper antibody or species-relevant non-specific IgG (n.s. IgG) for 4 h at 4°C. After the addition of protein A/G-sepharose, the mixture was incubated at 4°C for an additional 2 h. Sample were triple washed with HEPES buffer and eluted by sodium dodecyl sulfate – polyacrylamide gel electrophoresis (SDS–PAGE) loading buffer then boiled for 5 min. Western blot analysis was carried out on 4–20% SDS–PAGE and 50–100μg proteins were electrotransferred onto nitrocellulose filter (pore size, 0.45 μm). After blocking for 2 h in phsophate-buffered saline (PBS) with 0.1% Tween 20 (PBST) and 3% BSA, the membranes were incubated overnight with primary antibody (1:500) in PBST containing 3% BSA. Detection was carried out by the use of proper alkaline phosphatase conjugated IgG (1:20,000) and developed with NBT/BCIP assay kit (Promega). After immunoblotting, the density of the bands was scanned using an image analyzer (LabWorks Software, UVP Upland, CA, USA).
Rac1 assay
Rac activation assays were performed using color PAK1 PBD Agarose Beads (Rac/CDC42 assay kit; Upstate Biotechnology). Precipitated complexes were washed three times with magnesium-containing lysis buffer and boiled in sample buffer. Proteins were fractionated by SDS-PAGE and subjected to western blot analysis using anti-Rac1-specific antibody.
Cresyl violet and TUNEL staining
Rats were anesthetized with chloral hydrate and underwent transcardial perfusion with 0.9% saline followed by 4% paraformaldehyde in 0.1 M phosphate buffer. Brains were removed, postfixed overnight in paraformaldehyde, processed and embedded in paraffin. Coronal sections (6 μm) were cut on a microtome (Leica RM2155, Nussloch, Germany) and collected through the entire dorsal hippocampus (2.5–4.5 mm posterior from bregma) from animals at 5 d after ischemia or sham operation, and every fifth section was collected. Sections were de-paraffinized in xylene and rehydrated in a gradient of ethanol and distilled water and stained with cresyl violet then examined with a light microscope. The neuronal density of CA1 neurons per 1mm length of the medial CA1 pyramidal cell layer was counted bilaterally in four sections per animal. Cell counts from the right and left hippocampus on each of the four sections were averaged to provide mean value. TUNEL staining was performed using an ApopTag Peroxidase In Situ Apoptosis Detection Kit according to the manufacturer’s protocol with minor modifications. Briefly, sections were treated with protease K at 20 mg/ml for 15 min at room temperature. After incubation with reaction buffer containing TdT enzyme and at 37°C for 1 h, sections were washed with stop/wash buffer and treated with anti-digoxigenin conjugate for 30 min at room temperature and subsequently developed color in peroxidase substrate. The nuclei were lightly counterstained with haematoxylin and examined under a light microscope.
Statistical evaluation
Four or five independent animals were sampled at each time point for western blotting study and histology examination. Semi-quantitative analysis of the bands was performed with the Image J analysis software (Version 1.30v; Wayne Rasband, NIH, USA). All values are expressed as the means ± SD. Statistical analysis of the results was carried out by one-way analysis of variance (ANOVA) followed by the Dunnett’s test or Newman–Keuls test. Differences of p < 0.05 were considered significant.
Acknowledgments
This study was supported by a research grant from NINDS (NS050730) from the National Institutes of Health, United States of America.
References
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Benquet P, Gee CE, Gerber U. Transient brain ischemia: NMDA receptor modulation and delayed neuronal death. Med Sci (Paris) 2008;24:185–90. doi: 10.1051/medsci/2008242185. [DOI] [PubMed] [Google Scholar]
- Carboni S, Boschert U, Gaillard P, Gotteland JP, Gillon JY, Vitte PA. AS601245, a c-Jun NH2-terminal kinase (JNK) inhibitor, reduces axon/dendrite damage and cognitive deficits after global cerebral ischaemia in gerbils. Br J Pharmacol. 2008;153:157–63. doi: 10.1038/sj.bjp.0707574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehler E, van Leeuwen F, Collard JG, Salinas PC. Expression of Tiam-1 in the developing brain suggests a role for the Tiam-1-Rac signaling pathway in cell migration and neurite outgrowth. Mol Cell Neurosci. 1997;9:1–12. doi: 10.1006/mcne.1997.0602. [DOI] [PubMed] [Google Scholar]
- Gao Y, Dickerson JB, Guo F, Zheng J, Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci U S A. 2004;101:7618–23. doi: 10.1073/pnas.0307512101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelderblom M, Eminel S, Herdegen T, Waetzig V. c-Jun N-terminal kinases (JNKs) and the cytoskeleton--functions beyond neurodegeneration. Int J Dev Neurosci. 2004;22:559–64. doi: 10.1016/j.ijdevneu.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Gu Z, Jiang Q, Zhang G. c-Jun N-terminal kinase activation in hippocampal CA1 region was involved in ischemic injury. Neuroreport. 2001;12:897–900. doi: 10.1097/00001756-200104170-00006. [DOI] [PubMed] [Google Scholar]
- Guan QH, Pei DS, Zong YY, Xu TL, Zhang GY. Neuroprotection against ischemic brain injury by a small peptide inhibitor of c-Jun N-terminal kinase (JNK) via nuclear and non-nuclear pathways. Neuroscience. 2006;139:609–27. doi: 10.1016/j.neuroscience.2005.11.067. [DOI] [PubMed] [Google Scholar]
- Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, Davis RJ. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15:2760–70. [PMC free article] [PubMed] [Google Scholar]
- Harada N, Iimuro Y, Nitta T, Yoshida M, Uchinami H, Nishio T, Hatano E, Yamamoto N, Yamamoto Y, Yamaoka Y. Inactivation of the small GTPase Rac1 protects the liver from ischemia/reperfusion injury in the rat. Surgery. 2003;134:480–91. doi: 10.1067/s0039-6060(03)00256-3. [DOI] [PubMed] [Google Scholar]
- Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98:453–62. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- Irving EA, Bamford M. Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab. 2002;22:631–47. doi: 10.1097/00004647-200206000-00001. [DOI] [PubMed] [Google Scholar]
- Jin S, Ray RM, Johnson LR. Rac1 mediates intestinal epithelial cell apoptosis via JNK. Am J Physiol Gastrointest Liver Physiol. 2006;291:G1137–47. doi: 10.1152/ajpgi.00031.2006. [DOI] [PubMed] [Google Scholar]
- Jin S, Ray RM, Johnson LR. TNF-alpha/cycloheximide-induced apoptosis in intestinal epithelial cells requires Rac1-regulated reactive oxygen species. Am J Physiol Gastrointest Liver Physiol. 2008;294:G928–37. doi: 10.1152/ajpgi.00219.2007. [DOI] [PubMed] [Google Scholar]
- Kao YY, Gianni D, Bohl B, Taylor RM, Bokoch GM. Identification of a conserved Rac-binding site on NADPH oxidases supports a direct GTPase regulatory mechanism. J Biol Chem. 2008;283:12736–46. doi: 10.1074/jbc.M801010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–6. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- Kuan CY, Whitmarsh AJ, Yang DD, Liao G, Schloemer AJ, Dong C, Bao J, Banasiak KJ, Haddad GG, Flavell RA, Davis RJ, Rakic P. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc Natl Acad Sci U S A. 2003;100:15184–9. doi: 10.1073/pnas.2336254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukekov NV, Xu Z, Greene LA. Direct interaction of the molecular scaffolds POSH and JIP is required for apoptotic activation of JNKs. J Biol Chem. 2006;281:15517–24. doi: 10.1074/jbc.M601056200. [DOI] [PubMed] [Google Scholar]
- Linseman DA, Loucks FA. Diverse roles of Rho family GTPases in neuronal development, survival, and death. Front Biosci. 2008;13:657–76. doi: 10.2741/2710. [DOI] [PubMed] [Google Scholar]
- Lyons TR, Thorburn J, Ryan PW, Thorburn A, Anderson SM, Kassenbrock CK. Regulation of the Pro-apoptotic scaffolding protein POSH by Akt. J Biol Chem. 2007;282:21987–97. doi: 10.1074/jbc.M704321200. [DOI] [PubMed] [Google Scholar]
- Ma XM, Huang JP, Eipper BA, Mains RE. Expression of Trio, a member of the Dbl family of Rho GEFs in the developing rat brain. J Comp Neurol. 2005;482:333–48. doi: 10.1002/cne.20404. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Okazaki M, Zhao H, Asai S, Ishikawa K, Kitamura Y. Phosphorylation of c-Jun and its localization with heme oxygenase-1 and cyclooxygenase-2 in CA1 pyramidal neurons after transient forebrain ischemia. J Cereb Blood Flow Metab. 1999;19:1247–55. doi: 10.1097/00004647-199911000-00009. [DOI] [PubMed] [Google Scholar]
- Nikolova S, Lee YS, Kim JA. Rac1-NADPH oxidase-regulated generation of reactive oxygen species mediates glutamate-induced apoptosis in SH-SY5Y human neuroblastoma cells. Free Radic Res. 2005;39:1295–304. doi: 10.1080/10715760500176866. [DOI] [PubMed] [Google Scholar]
- Nozaki K, Nishimura M, Hashimoto N. Mitogen-activated protein kinases and cerebral ischemia. Mol Neurobiol. 2001;23:1–19. doi: 10.1385/MN:23:1:01. [DOI] [PubMed] [Google Scholar]
- Ozaki M, Deshpande SS, Angkeow P, Bellan J, Lowenstein CJ, Dinauer MC, Goldschmidt-Clermont PJ, Irani K. Inhibition of the Rac1 GTPase protects against nonlethal ischemia/reperfusion-induced necrosis and apoptosis in vivo. FASEB J. 2000;14:418–29. doi: 10.1096/fasebj.14.2.418. [DOI] [PubMed] [Google Scholar]
- Ozawa H, Shioda S, Dohi K, Matsumoto H, Mizushima H, Zhou CJ, Funahashi H, Nakai Y, Nakajo S, Matsumoto K. Delayed neuronal cell death in the rat hippocampus is mediated by the mitogen-activated protein kinase signal transduction pathway. Neurosci Lett. 1999;262:57–60. doi: 10.1016/s0304-3940(99)00034-8. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB. A new model of bilateral hemispheric ischemia in the unanesthetized rat. Stroke. 1979;10:267–72. doi: 10.1161/01.str.10.3.267. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–8. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Grooms SY, Bennett MV, Zukin RS. The AMPAR subunit GluR2: still front and center-stage. Brain Res. 2000;886:190–207. doi: 10.1016/s0006-8993(00)02951-6. [DOI] [PubMed] [Google Scholar]
- Tapon N, Nagata K, Lamarche N, Hall A. A new rac target POSH is an SH3-containing scaffold protein involved in the JNK and NF-kappaB signalling pathways. EMBO J. 1998;17:1395–404. doi: 10.1093/emboj/17.5.1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teramoto H, Coso OA, Miyata H, Igishi T, Miki T, Gutkind JS. Signaling from the small GTP-binding proteins Rac1 and Cdc42 to the c-Jun N-terminal kinase/stress-activated protein kinase pathway. A role for mixed lineage kinase 3/protein-tyrosine kinase 1, a novel member of the mixed lineage kinase family. J Biol Chem. 1996;271:27225–8. doi: 10.1074/jbc.271.44.27225. [DOI] [PubMed] [Google Scholar]
- Tolias KF, Bikoff JB, Burette A, Paradis S, Harrar D, Tavazoie S, Weinberg RJ, Greenberg ME. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron. 2005;45:525–38. doi: 10.1016/j.neuron.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Xu Z, Kukekov NV, Greene LA. POSH acts as a scaffold for a multiprotein complex that mediates JNK activation in apoptosis. EMBO J. 2003;22:252–61. doi: 10.1093/emboj/cdg021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Greene LA. Activation of the apoptotic JNK pathway through the Rac1-binding scaffold protein POSH. Methods Enzymol. 2006;406:479–89. doi: 10.1016/S0076-6879(06)06036-8. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Zhang G, Meng F, Tian H. Biphasic activation of apoptosis signal-regulating kinase 1-stress-activated protein kinase 1-c-Jun N-terminal protein kinase pathway is selectively mediated by Ca2+-permeable alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors involving oxidative stress following brain ischemia in rat hippocampus. Neurosci Lett. 2003;337:51–5. doi: 10.1016/s0304-3940(02)01295-8. [DOI] [PubMed] [Google Scholar]
- Zhang QG, Wang RM, Yin XH, Pan J, Xu TL, Zhang GY. Knock-down of POSH expression is neuroprotective through down-regulating activation of the MLK3-MKK4-JNK pathway following cerebral ischaemia in the rat hippocampal CA1 subfield. J Neurochem. 2005;95:784–95. doi: 10.1111/j.1471-4159.2005.03435.x. [DOI] [PubMed] [Google Scholar]
- Zhang QG, Wang XT, Han D, Yin XH, Zhang GY, Xu TL. Akt inhibits MLK3/JNK3 signaling by inactivating Rac1: a protective mechanism against ischemic brain injury. J Neurochem. 2006;98:1886–98. doi: 10.1111/j.1471-4159.2006.04020.x. [DOI] [PubMed] [Google Scholar]