

Abstract
Gold nanoparticles (AuNPs) functionalized with a short chain amine-terminated alkanethiol (HS-(CH2)2NH2 or C2 NH2-thiol) are prepared via a direct synthesis method and then ligand-exchanged with a long chain amine-terminated alkanethiol (HS-(CH2)11NH2 or C11 NH2-thiol). Transmission electron microscopy analysis showed the AuNPs were relatively spherical with a median diameter of 24.2±4.3 nm. X-ray photoelectron spectroscopy was used to determine surface chemistry of the functionalized and purified AuNPs. The ligand-exchange process was monitored within the time range from 30 min to 61 days. By the fourth day of exchange all the C2 NH2-thiol molecules had been replaced by C11 NH2-thiol molecules. C11 NH2-thiol molecules continued to be incorporated into the C11 NH2 self-assembled monolayer between days 4 and 14 of ligand-exchange. As the length of the exchange time increased, the functionalized AuNPs became more stable against aggregation. The samples were purified by a centrifugation and resuspension method. The C2 NH2 covered AuNPs aggregated immediately when purification was attempted. The C11 NH2 covered AuNPs could be purified with minimal or no aggregation. Small amounts of unbound thiol (∼15%) and oxidized sulfur (∼20%) species were detected on the ligand-exchanged AuNPs. Some of the unbound thiol and all of the oxidized sulfur could be removed by treating the functionalized AuNPs with HCl.
I. INTRODUCTION
The rapid increase in the use of nanoparticles in biotechnology applications has been driven by the unique properties of the nanomaterials provided by their high percentage of surface atoms.1–5 Size, shape and surface chemistry are all important properties for determining the performance of nanoparticles.6 By varying these properties one can tune the nanoparticle performance for a wide-range of applications.6 Special interest in gold nanoparticles (AuNPs) for in vivo nanomedicine studies can be attributed to their nontoxicity.7–9 AuNPs have also been used in areas such as microarray, biosensor, imaging, diagnostics, drug, and nucleic acid delivery, and fingerprinting applications.10–21 In these applications, the AuNPs are modified with surface ligands to allow subsequent biomolecule immobilization, or they are directly functionalized with the biomolecules. Among the common methods used to functionalize AuNPs is adsorbing a self-assembled monolayer (SAMs) of alkanethiols onto the AuNP surfaces.22 The preparation and characterization of SAMs on flat Au surfaces has been extensively studied for over three decades.22–36 Although SAMs are widely used to functionalize AuNPs, detailed, quantitative characterization of the SAM functionalized AuNPs is often lacking.4 For biomedical applications amine-terminated SAM functionalized AuNPs (NH2-SAM-AuNPs) are commonly used as carriers to deliver immobilized biomolecules such as DNA and siRNA into cells, and to perform colorimetric assays of enzymes such as hyaluronidase.37–39 Given the challenges of preparing well-defined, model amine SAMs on flat Au surfaces,40 it is especially important to characterize NH2-SAM-AuNPs.
AuNPs with diameters of ∼5 nm, commonly known as monolayer protected clusters, have been successfully functionalized with amine-terminated ligands by including the amine thiol in the synthesis solution.41,42 However, it has been difficult to functionalize large AuNPs by ligand-exchange with NH2-alkanethiols. We have observed that AuNPs with diameters >12 nm synthesized by the citrate reduction method43,44 aggregated severely and irreversibly when ligand-exchange of the citrate covered AuNPs was attempted with amine thiols. One-step ligand-exchange of the citrate covered AuNPs with OH and CH3 terminated alkanethiols have also been shown to lead to aggregation.45 A ligand-exchange method using thioctic acid and COOH-dithiol as an intermediate stabilizer and 11-amino-1-undecanethiol (C11 NH2-thiol) as the final thiol was reported by Lin et al. to convert citrate-AuNPs to NH2-SAM-AuNPs.46 Though the two-step functionalization improved the stability of the AuNPs, the final AuNPs were not completely covered with the C11 NH2-SAM.46
Niidome et al. developed a one-step one-phase synthesis and functionalization of AuNPs (∼34 nm diameter) with 2-aminoethanethiol (C2 NH2-thiol).37 Lee et al. reported using similar method to prepare 14 nm C2 NH2-SAM-AuNPs.38 These methods are a good starting point for preparing NH2-SAM-AuNPs. However, the stability of functionalized AuNPs against aggregation typically depends on both the charge and chain length of the molecule used to functionalize the AuNPs.45 For example, C2 NH2-thiols are typically too short to provide good AuNP stabilization against aggregation. Though Lee et al. reported purifying the C2 NH2-SAM-AuNPs using dialysis,38 we observed that using either dialysis or centrifugation for purification resulted in immediate aggregation of the C2 NH2-SAM-AuNPs. Niidome et al. did not report if the NH2-SAM-AuNPs were purified before DNA immobilization.37 In another study, we found for AuNPs that shorter-chain COOH-SAMs provided less stabilization against aggregation than longer-chain COOH-SAMs.47 Following similar reasoning, the short chain C2 NH2-SAM is not expected to provide much stability against AuNP aggregation. Since surface characterization results such as x-ray photoelectron spectroscopy (XPS) were not reported in previous studies of C2 NH2-SAM-AuNPs, the surface compositions and extent of purity of the C2 NH2-SAM-AuNPs were unknown.
In the present study, we followed the method of Niidome et al.37 to prepare C2 NH2-SAM-AuNPs, but then performed a ligand-exchange to ultimately functionalize the AuNPs with C11 NH2-SAMs. The AuNPs did not aggregate throughout the C2 to C11 amine thiol ligand-exchange process. Purification of the C11 NH2-SAM-AuNPs, after the ligand-exchange, resulted in some aggregation of the AuNPs. This aggregation decreased with ligand-exchange time and it was completely reversed by adding few drops of 1M HCl. Transmission electron microscopy (TEM) was used to determine the size and shape of C2 NH2-SAM-AuNPs, and XPS was used to determine surface chemistries of the AuNPs at various stages of the C11 amine thiol ligand-exchange process.
II. EXPERIMENT
A. Materials
The following chemicals were purchased from Sigma-Aldrich and used as received: gold (III) chloride hydrate (HAuCl4 · xH2O, x = ∼3, 99.999%), cysteamine hydrochloride (HS-(CH2)2NH2 · HCl, 98%), sodium borohydride (NaBH4, 99%), and 11-amino-1-undecanethiol hydrochloride (HS-(CH2)11NH2 · HCl, 99%). Additional reagents (company, concentration, and grade) included HCl (EMD Chemicals, Inc., 36.5–38%, ACS) and ethanol (AAPER, absolute 200 proof). Ultrapure water (resistivity >18.0 M Ω cm) was purified by a Modulab Analytical research grade water system. TEM grids (carbon type-A, 300 mesh, copper grids) were purchased from Ted Pella. Silicon wafers were purchased from Silicon Valley Microelectronics, diced into 0.6–1 cm × 1 cm pieces, then thoroughly cleaned by sonication in a series of organic solvents (dichloromethane, acetone and methanol; 2 × 5 min consecutive treatments in each solvent). A CHA 600 electron beam evaporator was used to deposit 10 nm titanium films at pressures below 1 × 10−6 Torr onto the flat, clean silicon wafer pieces.
B. Synthesis of C2 NH2-SAM-AuNPs
The method developed by Niidome et al. 37 was used to synthesize the C2 NH2-SAM-AuNPs. The main solutions were prepared as follows: 223.69 mg of HAuCl4 was dissolved into 400 mL of ultrapure water to produce a 1.42 mM solution, 96.80 mg of cysteamine hydrochloride was dissolved into 400 lL of ultrapure water to produce a 213 mM solution, and 7.57 mg of NaBH4 was dissolved into 20 mL of ultrapure water to produce a 10 mM stock solution. The HAuCl4 solution was added to a 500 mL-plastic beaker, covered with a glass plate and aluminum foil, and then stirring with a magnetic bar was commenced. The cysteamine solution was added to the HAuCl4 solution and the mixed solution was stirred for 20 min. Finally, 0.1 mL of the NaBH4 solution was added and stirring was continued for 2 h. The final product was used immediately after synthesis for ligand-exchange with the C11 NH2-thiol. Prior to the C11 NH2-thiol exchange about 1 mL of the C2 NH2-thiol sample was placed onto a TEM grid and allowed to air-dry on filter paper. This sample was then used for TEM analysis of the AuNP size and shape.
C. Ligand-exchange of C2 NH2-thiols with C11 NH2-thiols on AuNPs
The C2 NH2-SAM-AuNPs was ligand-exchanged with the C11 NH2-thiol as follows. First, a 5 mM solution of the C11 NH2-thiol was prepared in ethanol. Then, an excess amount of the thiol solution was added to the C2 NH2-SAM-AuNPs solution in an aluminum foil covered flask. For example, 8 mL of a 5 mM C11 NH2-thiol solution was added to a 400 mL of the C2 NH2-SAM-AuNP solution. The mixture was then stirred on a magnetic plate. Samples (~30 mL) were taken from the solution after the following times of ligand exchange: 30 min, 3 h, 12 h, 24 h, 2 days, 4 days, 7 days, 14 days, 21 days, 31 days, and 61 days.
Each of these samples were then divided into 20 1.5 mL microcentrifuge tubes and centrifuged at 10 000 RPM for 15 min at room temperature. After removing the supernatant, the precipitates were vortex-mixed and the contents from five tubes were combined into one tube. After adding ultrapure water and vortex-mixing, the samples were centrifuged again under the same conditions. Then, after removing the supernatant, the precipitates were vortex-mixed and the contents from two tubes were combined into one tube. Now, each sample was consolidated into two tubes and these tubes were further purified by repeating the centrifugation and vortex-mixing steps at least two times. Based on the XPS sulfur spectra, more rinsing was required for the longer ligand-exchange times to remove unbound thiols. Finally, the samples from the remaining two tubes were combined into a single tube and ∼100 μL of ultrapure water was added to the tube. Then the sample was vortex-mixed to produce a homogeneous solution. An additional step involved addition of HCl acid. In this case, after finishing the purification stage, a different number of drops (1, 6, 12, and 24) of a 1M HCl solution was added to 4.8 mL of 24 × diluted purified/concentrated samples. Adding one drop of the HCl solution did not result in a noticeable change in amine covered AuNPs. Adding 6, 12, or 24 drops of the HCl solution resulted in similar noticeable changes in the amine covered AuNPs. These HCl treated samples were analyzed by XPS before and after rinsing with water.
For XPS analysis, a small drop (∼20 μL) of the final concentrated product was placed onto a clean titanium coated substrate and allowed to dry in a vacuum desiccator. This step was repeated until a complete layer of AuNPs was formed on the substrate and the substrate Ti signal was minimized during XPS analysis. The samples were stored in petri-dishes backfilled with nitrogen gas and wrapped with parafilm. XPS measurements were performed approximately two hours after the sample drying was completed.
D. Transmission electron microscopy
TEM measurements were performed on Philips CM100 instrument operating at 100 kV accelerating voltage. It was equipped with a Galan Model 689 digital slow scan camera. Pictures with 128 × 128 pixels were taken of the AuNPs with typical magnifications in the range of 90 000–340 000 ×. IMAGEJ software was used to analyze average diameter, size distribution and circularity of AuNPs from the TEM images. The results were based on analysis of approximately 1300 nanoparticles from three batches of C2 NH2-SAM-AuNPs.
E. X-ray photoelectron spectroscopy
XPS measurements were performed on a Kratos AXIS Ultra DLD (Kratos, Manchester, UK) instrument in the “hybrid” mode using a monochromatic Al Ka x-ray source and a nominal photoelectron take-off angle of 0° (the take-off angle is defined as the angle between the substrate normal and the axis of the analyzer lens). All samples were run as insulators using a low-energy flood gun for charge neutralization. For each sample, a survey scan from 0–1100 eV binding energy (BE) and elemental scans of N1, O1s,S2p, and Ti2p were acquired using a pass energy of 80 eV on three spots to determine XPS compositions. High-resolution scans of C1s, N1s, S2p, and Au4f peaks were acquired from one spot on each sample using a 20 eV pass energy to examine the type of chemical species present. As a control, similar compositional measurements were acquired for the clean titanium substrates. Measurements were performed on three replicates for each sample. Data analysis was done using the Vision Processing data reduction software. All BEs were referenced to the C1s hydrocarbon peak at 285 eV.
III. RESULTS AND DISCUSSION
A. TEM analysis
A TEM image of the C2 NH2-SAM-AuNPs is shown in Fig. 1. IMAGEJ analyses of the size and shape distributions based on 1300 nanoparticles are shown in Fig. 2. The median diameter and standard deviation of the AuNPs was 24.2 ± 4.3 nm. The circularity index of 72% of the AuNPs was less than 1.1, where circularity index was calculated as the ratio of major axis to minor axis of a nanoparticle.
Fig. 1.

TEM image of the C2NH2-SAM-AuNPs.
Fig. 2.
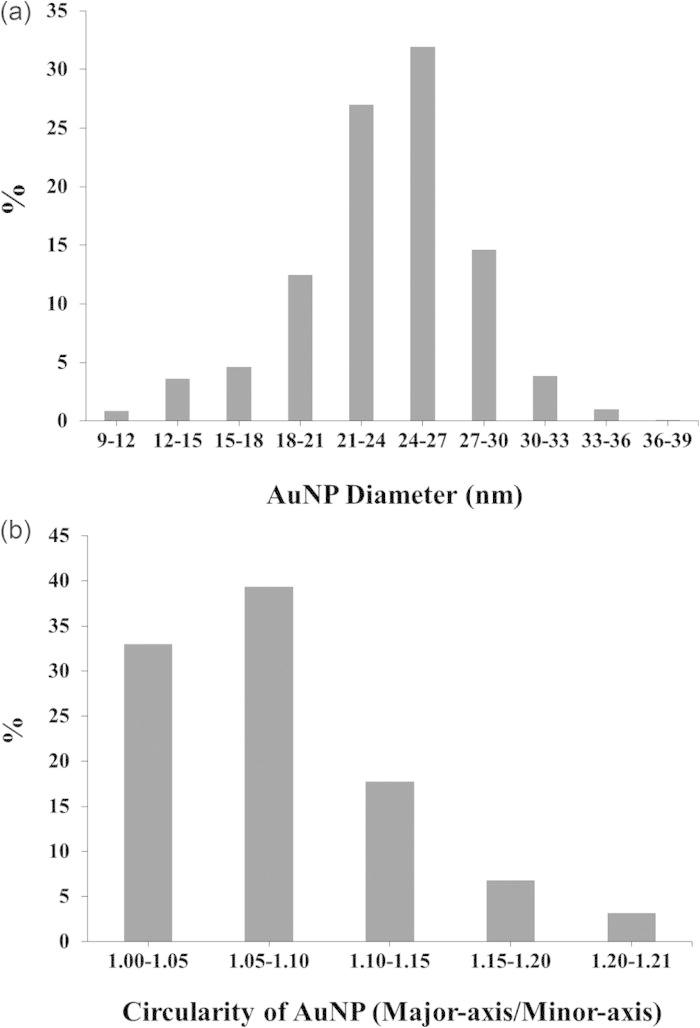
imagej results for the size distribution (median diameter = 24.2 ± 4.3 nm) and circularity of C2 NH2-SAM-AuNPs based on analysis of 1300 nanoparticles. The circularity is represented by the ratio of the major axis to the minor axis of a particle.
B. XPS analysis
To prepare stable NH2-SAM-AuNPs, a one-step synthesis and functionalization method was performed using C2 NH2-thiols followed by ligand exchange using C11 NH2-thiols. We observed that the unpurified C2 NH2-SAM-AuNPs were stable in solution for only a few days. When similar ligand exchange was performed on citrate-covered AuNPs or AuNPs that had been functionalized with C6 COOH-SAMs (mercaptohexanoic acid SAMs), the AuNP aggregated irreversibly. This could be due to reversal of the surface charge from the negatively charged citrate or COOH-SAM covered AuNPs to the positively charged NH2-SAM covered AuNPs. Therefore, it appears to be advantageous to have the same surface functionalities on the AuNP surfaces before and after and the exchange with the final ligand. Our results indicate that this prevents aggregation of the amine functionalized AuNPs during final functionalization step.
Composition results from XPS analysis of the NH2-SAM-AuNPs after various ligand-exchange times are compiled in Table I. All samples showed the presence C, N, and S from the NH2-SAMs overlayer, as well as Au from the NPs. In addition to these expected elements, O was detected. Previously it has been reported that oxygen is a common contaminant observed on NH2-SAMs formed on flat Au surfaces.40,48,49 Small amounts of Ti from the underlying Ti coated substrate were also observed on some of the samples. The Ti atomic concentrations for those samples were as follows at the various ligand-exchange times: 0.2% (3 h), 1.5% (24 h) and 0.1% (14, 21, and 31 days). To remove the contributions from the Ti substrate, proportional values from the measured composition of a bare Ti surface (at. %: Ti = 30.2, O = 40.6, C = 21.0, and N = 2.3) were subtracted from the NH2-SAM-AuNP samples, and the data was then renormalized.
Table I.
XPS determined compositions of the NH2-SAMs on 24 nm AuNPs after undergoing a C11 NH2-thiol ligand-exchange with a C2 NH2-SAM for different lengths of time (30 min to 61 days). The data was normalized without the Ti signal as discussed in the text.
| at. % (std. dev.) | |||||
|---|---|---|---|---|---|
| Time of ligand-exchange | C1s | Au4f | N1s | O1s | S2p |
| 30 min | 23.6(3.8) | 64.6(3.1) | 5.3(0.8) | 2.2(0.5) | 4.4(0.4) |
| 3 h | 23.5(2.6) | 63.7(2.4) | 5.4(0.8) | 2.3(0.4) | 4.4(0.5) |
| 12 h | 25.9(3.7) | 61.9(3.6) | 5.4(0.8) | 2.3(0.3) | 4.5(0.4) |
| 24 h | 27.3(7.6) | 56.8(6.0) | 4.2(1.0) | 2.6(0.4) | 4.0(0.5) |
| 2 days | 36.2(5.1) | 52.5(4.8) | 4.5(0.4) | 2.8(0.3) | 4.0(0.3) |
| 4 days | 43.9(3.9) | 45.7(4.1) | 4.0(0.4) | 2.4(0.3) | 4.0(0.2) |
| 7 days | 47.5(4.2) | 42.3(4.0) | 4.2(0.5) | 2.1(0.3) | 3.9(0.4) |
| 14 days | 51.3(2.6) | 37.0(2.9) | 4.6(0.4) | 2.9(0.5) | 3.9(0.3) |
| 21 days | 51.6(1.2) | 36.4(0.9) | 4.7(0.4) | 3.5(0.6) | 3.6(0.4) |
| 31 days | 50.6(1.5) | 36.9(1.3) | 4.8(0.4) | 3.6(0.1) | 3.8(0.3) |
| 61 days | 52.3(1.5) | 35.5(1.1) | 4.8(0.3) | 3.3(0.5) | 3.9(0.3) |
As shown in Table I, the C concentration increased from ∼24 at. % to ∼52% while the Au concentration correspondingly decreased from ∼65% to ∼35 at. % (an increase of the C/Au atomic ratio from 0.4 to 1.5) when the ligand-exchange was increased from 30 min to 61 days. The changes in the C and Au signals with ligand-exchange time are consistent with increasing replacement of the shorter C2 NH2-thiol molecules with the longer C11 NH2-thiol molecules. The C11 NH2 SAM contains more carbon atoms and the resulting thicker SAM attenuates the Au signal more.50 As shown in Table I, the N concentration also decreased slightly with increased ligand-exchange time. The shortest ligand-exchange times had the highest N concentration, while the longest ligand-exchange times had the lowest N concentration. The N/C atomic ratio, as well as the S/C atomic ratio, decreased from 0.2 to 0.1 when ligand-exchange time increased from 30 min to 61 days. These results are consistent with more C2 NH2-thiols on the AuNPs at shorter exchange times and more C11 NH2-thiols on the AuNPs at longer exchange times as the relative concentrations of N and S are higher in the C2 NH2-thiol than in the C11 NH2-thiol. The O concentration increased slightly with time. While it has been shown that oxygen-containing contaminants are present in amine SAMs,40 high-resolution XPS S2p spectra showed that some of the detected oxygen from the NH2-SAM-AuNP samples was associated with the presence of oxidized sulfur, as discussed below.
In Fig. 3 the surface compositions of just the SAM overlayers, renormalized without the Au and O concentrations, were compared with the stoichiometric compositions of the two thiols to examine the replacement of the C2 NH2-thiol with the C11 NH2-thiol. The carbon concentration increases, while both the nitrogen and sulfur concentrations decrease with increasing ligand exchange time. At first time point the carbon concentration is higher and the nitrogen and sulfur concentrations lower than those expected for a pure C2 NH2-thiol. This indicates a significant amount of the C2 NH2-thiol is already replaced by C11 NH2-thiol within the first 30 min of ligand-exchange. The composition of the C2 NH2 covered AuNPs before ligand exchange could not be measured since this sample immediately aggregated upon trying to purify it for analysis. As the ligand-exchange is increased to 4 days and beyond, the renormalized composition of C, N, and S became constant and the error bars decreased, showing that the sample compositions became more reproducible. The measured C, N, and S concentrations at exchange times longer than 4 days are consistent with the presence of a fully exchanged C11 NH2-SAM on the AuNPs. However, the data before renormalization indicates that the sample composition continues to change until 14 days of exchange time. Most notably the Au atomic percentage continued to decrease until 14 days, indicating the Au signal is increasingly attenuated by further changes in the SAM overlayer. This suggests a mechanism where during the first 4 days of exchange the C11 NH2-thiols have completely replaced the C2 NH2-thiols on the surface of the AuNPs, then from 4 to 14 days of exchange additional C11 NH2-thiols are incorporated in the SAM. This second step would result in a more densely packed SAM that would further attenuate the Au signal from the NP core.
Fig. 3.
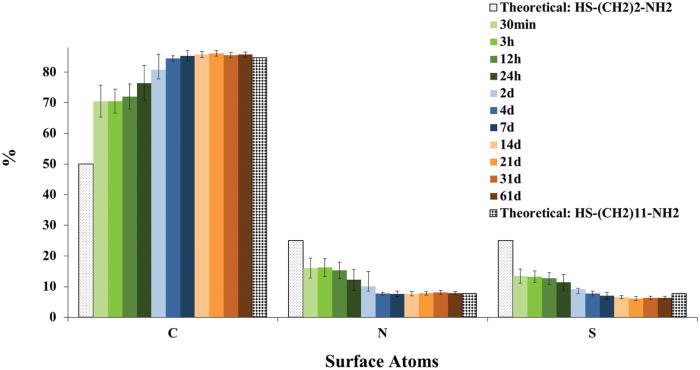
(Color) XPS determined compositions of NH2-SAMs on the 24 nm AuNP surface after undergoing a C11 NH2-thiol ligand-exchange for different lengths of time (30 min to 61 days). The theoretical stoichiometric compositions for the C2 NH2-thiol and C11 NH11-thiol are also shown. The experimental data was renormalized without the Au, O, and Ti signals as discussed in the text.
Figure 4 shows representative XPS high-resolution C1s and N1s spectra from the 21-day, ligand-exchanged NH2-SAM-AuNPs. The C1s spectrum has two peaks. One peak has a BE of 285.0 eV (C–H and C–C) and the other peak has a BE of 286.6 eV (C–N and C–S), with each peak containing approximately 87 and 13% of the total C1s intensity, respectively. These results are comparable with those found for the C11-NH2 SAM on flat gold surfaces.40 The N1s spectrum contained two peaks at 399.8 eV (NH2–C) and 401.2 eV (+NH3-C). In Fig. 4(b) these peaks have intensities that are 40 and 60% of the total N1s intensity, respectively. The intensity ratio between the two N1s peaks was not consistent, even for different replicates of the same samples likely due to lack of potential control as the samples are removed from solution. Similar results been reported for the C11-NH2 SAM on flat gold surfaces.40
Fig. 4.
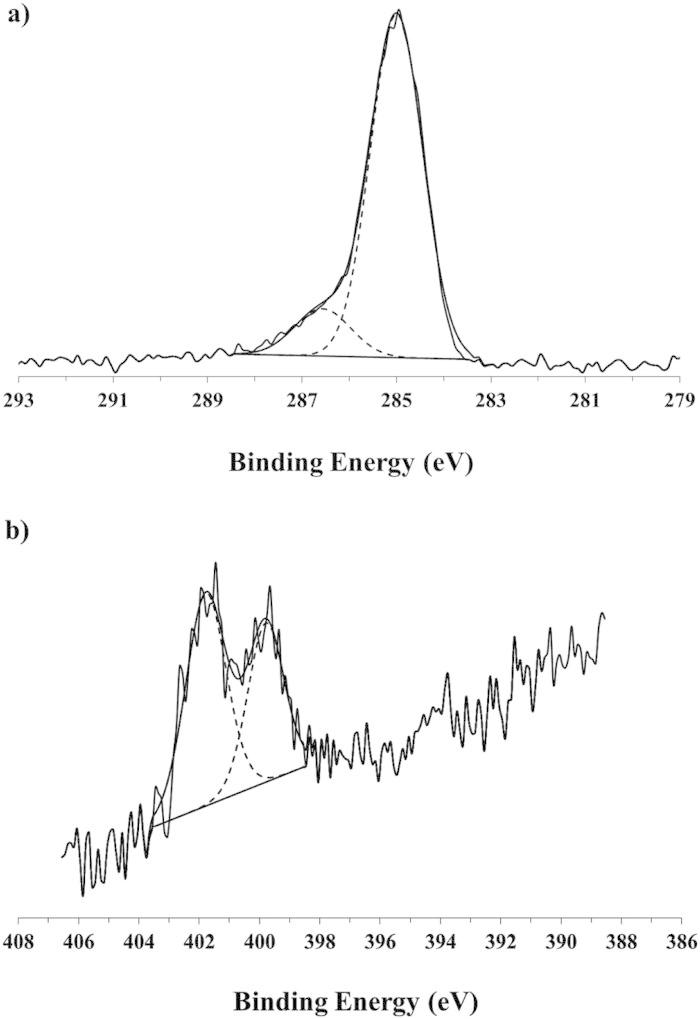
XPS high-resolution spectra of (a) C1s and (b) N1s for NH2-SAMs on 24 nm AuNPs after 21 days of ligand-exchange.
Figure 5(a) shows a representative XPS high-resolution S2p spectrum from the 21-day, ligand-exchanged NH2-SAM-AuNPs. The S2p peaks were fit using doublet peaks with a 2p1/2/2p3/2 ratio of 0.5 and separation of 1.2 eV, as described previously.32 The spectrum in Fig. 5(a) contained three sets of doublets. The most intense doublet is attributed to a surface bound Au-thiolate species (162 eV S2p3/2 BE).32 The two smaller doublets are attributed to unbound thiols (S2p3/2 BE near 163.5 eV) and oxidized sulfur (S2p3/2 BE near 168 eV).32 Typically it was observed in most samples that ∼65, 10–20, and 15–20% of sulfur atoms were present as bound Au-thiols, unbound thiols and oxidized sulfur species, respectively. The typical spectrum shown here in Fig. 5(a) had 67% bound Au-thiolate, 16% unbound thiol, and 17% oxidized S.
Fig. 5.
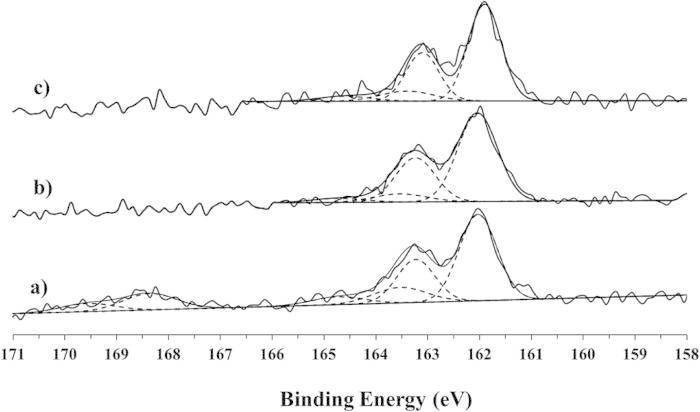
XPS high-resolution S2p spectra from NH2-SAMs on 24 nm AuNPs that were purified after 21 days of ligand-exchange for samples analyzed— (a) before adding HCl, (b) after adding 24 drops of the HCl solution but before rinsing, and (c) after adding 24 drops of the HCl solution and rinsing.
An additional HCl treatment was done to better disperse the nanoparticles. A comparison of the XPS data before and after treatment with 24 drops of the HCl solution showed that the addition of HCl also resulted in the removal of the some of the unbound thiol and all of the oxidized sulfur. The 21-day, ligand-exchanged NH2-SAM-AuNPs had ∼7% unbound thiols after HCl treatment and ∼11% unbound thiols after rinsing the HCl treated samples. However, oxidized sulfur was not detected on the samples treated with HCl, even if the acid treatment was followed by a final rinsing step. Figure 5(b) and 5(c) show representative high-resolution XPS S2p spectra after the addition of 24 drops of the HCl solution to the NH2-SAM-AuNP solution. The rinsing was performed by centrifuging the sample under the conditions noted in the experimental section with a 3 × water change. To remove more of the unbound thiol species, additional steps of centrifugation, rinsing in the presence of HCl to prevent aggregation, then final rinsing to remove the HCl is recommended. The surface compositions of the three types of samples (before adding HCl, after adding HCl, and after adding HCl and then rinsing) are shown in Table II. A contribution from Cl of 3–4 at. % was observed on samples that were treated with HCl and not rinsed. Cl was not detected after the final rinsing with water. The surface composition of the “with HCl” sample shown in Table II has been renormalized without the Cl contribution to better compare the composition of that sample to the compositions of the “before HCl” and “after rinsing” samples. As seen in Table II, the O1s concentration decreased from 3.5 at. % (before HCl) to 1.4 at. % (after rinsing). The amount of sulfur also decreased from 3.6 to 2.7 at. % after the HCl treatment and a final rinse. These results are consistent with the removal of the oxidized sulfur species. Interestingly, the Au, N, and C percentages are not significantly changed after the HCl treatment and rinsing. This may indicate that the oxidized sulfur species removed was a sulfate ion and not an oxidized amine thiol molecule.
TABLE II.
XPS determined compositions of NH2-SAMs on 24 nm AuNPs after undergoing a C11 NH2-thiol ligand-exchange with a C2 NH2-SAM for 21 days followed by purification (before adding HCl), purification then adding 24 drops of HCl solution (with HCl), or purification then adding 24 drops of HCl solution and rinsing (after rinsing HCl). Data for the sample with the unrinsed acid treatment was renormalized without the Cl signals.
| at. % (std. dev.) | |||||
|---|---|---|---|---|---|
| 21 days ligand-exchanged | C1s | Au4f | N1s | O1s | S2p |
| Before adding HCl | 51.6(1.2) | 36.4(0.9) | 4.7(0.4) | 3.5(0.6) | 3.6(0.4) |
| With HCl | 51.5(1.2) | 39.7(1.1) | 4.8(0.3) | 1.3(0.1) | 2.8(0.2) |
| After rinsing HCl | 54.8(1.4) | 36.0(2.0) | 4.8(0.4) | 1.4(0.6) | 2.7(0.2) |
The XPS results reported in this study are the first step to preparing well-defined and well-characterized AuNPs covered with amine SAMs. Further studies to obtain more detail about the SAM thickness and structure on AuNPs can be obtained using by combining simulated electron spectra for surface analysis calculations with the experimental XPS measurements.51 Time-of-flight secondary ion mass spectrometry can provide additional structural insights into SAM covered AuNPs.47 Additional complementary analysis techniques such as sum frequency generation vibration spectroscopy can provide further information such as the density of gauche defects in the alkyl chains as well as the orientation of those chains.40
IV. CONCLUSIONS
C11 NH2-SAM-AuNPs were prepared by starting with a previously reported one-step C2 NH2-SAM-AuNPs synthesis/ functionalization method followed by a ligand-exchange to replace the C2 NH2-thiol molecules with C11 NH2-thiol molecules. TEM showed the starting C2 NH2-SAM-AuNPs were relatively spherical and reasonably monodispersed (average diameter of 24.2 ± 4.3 nm). The C11 NH2-SAM-AuNPs were more stable than the C2 NH2-SAM-AuNPs, which exhibited significant aggregation during attempts to purify them. The C11 NH2-SAM-AuNPs could be purified with a centrifugation/resuspending method. XPS analysis showed that by day 4 of exchange most of the shorter C2 NH2-thiol molecules were replaced by the longer C11 NH2-thiol molecules. However, the longer thiols continued to be incorporated into the SAM until day 14 of exchange. XPS results detected the presence of bound, unbound, and oxidized sulfur species. Partial removal of the unbound thiol molecules and complete removal of the oxidized sulfur species was achieved with treatment of the samples with HCl. The final C11 NH2-SAM-AuNPs are stable nanoparticles that can be used as a model surfaces for biomolecule immobilization.
ACKNOWLEDGMENTS
This research was supported by NIH Grants Nos. GM-074511 and EB-002027 (NESAC/Bio). S.D.T. thanks NSF for an IGERT fellowship.
REFERENCES
- 1. Pissuwan D., Niidome T. and Cortie M.B.J., J. Controlled Release 149, 65 (2011) 10.1016/j.jconrel.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 2. Katz E. and Willner I., Angew. Chem., Int. Ed. Engl. 43, 6042 (2004) 10.1002/anie.200400651. [DOI] [PubMed] [Google Scholar]
- 3. Kumar C. S. S. R., Nanotechnologies for the Life Sciences, 1st ed. (Wiley-VCH Verlag, Weinheim, 2005). [Google Scholar]
- 4. Grainger D. W. and Castner D. G., Adv. Mater. 20, 867 (2008) 10.1002/adma.200701760. [DOI] [Google Scholar]
- 5. Baer D. R., Gaspar D. J., Nachimuthu P., Techane S. D. and Castner D. G., Anal. Bioanal. Chem. 396, 983 (2010) 10.1007/s00216-009-3360-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Daniel M.-C. and Didier A., Chem. Rev. 104, 293 (2004) 10.1021/cr030698+. [DOI] [PubMed] [Google Scholar]
- 7. El-Sayed I. H., Huang X. and El-Sayed M. A., Nano Lett. 5, 829 (2005) 10.1021/nl050074e. [DOI] [PubMed] [Google Scholar]
- 8. Eustis S. and El-Sayed M. A., Chem. Soc. Rev. 35, 209 (2006) 10.1039/b514191e. [DOI] [PubMed] [Google Scholar]
- 9. Jain P. K., Lee K. S., El-Sayed I. H. and El-Sayed M. A., J. Phys. Chem. B 110, 7238 (2006) 10.1021/jp057170o. [DOI] [PubMed] [Google Scholar]
- 10. Wang Z., Lee J., Cossins A. R. and Brust M., Anal. Chem. 77, 5770 (2005) 10.1021/ac050679v. [DOI] [PubMed] [Google Scholar]
- 11. Taton T. A., Lu G. and Mirkin C. A., J. Am. Chem. Soc. 123, 5164 (2001) 10.1021/ja0102639. [DOI] [PubMed] [Google Scholar]
- 12. Taton T. A., Mirkin C. A. and Letsinger R. L., Science 289, 1757 (2000) 10.1126/science.289.5485.1757. [DOI] [PubMed] [Google Scholar]
- 13. Sokolov K., Follen M., Aaron J., Pavlova I., Malpica A., Lotan R. and Richards-Kortum R., Cancer Res. 63, 1999 (2003). [PubMed] [Google Scholar]
- 14. Shenoy D., Fu W., Li J., Crasto C., Jones G., DiMarzio C., Sridhar S. and Amiji M., Int. J. Nanomedicine 1, 51 (2006) 10.2147/nano.2006.1.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mirkin C. A., Letsinger R. L., Mucic R. C. and Storhoff J. J., Nature (London) 382, 607 (1996) 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]
- 16. Leggett R., Lee-Smith E. E., Jickells S. M. and Russell D. A., Angew. Chem., Int. Ed. 46, 4100 (2007) 10.1002/anie.200700217. [DOI] [PubMed] [Google Scholar]
- 17. Lee J.-S., Stoeva S. I. and Mirkin C. A., J. Am. Chem. Soc. 128, 8899 (2006) 10.1021/ja061651j. [DOI] [PubMed] [Google Scholar]
- 18. Bergen J. M., Recum H. A., Goodman T. T., Massey A. P. and Pun S. H., Macromol. Biosci. 6, 506 (2006) 10.1002/mabi.200600075. [DOI] [PubMed] [Google Scholar]
- 19. Aslan K., Lakowicz J. R. and Geddes C. D., Anal. Chem. 77, 2007 (2005) 10.1021/ac0484880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aslan K., Luhrs C. C. and Perez-Luna V. H., J. Phys. Chem. B 108, 15631 (2004) 10.1021/jp036089n. [DOI] [Google Scholar]
- 21. Alivisatos A. P., Johnsson K. P., Peng X., Wilson T. E., Loweth C. J., Bruchez M. P. Jr. and Schultz P. G., Nature (London) 382, 609 (1996) 10.1038/382609a0. [DOI] [PubMed] [Google Scholar]
- 22. Love J. C., Estroff L. A., Kriebel J. K., Nuzzo R. G. and Whitesides G. M., Chem. Rev. 105, 1103 (2005) 10.1021/cr0300789. [DOI] [PubMed] [Google Scholar]
- 23. Nuzzo R. G. and Allara D. L. J., Am. Chem. Soc. 105, 4481 (1983) 10.1021/ja00351a063. [DOI] [Google Scholar]
- 24. Lenk T. J., Hallmark V. M., Hoffman C. L., Rabolt J. F., Castner D. G., Erdelen C. and Ringsdorf H., Langmuir 10, 4610 (1994) 10.1021/la00024a037. [DOI] [Google Scholar]
- 25. Strong L. and Whitesides G. M., Langmuir 4, 546 (1988) 10.1021/la00081a009. [DOI] [Google Scholar]
- 26. Bain C. D., Troughton E. B., Tao Y. T., Evall J., Whitesides G. M. and Nuzzo R. G. J., Am. Chem. Soc. 111, 321 (1989) 10.1021/ja00183a049. [DOI] [Google Scholar]
- 27. Tsao M.-W., Hoffman C. L., Rabolt J. F., Johnson H. E., Castner D. G., Erdelen C. and Ringsdorf H., Langmuir 13, 4317 (1997) 10.1021/la970293i. [DOI] [Google Scholar]
- 28. Dubois L. H. and Nuzzo R. G., Annu. Rev. Phys. Chem. 43, 437 (1992) 10.1146/annurev.pc.43.100192.002253. [DOI] [Google Scholar]
- 29. Pan S., Castner D. G. and Ratner B. D., Langmuir 14, 3545 (1998) 10.1021/la971212l. [DOI] [Google Scholar]
- 30. Ulman A., Chem. Rev. 96, 1533 (1996) 10.1021/cr9502357. [DOI] [PubMed] [Google Scholar]
- 31. Graham D. J. and Ratner B. D., Langmuir 18, 5861 (2002) 10.1021/la0113062. [DOI] [Google Scholar]
- 32. Castner D. G., Hinds K. and Grainger D. W., Langmuir 12, 5083 (1996) 10.1021/la960465w. [DOI] [Google Scholar]
- 33. Tao F. and Bernasek S. L., Chem. Rev. 107, 1408 (2007) 10.1021/cr050258d. [DOI] [PubMed] [Google Scholar]
- 34. Cheng F., Gamble L. J. and Castner D. G., Anal. Chem. 80, 2564 (2008) 10.1021/ac702380w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee C.-Y., Gong P., Harbers G. M., Grainger D. W., Castner D. G. and Gamble L. J., Anal. Chem. 78, 3316 (2006) 10.1021/ac052137j. [DOI] [PubMed] [Google Scholar]
- 36. Xia N., Hu Y., Grainger D.W. and Castner D. G., Langmuir 18, 3255 (2002) 10.1021/la011423x. [DOI] [Google Scholar]
- 37. Niidome T., Nakashima K., Takahashi H. and Niidome Y., Chem. Commun. 17, 1978 (2004) 10.1039/b406189f. [DOI] [PubMed] [Google Scholar]
- 38. Lee S. H., Bae K. H., Kim S. H., Lee K. R. and Park T. G., Int. J. Pharm. 364, 94 (2008) 10.1016/j.ijpharm.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 39. Kim J.-W., Kim J. H., Chung S. J. and Chung B. H., Analyst (Cambridge, U. K.) 134, 1291 (2009) 10.1039/b822917a. [DOI] [PubMed] [Google Scholar]
- 40. Baio J. E., Weidner T., Brison J., Graham D. J., Gamble L. J. and Castner D. G., J. Electron Spectrosc. Relat. Phenom. 172, 2 (2009) 10.1016/j.elspec.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Templeton A. C., Hostetler M. J., Warmoth E. K., Chen S., Hartshorn C. M., Krishnamurthy V. M., Forbes M. D. E. and Murray R. W., J. Am. Chem. Soc. 120, 4845 (1998) 10.1021/ja980177h. [DOI] [Google Scholar]
- 42. Cutler E. C., Lundin E., Garabato B. D., Choi D. and Shon Y.-S., Mater. Res. Bull. 42, 1178 (2007) 10.1016/j.materresbull.2006.08.033. [DOI] [Google Scholar]
- 43. Turkevich J., Stevenson P. C. and Hillier J., Discuss. Faraday Soc. 11, 55 (1951) 10.1039/df9511100055. [DOI] [Google Scholar]
- 44. Frens G., Nature (London) 241, 20 (1973). [Google Scholar]
- 45. Weisbecker C. S., Merritt M. V. and Whitesides G. M., Langmuir 12, 3763 (1996) 10.1021/la950776r. [DOI] [Google Scholar]
- 46. Lin S.-Y., Tsai Y.-T., Chen C.-C., Lin C.-M. and Chen C. -H., J. Phys. Chem. B 108, 2134 (2004) 10.1021/jp036310w. [DOI] [Google Scholar]
- 47. Techane S. D., Gamble L. J. and Castner D. G., J. Phys. Chem. C 115, 9432 (2011) 10.1021/jp201213g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang H., Chen S., Li L. and Jiang S., Langmuir 21, 2633 (2005) 10.1021/la046810w. [DOI] [PubMed] [Google Scholar]
- 49. Wang H., Castner D. G., Ratner B. D. and Jiang S., Langmuir 20, 1877 (2004) 10.1021/la035376f. [DOI] [PubMed] [Google Scholar]
- 50. Ratner B. D. and Castner D.G., Electron Spectroscopy for Chemical Analysis, 2nd ed., edited by Gilmore J. C. V. (Wiley, Chichester, 2009) p. 47. [Google Scholar]
- 51. Techane S. D., Baer D. R. and Castner D. G., “Simulation and Modeling of Self-Assembled Monolayers of Carboxylic Acid Thiols on Flat and Nanoparticle Gold Surfaces,” Anal. Chem. (in press). [DOI] [PMC free article] [PubMed]