

Abstract
Chimeric polypeptides (CPs) that are derived from elastin-like polypeptides (ELPs) can self-assemble to form nanoparticles by site-specific covalent attachment of hydrophobic molecules to one end of the biopolymer backbone. Molecules with a distribution coefficient greater than 1.5 impart sufficient amphiphilicity to drive self-assembly into sub-100 nm nanoparticles.
Keywords: elastin-like polypeptides, thermoresponsive, self-assembly, nanoparticle, biopolymer, distribution coefficient
Most small molecule therapeutics utilized in the clinic have poor bioavailability and suboptimal pharmacokinetics because of their hydrophobicity and low molecular weight. Engineered drug delivery vehicles seek to improve the efficacy of these therapeutics by increasing their solubility, extending their plasma half-life, increasing the amount of drug deposited in the desired tissue, and decreasing their exposure to healthy tissues.[1] Repackaging hydrophobic drugs by sequestering them within the core of soluble polymeric nanoparticles can overcome these limitations by increasing drug solubility; the appropriate choice of polymer can also lead to long in vivo circulation and improved tissue distribution as compared to the free drug.[1–4] Furthermore, the choice of stimulus responsive polymers as the carrier suggests the intriguing possibility of endowing these nanoparticles with thermal responsiveness in the clinically relevant temperature range of 37–42ºC that would allow them to be targeted in vivo to a site of disease by externally applied, focused mild hyperthermia. To our knowledge, no such thermally responsive, drug loaded nanoparticles currently exist.
The launching point of our attempt to rationally design drug-loaded, thermally targeted nanoparticles was our recent observation that the site-specific (C-terminal), covalent attachment of multiple copies of doxorubicin –a small molecule chemotherapeutic– to a chimeric polypeptide (CP) resulted in the formation of near-monodisperse micelles.[2, 5] This observation prompted three questions that provided the roadmap for this study: (1) is the conjugation triggered self-assembly of a CP observed in the previous study restricted to a small set of compounds or does it reflect a more general propensity of CPs to undergo self-assembly upon conjugation to small molecules? (2) If this is indeed a general phenomenon, what is the mechanism that drives their self-assembly? (3) If we can uncover the rules that drive self-assembly, can we use this information to rationally design drug-loaded nanoparticles that also exhibit thermal responsiveness in the clinically relevant temperature range of 37–42ºC under physiologically relevant conditions?
To explore these questions, 14 different maleimide derivatives of small molecules spanning a large range of hydrophobicity were covalently attached to a CP. These model compounds were chosen with two considerations in mind: first, they display a range of hydrophobicity, as reflected by their octanol-water distribution coefficient, Log(D).[6] Second, they all contain a reactive maleimide moiety so that they could be covalently coupled to the CP. The CP used in this study consists of two segments: a hydrophilic, biodegradable elastin-like polypeptide (ELP) segment with a MW of 62 kDa, and a short, 1.6 kDa cysteine-rich Cys(Gly-Gly-Cys)7 segment that provides eight thiol groups for conjugation of the maleimide derivatives (Figure 1A).
Figure 1. A) Sequence of the chimeric polypeptide.
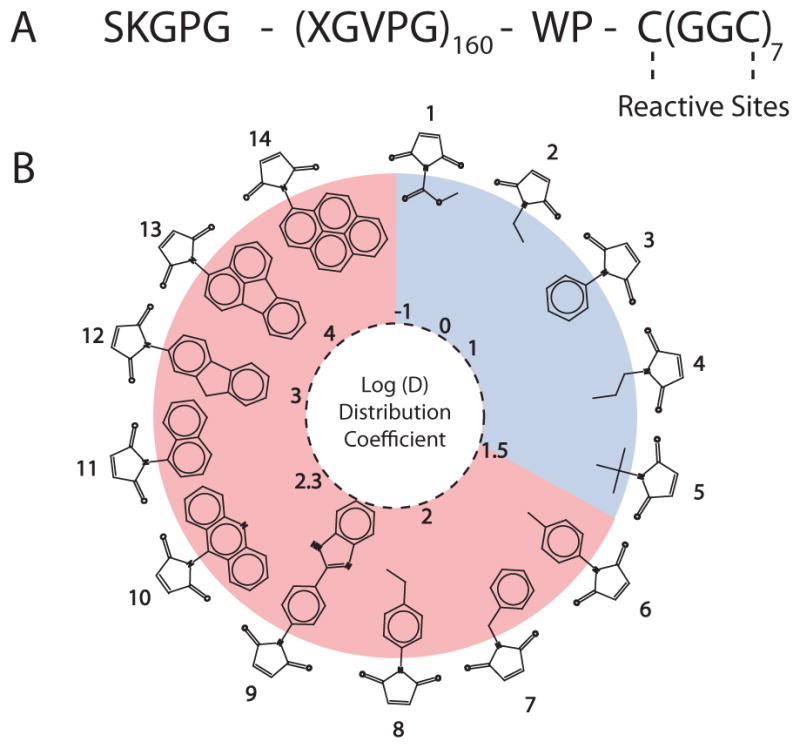
The 62 kDa ELP segment of the CP consists of 160 repeats of VPGXG with the guest residue X having the composition Val1Gly7Ala8. The 1.6 kDa cysteine-rich sequence at the C-terminus provides sites for covalent conjugation of maleimide derivatives of model compounds, shown in (B). B) Structure of the model compounds. The circle represents a visual map of the model compounds and their hydrophobicity as measured by the distribution coefficient at pH 7.4. The attachment of compounds with a Log(D) ≤ 1.5 (shown in blue) did not trigger self-assembly of the CP, whereas compounds with a Log(D) > 1.5 (shown in pink) triggered the CP to self-assemble into nanoparticles.
ELPs are biopolymers consisting of repeats of the peptide sequence VPGXG, derived from tropoelastin,[7] where the guest residue ‘X’ can be selected from all amino acids (except Pro) to tune the biophysical properties of the polypeptide. An ELP was selected as the hydrophilic segment of the CP because it has many properties desirable for a drug carrier: ELPs can be encoded at the gene level and recombinantly overexpressed from a heterologous host, which allows precise control over the number and location of reaction sites along the backbone; they are monodisperse because they are recombinantly synthesized; they exhibit a sharp hydrophilic to hydrophobic phase transition in response to environmental variables such as temperature and ionic strength, which can be tuned by modulating the biopolymer composition and molecular weight; [8, 9] they are biodegradable[10] and nontoxic;[11] and they display favorable pharmacokinetics.[2, 12–14] ELPs also provide the important practical attributes that they can be expressed with high yield in bacterial expression systems[15] and they can be conveniently purified by exploiting their environmental sensitivity.[16, 17]
The CP was overexpressed from a plasmid-borne gene in E. coli (see supporting information) and was purified by a nonchromatographic method, inverse transition cycling.[16, 17] The model compounds with Log(D) values ranging from −1 to 4 were selectively conjugated to the Cys residues in the CP by a Michael addition reaction. Figure 1B displays the structure of the model compounds, as defined by their Log(D) at pH 7.4, where higher values indicate greater hydrophobicity. The Log(D) was calculated with the ACD Labs/PhysChem Suite,[18] which fragments a molecule into non-overlapping structures. The Log(D) value is then calculated through a summation of the hydrophobicities of the individual components and their correction factors.[6, 19] Following attachment, the degree of conjugation was assessed by measuring the ratio of free, residual cysteine residues, quantified with Ellman’s reagent, and the polypeptide concentration, measured by the bicinchoninic acid assay (experimental details in supporting information), and ranged from 3.3 – 6.4 for all of the molecules, as listed in SI Table II.
Next, the spontaneous self-assembly of the conjugates were investigated by dynamic and static light scattering (DLS, SLS), temperature-programmed turbidimetry, and fluorescence spectroscopy (see SI Figure 2 for fluorescence data). The attachment of 3–6 copies of compounds with a Log (D) < 1.5 (shown in blue in Figure 1B) did not trigger self-assembly of the CP, whereas compounds with a Log(D) > 1.5 (shown in pink) imparted sufficient amphiphilicity to the CP to trigger their self-assembly into nanoparticles with the conjugated molecules presumably comprising the hydrophobic core. The conjugates that did not trigger self-assembly had an average hydrodynamic radius (Rh) of 5.9 nm ± 0.7, which was similar to the Rh of the unmodified, control CP. In contrast, the conjugates that formed nanoparticles had an Rh ranging from 30 nm to 58 nm, with an average standard deviation of ~15% within each population (Figure 2B). There was no correlation between the hydrodynamic size of the nanoparticles and the number of molecules conjugated per CP (R2 = 0.0009).
Figure 2. Physical properties of CP nanoparticles.

(A) CP thermal characterization. Transition temperature (Tt) as a function of CP concentration of CPs conjugated to hydrophilic compounds (blue; unimer; compounds 1–5), and hydrophobic compounds (red; nanoparticle; compounds 6–10, 14) compared with an unconjugated control (black; unimer). The thermal behavior of all 6 CP-small molecule conjugates that formed nanoparticles was identical, and is hence plotted together as the mean of the Tt of each of the CP-small molecule conjugate, and the error bars are the standard deviation. The lines are linear fits to the data. (B) DLS results of the CP-conjugate of compound 8 with a Log(D) of 2.1, which shows the increase in Rh from ~6 nm corresponding to unimers prior to conjugation to the formation of nanoparticles with a Rh of ~33 nm after conjugation. (C) Relationship between the Tt (left Y-axis, data in red) and Rh (right Y-axis, data in black) as a function of Log(D) for all 14 conjugates. As the Log(D) increases to greater than 1.5, the particle Rh increases from the unimer size of 6 nm to nanoparticles with an Rh of 30 – 55 nm for different conjugates. The concentration dependence of the Tt (slope from A) decreases from an average value of −5.5 to −1.0 °C/Log(concentration). The curves in (C) are solely a guide to the eye.
Each nanoparticle-forming conjugate was next analyzed by SLS to determine the number of CPs per nanoparticle and the shape factor (ρ = Rg/Rh) that describes the distribution of mass within the nanoparticle. The shape factors ranged from 0.69 to 1, which indicate that there is likely a significant difference in the morphology of some of these nanoparticles. The precise attribution of the morphology of nanoparticles by light scattering is subject to some ambiguity, as the shape factor is subject to deviations arising from polydispersity and shape diversity within the ensemble of nanoparticles. Shape factors of 0.775 are indicative of spherical micelles, which is likely to be the case for nanoparticles with ρ values in the 0.69 to 0.8 range, while nanoparticles with shape factors of ~1.0 could instead be vesicles, as spherical shells have a ρ of 1.0, or polydisperse rods with relatively low aspect ratios.[20] The apparent coordination number (Z) also varied significantly, ranging from ~10–60 for the different conjugates. We note that the apparent coordination numbers were not corrected for the critical aggregation concentration (CAC) of the nanoparticles, which suggests that the numbers shown in Figure 4 are the minimum coordination numbers for each nanoparticle. There was a trend toward larger apparent coordination numbers as the hydrophobicity of the conjugated molecules increased (Figure 4).
Figure 4.
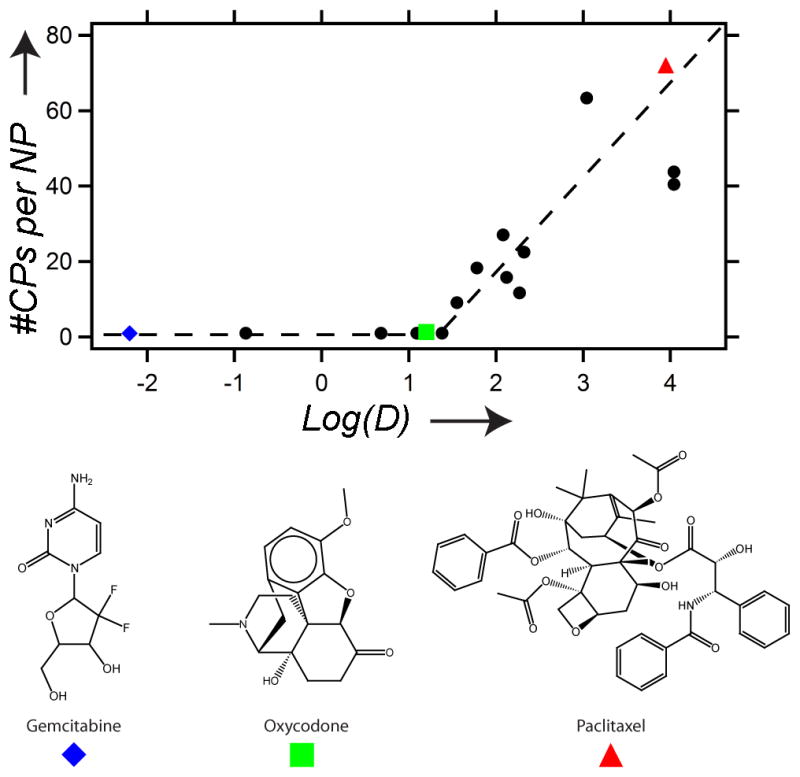
The apparent coordination number (#CPs per NP) versus the Log(D) of the small molecules conjugated to each CP. Above the threshold of Log(D) = ~1.5, the number of CPs per nanoparticle (#CPs/NP) increases with hydrophobicity of the conjugated small molecule [Log(D)]. The blue diamond, green square, and red triangle markers indicate gemcitabine, oxycodone, and paclitaxel, respectively. The lines are drawn solely as a guide to the eye.
To directly visualize the morphology and shape diversity of the nanoparticles, selected conjugates were imaged via cryogenic transmission electron microscopy (cryo-TEM). The samples were imaged at 80 keV to increase the contrast between the environment and the nanoparticle core, which exhibited a relatively high aqueous content. Figure 3 shows that the conjugates with a Log(D) > 1.5 formed nanoparticle cores distributed throughout the ice at regular intervals (Figure 3B–F) whereas the one hydrophilic conjugate displayed a film devoid of any structures (Figure 3A). In agreement with the light scattering data, the selected conjugates primarily consisted of spherical nanoparticles, though there were subpopulations of worm-like micelles and stiff rods present in a few of the samples that may explain the variability of the shape factor measured by light scattering (SI Figure 1).
Figure 3. Cryo-TEM.
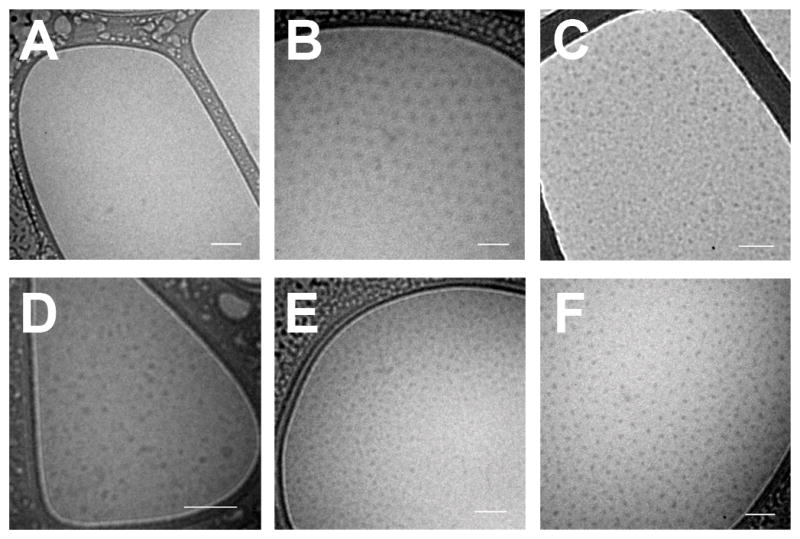
CP conjugates were imaged via cryo-TEM in phosphate buffered saline. A) N-methoxycarbonylmaleimide (Compound 1) did not form nanoparticles and is displayed as a negative control. The remaining conjugates spontaneously formed nanoparticles: B) n-benzylmaleimide (Compound 7); C) n-[4-(2-benzimidazolyl)phenyl]maleimide (Compound 9); D) 2-maleimido fluorene (Compound 12); E) n-(1-pyrenyl)maleimide (Compound 14); and F) paclitaxel. Scale bars represent 100 nm.
These data clearly demonstrate that attachment triggered self-assembly of a CP is exhibited by a diverse range of hydrophobic small molecules. These results are also notable because they clearly reveal a simple predictive rule that governs the self-assembly of CPs based on a threshold hydrophobicity of the conjugated small molecule. Although the threshold of Log(D) > 1.5 predicts whether self-assembly will occur, the Log(D) of the conjugated molecule does not predict the size or shape of the nanoparticle that is formed, as we observed significant differences in both parameters based on the light scattering results that are likely related to structural differences between these molecules.
We also found that the phase transition behavior of the CP was altered following conjugation of the model compounds (Figure 2A). CPs, similar to the ELPs from which they are derived, display a characteristic transition temperature (Tt), below which they are soluble in aqueous solutions, and above which they form polydisperse micron-sized aggregates. This Tt is typically modulated by varying the hydrophobicity of the guest residue (X), with hydrophobic guest residues depressing the Tt, and hydrophilic residues elevating the Tt.[9] In a similar manner, as the hydrophobicity of the conjugated molecules increased to the assembly threshold as defined by Log(D) = 1.5, the Tt shifted downward, though the dependence upon concentration (the slope) remained uniform (Figure 2A). Upon reaching this threshold, however, the Tt was immediately reduced to a temperature that was near independent of concentration, though we note that this transition occurs from a nanoparticle to micron-sized aggregates in contrast to unmodified CPs, which undergo a transition from soluble unimers to micron-sized aggregates. Notably, all conjugates that form nanoparticles display the same thermal behavior that is described by the same quantitative relationship between the Tt and unimer CP concentration. The fact that all self-assembled CP nanoparticles display the same functional relationship between their Tt and their solution concentration (on a unimer basis) strongly suggests that their phase behavior is controlled by the high and invariant local ELP concentration within the nanoparticles and not by the total concentration of the CP in solution.
This finding is significant because this near-independence of Tt from CP concentration enables the nanoparticles to maintain a very stable Tt (within 2°C) over a 100-fold decrease in concentration that would arise from physiological effects such as clearance from circulation. In contrast, the CP conjugates and the unmodified CP that exist as unimers show a Tt shift of over 20°C within the same concentration range (Figure 2A). The ability of these thermoresponsive nanoparticles to maintain a constant thermal response over a range of concentration eliminates the need to compensate for the effect of dilution and clearance that would occur upon their injection into systemic circulation and is hence likely to be an extraordinarily useful feature in future attempts to thermally target these CP nanoparticles to specific tissues in vivo by the application of external focused hyperthermia to disease sites.
These experiments with model compounds provide a simple physical model for self-assembly, in that molecules with a Log(D) > 1.5 will drive self-assembly of the conjugate into nanoparticles. To further investigate the predictive validity of this model, three small molecule therapeutics were selected for conjugation to the CP through heterobifunctional linkers, where one end of the linker was attached to the CP and the other end to a reactive moiety on the drug (see SI for methods). We chose three drugs that spanned a range of logD at pH 7.4, estimated by ACD Labs/PhysChem Suite [18]: gemcitabine (−2.2), oxycodone (1.2), and paclitaxel (4.0). Conjugation of gemcitabine and oxycodone did not trigger self-assembly of the CP, while paclitaxel conjugation led to the spontaneous formation of nanoparticles that were similar in size to the nanoparticles that self-assembled by the attachment of hydrophobic small molecules to the CP (Table 1 and Figure 4). Conjugation of paclitaxel to the hydrophilic CP also significantly increased the solubility of paclitaxel in excess of 2 mM paclitaxel equivalents in PBS, which was ultimately limited by the viscosity of the CP solution. Cryo-TEM of the CP-paclitaxel nanoparticles displayed close-packed spherical nanoparticles with a dense electron core, whose size (measured by the core-to-core distance) was smaller than the Rh determined by dynamic light scattering (RTEM = 22 ± 4 nm; n = 50) (Figure 3F). This discrepancy is likely due to the fact that the soft CP nanoparticle corona can become significantly compressed under the conditions necessary to visualize the particles with cryo-TEM (the high sheer stressed induced by blotting and the high polymer concentration) when compared to the very dilute conditions necessary for light scattering measurements. The CP-paclitaxel nanoparticles were also highly stable with a CAC ≤ 10 μM (SI Figure 3). These results are consistent with the model: gemcitabine and oxycodone, as predicted by the model, were too hydrophilic to drive the assembly of nanoparticles at pH 7.4, whereas paclitaxel, with a Log(D) of 4.0, is above the threshold of hydrophobicity needed to trigger self-assembly.
Table 1.
CP nanoparticles assembled through drug conjugation
| Drug | Log(D)[a] | Rh (nm) | Drug/CP | #CPs/NP |
|---|---|---|---|---|
| Gemcitabine | −2.2 | 5.7 | 5 | 0.9 |
| Oxycodone | 1.2 | 9.7 | 4 | 1.3 |
| Paclitaxel | 4.0 | 53.3 | 2 | 72.0 |
Log(D) was estimated using ACD Labs/PhysChem Suite.
Having shown that we can rationally design and synthesize a drug-loaded CP nanoparticle, we next turned our attention to constructing a thermally responsive CP-nanoparticle that is useful for in vivo targeting. To do so, we resynthesized the CP –termed CP2– with a V1A9 guest residue composition that is slightly more hydrophobic than the V1G7A8 sequence used throughout the rest of this paper (CP1) to decrease its Tt to the desired range of 37–42ºC, based on a large body of structure-property behavior that we have assembled for ELPs in the past decade. CP2 exhibited a Tt between 38 and 42°C in 90% fetal bovine serum, a medium that closely mimics in vivo conditions. Figure 5 shows that the Tt is invariant between three different CP2 nanoparticle conjugates ranging in Log(D) from 1.5 (Compound 6) to 4.0 (Compound 14), and that these nanoparticles are stable in serum as the Tt remains characteristically near-independent from the CP solution concentration.
Figure 5. Design of thermally sensitive CP nanoparticles.
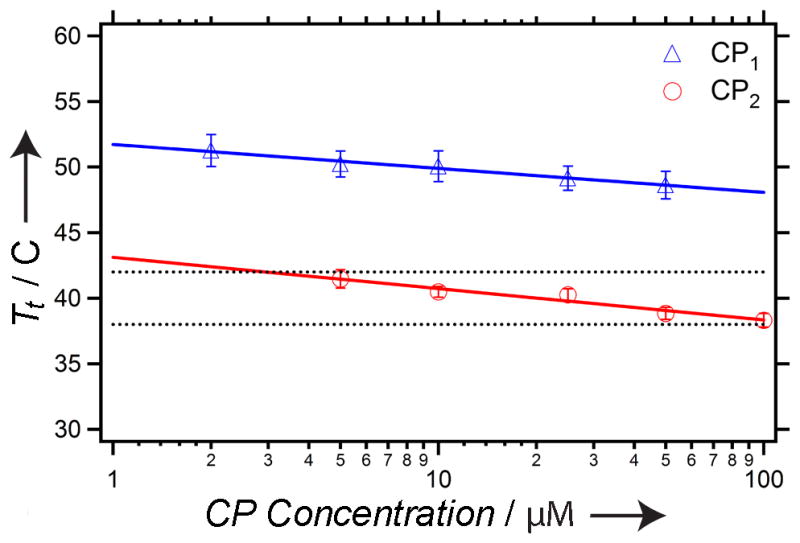
The transition temperature (Tt) as a function of CP1 and CP2 concentration. The CP1 data represents an average of conjugates 6–10 and 14, whereas the CP2 data represents an average of conjugates 6, 7, and 14. The black lines represent the targeted range of hyperthermia (38–42°C). The error bars are the standard deviation. The lines are linear fits to the data.
Our results are notable for several reasons. Although attachment of drugs to polymers has been used to develop self-assembling therapeutic formulations, this work departs significantly both in its conceptual novelty and scope from these previous studies: adriamycin was covalently conjugated to the aspartate groups in PEG4300-b-polyaspartate (NK911), thereby assembling into 50 nm micelles;[21] the biologically active metabolite of irinotecan, SN-38, was attached to the glutamate chain in PEG5000-b-polyglutamate (NK012) to form micelles;[22] and the polyaspartate chain of PEG12000-b-polyaspartate was modified with 4-phenyl-1-butanol, which induced assembly and facilitated the noncovalent incorporation of paclitaxel into (NK105).[23] In these studies, a general route to the conjugation triggered self-assembly of the polymer by a range of molecules was not shown, nor did these and related studies provide a model that enabled a priori prediction of the propensity –or lack thereof– of a molecule to trigger the self-assembly of the polymer.[21, 24–26] Furthermore, none of these systems exhibit the thermal responsiveness observed with these CPs over a range of concentrations relevant for drug delivery, a feature that can be exploited for in vivo thermal targeting of CP-drug nanoparticles
In conclusion, this paper is the first demonstration of a roadmap for the rational design of highly soluble, thermally responsive drug-loaded nanoparticles whose properties can be tailored at the molecular level. We show that attachment of small molecules above a critical threshold of hydrophobicity trigger self-assembly of the CP into soluble nanoparticles ranging from 60 nm to ~100 nm in diameter. These nanoparticles are soluble at concentrations greater than 100 μM of CP and ~3–6 fold higher for the small molecules, and are stable upon dilution up to low micromolar concentrations of the CP, a concentration regime that is of great utility for drug delivery. These results provide a simple predictive model for nanoparticle formation that permits a priori screening of the propensity of drugs to be sequestered into the nanoparticle core solely based on their Log(D) value and demonstrate that conjugation-triggered self-assembly can package a hydrophobic drug into soluble nanoparticles. Finally, we demonstrate that we can rationally tune the nanoparticle-to-aggregate Tt to occur between 38 and 42ºC, which provides a nanoparticle drug delivery system that can be targeted to diseased tissue by external applied, focused mild hyperthermia.
Supplementary Material
Footnotes
This work was supported by a grant from the National Institutes of Health (R01 EB000188) to A.C and by the NSF’s Research Triangle MRSEC (DMR-1121107). J.R.M. acknowledges the financial support of a NIH Biotechnology predoctoral fellowship (T32 GM 8555).
References
- 1.Duncan R. Nat Rev Cancer. 2006;6:688–701. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]
- 2.MacKay JA, Chen MN, McDaniel JR, Liu WG, Simnick AJ, Chilkoti A. Nature Mater. 2009;8:993–999. doi: 10.1038/nmat2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maeda H, Seymour LW, Miyamoto Y. Bioconjugate Chemistry. 1992;3:351–362. doi: 10.1021/bc00017a001. [DOI] [PubMed] [Google Scholar]
- 4.Veronese FM, Monfardini C. Bioconjugate Chemistry. 1998;9:418–450. doi: 10.1021/bc970184f. [DOI] [PubMed] [Google Scholar]
- 5.McDaniel JR, Macewan SR, Dewhirst M, Chilkoti A. J Control Release. 2012;159:362–7. doi: 10.1016/j.jconrel.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Livingstone DJ. Current Topics in Medicinal Chemistry. 2003;3:1171–1192. doi: 10.2174/1568026033452078. [DOI] [PubMed] [Google Scholar]
- 7.Urry DW, Trapane TL, Prasad KU. Biopolymers. 1985;24:2345–2356. doi: 10.1002/bip.360241212. [DOI] [PubMed] [Google Scholar]
- 8.Meyer DE, Chilkoti A. Biomacromolecules. 2004;5:846–851. doi: 10.1021/bm034215n. [DOI] [PubMed] [Google Scholar]
- 9.Urry DW. Journal of Physical Chemistry B. 1997;101:11007–11028. [Google Scholar]
- 10.Shamji MF, Betre H, Kraus VB, Chen J, Chilkoti A, Pichika R, Masuda K, Setton LA. Arthritis Rheum. 2007;56:3650–61. doi: 10.1002/art.22952. [DOI] [PubMed] [Google Scholar]
- 11.Urry DW, Parker TM, Reid MC, Gowda DC. Journal of Bioactive and Compatible Polymers. 1991;6:263–282. [Google Scholar]
- 12.Betre H, Liu W, Zalutsky MR, Chilkoti A, Kraus VB, Setton LA. J Control Release. 2006;115:175–182. doi: 10.1016/j.jconrel.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 13.Liu W, Dreher MR, Chow DC, Zalutsky MR, Chilkoti A. J Control Release. 2006;114:184–192. doi: 10.1016/j.jconrel.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 14.Liu WE, Dreher MR, Furgeson DY, Peixoto KV, Yuan H, Zalutsky MR, Chilkoti A. J Control Release. 2006;116:170–178. doi: 10.1016/j.jconrel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 15.Chilkoti A, Chow DC, Dreher MR, Trabbic-Carlson K. Biotechnol Prog. 2006;22:638–646. doi: 10.1021/bp0503742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meyer DE, Chilkoti A. Nat Biotechnol. 1999;17:1112–5. doi: 10.1038/15100. [DOI] [PubMed] [Google Scholar]
- 17.Trabbic-Carlson K, Liu L, Kim B, Chilkoti A. Protein Sci. 2004;13:3274–84. doi: 10.1110/ps.04931604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Advanced chemical development inc., 133 richmond street west, suite 605, toronto, canada m5h 2l3.
- 19.Petrauskas AA, Kolovanov EA. Perspectives in Drug Discovery and Design. 2000;19:99–116. [Google Scholar]
- 20.Manners I, Massey J, Power KN, Winnik MA. J Am Chem Soc. 1998;120:9533–9540. [Google Scholar]
- 21.Yokoyama M, Miyauchi M, Yamada N, Okano T, Sakurai Y, Kataoka K, Inoue S. Cancer Res. 1990;50:1693–1700. [PubMed] [Google Scholar]
- 22.Koizumi F, Kitagawa M, Negishi T, Onda T, Matsumoto S, Hamaguchi T, Matsumura Y. Cancer Res. 2006;66:10048–10056. doi: 10.1158/0008-5472.CAN-06-1605. [DOI] [PubMed] [Google Scholar]
- 23.Hamaguchi T, Matsumura Y, Suzuki M, Shimizu K, Goda R, Nakamura I, Nakatomi I, Yokoyama M, Kataoka K, Kakizoe T. Br J Cancer. 2005;92:1240–1246. doi: 10.1038/sj.bjc.6602479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kataoka K, Bae Y, Fukushima S, Harada A. Angewandte Chemie International Edition. 2003;42:4640–4643. doi: 10.1002/anie.200250653. [DOI] [PubMed] [Google Scholar]
- 25.Kataoka K, Nishiyama N, Okazaki S, Cabral H, Miyamoto M, Kato Y, Sugiyama Y, Nishio K, Matsumura Y. Cancer Res. 2003;63:8977–8983. [PubMed] [Google Scholar]
- 26.Kwon G, Naito M, Yokoyama M, Okano T, Sakurai Y, Kataoka K. Langmuir. 1993;9:945–949. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.