

Abstract
Gastric epithelial cells (GECs) are the primary target for Helicobacter pylori (H. pylori) infection and may act as antigen presenting cells (APC) regulating local T cell responses. We previously reported that H. pylori infection of GECs induces the expression of the T cell co-inhibitory molecule B7-H1 on GECs. This process contributes to the hyporesponsiveness of CD4+ effector T cells and accumulation of T regulatory cells. In the studies presented herein we investigated the impact of H. pylori cytotoxin CagA on the modulation of the expression of the T cell co-stimulator B7-H2 by GEC. B7-H2 is involved in promoting Th17 type responses. H. pylori infection downregulates B7-H2 expression by GECs in a CagA dependent manner. IFNγ, which is increased in the H. pylori infected gastric mucosa, synergizes with H. pylori in downregulating B7-H2 expression by GECs. CagA-mediated modulation of B7-H2 on GEC involves p70 S6 kinase phosphorylation. The CagA-dependent B7-H2 downregulation in GEC correlates with a decrease in Th17 type responses in vitro and in vivo. Further, CagA-dependent modulation of Th17 responses inversely correlated with the H. pylori colonization levels in vivo. Our data suggest that CagA contributes to the ability of H. pylori to evade Th17 mediated clearance by modulating expression of B7-H2 and, thus, to the establishment of the H. pylori chronic infection.
Introduction
Helicobacter pylori (H. pylori) is a Gram-negative, spiral shaped bacterium that infects the gastric mucosa of more than 50% of the world’s population. H. pylori infection initially occurs in childhood and becomes persistent. This chronic infection leads to gastric inflammation (1) and is the major cause of gastritis, gastric and duodenal ulcers as well as gastric adenocarcinoma (2–8).
The CagA (cytotoxin associated gene A) protein is a major virulence factor of H. pylori. Patients infected with cagA+ strains have higher levels of inflammatory responses and are at a higher risk of developing peptic ulcer or gastric cancer (4, 9). CagA is encoded by the cagA gene within the cag pathogenicity island (cag PAI), a 40-kilobase chromosomal region that encodes for a type four secretion system (T4SS).
Gastric epithelial cells (GECs) are the primary target for H. pylori infection. After H. pylori adhere to GECs, the CagA protein is translocated into their cytosol via a T4SS (10). Once inside GECs, CagA becomes phosphorylated and elicits multiple cell responses, including disruption of epithelial tight junctions, cytoskeleton rearrangement, changes in cellular adhesion properties and polarity as well as secretion of proinflammatory mediators (11, 12). Despite the marked inflammatory response within the H. pylori-infected gastric mucosa the host immune response is unable to clear H. pylori resulting in persistent infection and development of chronic gastric inflammation (13, 14). Studies by us and others suggested that the imbalance in CD4+ T cells responses to H. pylori is responsible for the host’s inability to clear the infection (15–18). Th17 cells, whose hallmark cytokine is IL-17A, are crucial in the clearance of extracellular bacteria (19). IL-17A is primarily associated with gastric inflammation during H. pylori infection and, if chronically present, may contribute to the inflammation associated carcinogenesis (19–21). On the other hand, IL-17A-initiated recruitment of neutrophils is critical for the clearance of the bacteria (22). Although increased IL-17A expression is observed during chronic gastric inflammation, the levels produced are not sufficient to clear the infection. The mechanisms responsible for the reduced Th17 responses during the establishment of H. pylori persistence in the gastric mucosa remain poorly understood. DC-mediated skewing of T cell balance toward suppressive regulatory T cells (Treg) has been suggested to be important in the down regulation of Th17 and the establishment of the H. pylori persistence (23). However, it is unknown whether and how GEC, as primary targets for H. pylori infection, contribute to suboptimal Th17 cell responses during the establishment of the chronic infection.
During H. pylori infection GEC express MHC class II and may act as local APCs (24, 25). In addition to the recognition of MHC-bound peptides on APCs by T cell receptor (TCR), the outcome of APC-T cell interactions depends on a second signal provided by engagement of the B7-co-stimulatory receptor (26). Our lab has previously shown that GECs express the classical B7 co-stimulators B7–1 and B7–2, whose expression is increased during H. pylori infection (25). We also demonstrated that GECs express B7-H1, whose expression also increases during H. pylori infection and contributes to the suppression of CD4+ effector T cell activity and upregulation of Treg cells (18, 27).
B7-H2 (ICOS-L) is among the newer members of the B7-family of receptors and is known to have a co-stimulatory function on T cell activity upon binding to its receptor, ICOS (28). Recent studies have implicated B7-H2/ICOS interaction in Th17 cell development, maintenance and function (29–31). However, the role of B7-H2 in immune responses to H. pylori is unknown. Thus, in this study we investigated the impact of H. pylori and its major virulence factor CagA on the modulation of B7-H2, as an important regulator of Th17 cell responses. Our in vitro and in vivo studies showed that H. pylori causes down-regulation of B7-H2 on GECs and this effect depends on the presence of H. pylori CagA via a process involving p70 S6 kinase activation. CagA-dependent B7-H2 downregulation on GEC correlated with the decrease in Th17 responses and was inversely correlated with the level of H. pylori colonization in vivo. Thus, this study points out a novel strategy used by H. pylori to impair Th17 responses, and this impairment could contribute to the establishment of persistent infection in the host.
Materials and Methods
Animals
Female C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Six-to-eight-week old mice, which were tested negative for intestinal Helicobacter spp., were used in the experiments. The UTMB Institutional Animal Care and Use Committee approved protocol was followed.
Human tissue and cell lines
Gastric epithelial cells were isolated from biopsy specimens as described previously (32). Briefly, biopsy specimens of the gastric antrum were obtained from consenting patients undergoing gastro-esophageal-duodenoscopy for various clinical indications in accordance with Institutional Review Board approved protocol. Patients were considered infected if H. pylori was detected by both rapid urease testing and histopathology, and for these studies were confirmed to be infected by culture of H. pylori from biopsies. Biopsy tissue was placed in calcium- and magnesium-free HBSS, supplemented with 5% FCS and penicillin plus streptomycin, and transported immediately to the laboratory. The tissue was then placed in HBSS containing 0.1 mM EDTA and 0.1 mM DTT and agitated at 37°C for 15–30 min to remove the mucus. The biopsy tissue was placed in dispase solution (2.4 U/ml; Boehringer Mannheim, Mannheim, Germany) and agitated at 37°C for 30 min with a change of fresh dispase solution after 15 min. The supernatant was collected and the cells were pelleted by centrifugation at 200 g for 5 min. The cells isolated were mostly (>90%) epithelial cells according to the morphology with May-Grunwald-Giemsa staining (Sigma Chemical Co., St. Louis, MO) and flow cytometry with immunofluorescence staining of anticytokeratin mAb. Human gastric carcinoma epithelial cells (GECs) N87 and AGS as well as Hs738.st/int human gastric epithelial cells were obtained from the American Type Culture Collection (ATCC). HGC-27 was obtained from RIKEN, The Institute of Physical and Chemical Research, Japan, and was maintained in RPMI 1640 with 10% fetal bovine serum (FBS) and 2 mM L-glutamine.
Bacterial cultures and infection of GEC
H. pylori LC11 (cagA+) and RD26 (cagA+) strains were originally isolated from duodenal ulcer and peptic ulcer patients, respectively (33, 34). H. pylori strain Sydney strain 1 (SS1) and PMSS1 (pre-mouse SS1) (35) used to infect mice were kind gifts from Drs. J. Pappo (Astra) and Dr. Richard Peek (Vanderbilt Univ.), respectively. These bacterial strains were grown on tryptic soy agar (TSA) plates supplemented with 5% sheep’s blood (Becton Dickinson, San Jose, CA) or on blood agar plates with 2.5 μg/ml of chloramphenicol (Technova, Hollister, CA) to maintain cagA− strains at 37°C under microaerophilic conditions. Bacteria were transferred after 48 hour into Brucella broth containing 10% FBS for overnight. After centrifugation at 3000 RPM for 10 min, bacteria were resuspended in normal saline. The concentration of bacteria was determined by measuring the OD530 using a spectrophotometer (DU-65; BD Biosciences) and comparing the value to a standard curve generated by quantifying viable organisms from aliquots of bacteria at varying concentrations that were also assessed by OD and colony formation. For specific MOI the numbers of GEC were determined using trypan blue staining and the required number of bacteria was added after calculation. GEC were treated with IFNγ (100 U/ml) for 48 hour, then washed and incubated an additional day in regular medium without IFNγ before infection. When IFNγ needed to be neutralized, anti-IFNγ-neutralizing Ab was added at an optimal concentration (10 μg/ml). As an isotype control, mouse IgG1, κ Ab was used in the same concentration. B7-H2 expression was measured after coculture with IFNγ or H. pylori-infected GEC in the presence of either anti- IFNγ-neutralizing Ab or isotype control Ab.
Construction of cagA isogenic mutant
For isogenic cagA mutants, portions of the genes were amplified by PCR and the amplified fragment was inserted into the pBluescript SK (+) (Stratagene, La Jolla, CA). After mutagenesis by insertion of a chloramphenicol resistance gene cassette (a gift from Dr. D. E. Taylor, University of Alberta, Edmonton, Canada) in the cagA gene, the obtained plasmids (1 to 2 μg) were used for inactivation of chromosomal genes by natural transformation as previously described (36). Correct integration of the chloramphenicol resistance gene cassette into the H. pylori chromosome by double crossover recombination was confirmed by PCR amplification followed by Southern blot hybridization.
Antibodies, recombinant proteins and cell signaling inhibitors
PE-conjugated anti-human B7-H2 (clone M1H12), PE-conjugated anti-murine B7-H2 (clone HK5.3), APC-conjugated anti-murine epithelial cell marker EpCAM (clone G8.8), and PE-conjugated RORγτ (clone AFKJS-9) were purchased from eBioscience as were the isotype controls. The viability dye eFluor 780 (eBioscience, San Diego, CA, USA) was included in the experiments to control cell viability. Human rIFNγ (Roche) was used at 100 U/ml. Neutralizing Abs for IFNγ included the purified functional grade anti-human IFNγ from eBioscience. The isotype control Ab used for IFNγ studies was functional grade mouse IgG1 κ from eBioscience. For cell signaling inhibition the following inhibitors were used: CAY10512 (10 μM; Cayman chemical, Michigan, USA), AG-490 (100 ng/mL; Enzo Life Sciences, Farmingdale, NY), Wortmannin (100 nM; Calbiochem, Billerica, MA), and Rapamycin (100 ng/mL; Calbiochem, Germany).
GEC:T cell co-culture
For GEC:T cells co-culture experiments naïve CD4+ T cells were isolated from peripheral blood as previously described (17). Briefly, heparinized venous blood samples were collected from healthy volunteers negative for H. pylori (IRB-approved protocol 06-122 at the University of Texas Medical Branch). Peripheral blood mononuclear cells (PBMC) were prepared from collected blood by density gradient centrifugation over Ficoll-Paque Plus. Naïve CD4+ T cells were isolated from the peripheral blood by negative selection using MACS Naïve CD4+ T isolation kit II (Miltenyi Biotec, Germany). GEC where exposed to H. pylori strains for 8 hours, then supernatants from H. pylori-treated cultures were filtered to remove bacteria, and GEC cells were washed twice with PBS to remove attached bacteria and replaced with filtered supernatants till 24 hours. CD4+ T cells were preactivated for 1 hour with anti-CD3/CD28 beads (Invitrogen) according to the manufacturer’s instructions and added to each well at 3:1 T cell:GEC ratio, then incubated at 37°C with 5% CO2 for 2 days.
Western blot analysis
Western blot analysis was performed as previously described (37).
Infection of mice, processing of stomach and detection of H. pylori in murine stomach
Experimental animal groups were inoculated by orogastric gavage with 108 CFU (in 100 μL of PBS) of H. pylori SS1 or PMSS1 strains, three times over a week. Placebo group was inoculated with PBS. Four weeks later animal serum was collected, mice were euthanized and stomach was removed. Stomachs were dissected longitudinally in 2–4 pieces and used for analysis of the H. pylori load, histopathology, RT-PCR and flow cytometry analysis. For histopathology analysis one longitudinal strip of stomach was placed in 10% normal buffered formalin for 24 hour at 4°C, transferred into 70% ethanol solution and stored at 4°C until needed. Tissue was then embedded in paraffin and processed by hematoxylin and eosin (H&E) staining. Stomach tissue dissociation and enzymatic digestion was performed using gentleMACS™ Dissociator (Milteneyi Biotec, Auburn, CA) according to the manufacturer’s instructions. Briefly, DTT/EDTA treatment was performed similarly as described above for human tissue. Cells were then washed twice with HBSS followed by treating with Collagenase I, II, IV (100 U/mL). After first round of dissociation cells were incubated with enzymatic mixture at 37°C for 30 min. Tissue was then subjected to the second round of dissociation and treated with DNAse (10 ug/mL, Worthington) at 37°C for 15 min. Finally, tissue was processed using the dissociator, and washed twice with HBSS. The digested tissue was passed through a 40 μm cell strainer. Recovered cells were counted and used for immunostaining followed by flow cytometry analysis.
Murine gastric tissue was homogenized and DNA was extracted using DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA) followed by a purification of the DNA according to the manufacturer’s protocol. Extracted DNA was used for the detection of H. pylori by real time PCR using a protocol originally described by Rouessel et. al. A primer/probe set 16SHP229BP for 16S gene was used for the quantification of the H. pylori bacterial load. To determine bacterial load standard curves were generated by PCR of serial dilutions of extracted H. pylori DNA. Quantification of H. pylori in murine gastric mucosa and determination of absolute genome copy number was calculated according to the method described by Rouessel et. al. (38). Murine GADPH gene amplification was used to control the equal loading of total DNA used in PCR reaction.
Flow cytometry
Flow cytometry was performed as we described previously (17).
Real-Time RT-PCR
Real-time RT-PCR was performed according to the Applied Biosystems’s two-step Real-time RT-PCR protocol (Applied Biosystems, Foster City, CA). The appropriate assays-on-demand™ gene expression assay primers/probe mix (Applied Biosystem) for human 18S and gene of interest (a 20X mix of unlabeled PCR primers and TaqMan® MGB probe, FAM™ dye-labeled) and 2 μL of cDNA was added to the PCR reaction step. The reactions were carried out in 20 μL final volume using BioRad Q5 real-time PCR machine according to the following protocol: 2 min at 50°C, 10 min at 95°C (1 cycle) and 15 sec 95°C and one min at 60°C (40 cycles). Triplicate cycle threshold values were analyzed in Microsoft Excel using the comparative CT (ΔΔCT) method as described by the manufacturer (Applied Biosystems).
Bio-Plex
The levels of total and phosphorylated cell signaling proteins in N87 cells infected with H. pylori strains; IL-17A from T cell-GEC co-culture and IL-17, IL-21, IL-22, IL-23 and IL-6 from murine serum collected from H. pylori infected mice was measured using Luminex array (Millipore, Billerica, MA, USA) according the manufactures instruction. Samples were analyzed using Bio-Plex Manager software (Bio-Rad).
Statistical analysis
The results were expressed as the mean ± SD of data obtained from at least three independent experiments done with triplicate sets in each experiment, unless otherwise indicated, Differences between means were evaluated by ANOVA using Student’s t-test for multiple comparisons. Values of P<.05 were considered statistically significant.
Results
H. pylori downregulate B7-H2 expression on Gastric Epithelial Cells
To assess B7-H2 expression during H. pylori infection, mRNA expression was examined in GECs isolated from gastric mucosa biopsy samples from H. pylori infected and uninfected individuals using real time RT-PCR. B7-H2 mRNA expression was significantly decreased in GECs from H. pylori positive subjects when compared to GECs from uninfected controls (Fig. 1A). To determine whether H. pylori directly induced a reduction of B7-H2 by GECs, a human GEC line (N87) was infected with H. pylori 51B, LC-11 or RD26 strains. A significant decrease in surface B7-H2 expression on GECs was observed at 24 hours post-infection with all H. pylori strains (Fig. 1B, C). Fig 1B shows differential regulation of B7-H1 and B7-H2 by N87 cells infected with H. pylori 51B. This effect was dose-dependent (Fig. 1D). Similar results were observed when other human GEC lines (e.g. AGS, HGC-27 and HS-738 cells) were infected (not shown). Thus, our results indicate that H. pylori infection down-regulates B7-H2 expression on human GEC in a dose-dependent manner.
Figure 1.

Decreased expression of B7-H2 in H. pylori infected GEC. (A) Gastric biopsy samples were collected from five H. pylori negative and five positive patients, GEC was isolated and were analyzed for the B7-H2 mRNA expression by real-time RT-PCR. (B) Modulation of the B7-H2 surface expression on GEC N87 by the infection with H. pylori 51B and LC11 (24 hour post-infection, H. pylori:GEC ratio 10:1) as determined by flow cytometry analysis. Upregulation of B7-H1 on N87 cells after treating with H. pylori 51B was shown as a positive control. (C) One of representative flow cytometry histogram obtained for GEC N87 infected with H. pylori RD26. (D) Dose-response analysis. N87 cells were infected with H. pylori RD26 strain at different ratio of H. pylori:GEC (10:1, 30:1 and 100:1) for 24 hour and analyzed by flow cytometry. The data expressed as a mean of fluorescence intensity (MFI). Data represents the mean ± SD (n=8); P < .05.
Downregulation of B7-H2 expression depends on the presence of H. pylori CagA
Since CagA is an important H. pylori virulence factor capable of eliciting multiple host cell responses, (39) we sought to determine whether downregulation of B7-H2 is influenced by H. pylori CagA. Infection of GEC with H. pylori 51B wild type (WT) strain (Fig. 2A) led to as much as 50% decrease of B7-H2 mRNA as compared to uninfected controls at the time points examined (2 and 4 hours). In contrast, H. pylori cagA− mutant had a very limited effect on B7-H2 expression compared to the controls. These data were confirmed at the protein level. In contrast to the cagA+ H. pylori strain, infection with cagA− mutant had a minor effect on the reduction of B7-H2 surface expression by GECs (Fig. 2B–D). Western blot analysis of N87 cells treated under the same conditions provided an independent approach to validate decreased B7-H2 protein levels in cells infected with H. pylori WT as compared to cells infected with cagA− strain (Fig. 2E–F). These results suggested that the major H. pylori virulence factor CagA is involved in the B7-H2 downregulation on GEC.
Figure 2.
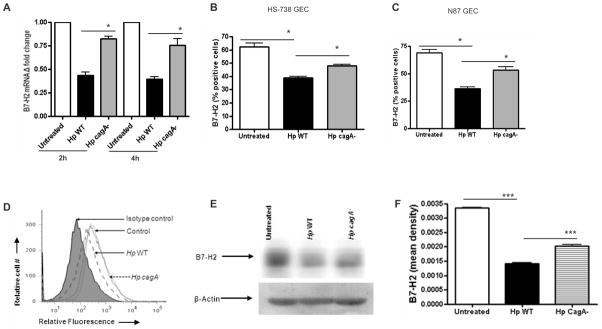
H. pylori mediated downregulation of B7-H2 on GEC involves CagA. (A) B7-H2 mRNA expression in N87 cells was analyzed using real-time RT-PCR. RNA was isolated from uninfected and 2 hour, 4 hour H. pylori cagA+ (WT) and cagA− strain infected GEC. mRNA level for B7-H2 was normalized to 18S and compared to the level of B7-H2 mRNA of untreated N87 cells. N=9, P < .05. (B) Surface B7-H2 expression was analyzed by flow cytometry on HS-738 and (C) N87 GEC cell lines after 24 hour of infection with H. pylori cagA+ (WT) or cagA− mutant. The data expressed as percent of positive cells expressing B7-H2. (D) One representative histogram is shown for GEC N87 infected with H. pylori 51B WT or cagA− mutant. N=8, P<.05. (E) B7-H2 expression was analyzed by western blot in N87 cells treated with H. pylori WT and cagA− strain after 24 hour infection. (F) Quantitative analysis of B7-H2/beta-actin ratio in GEC infected sample is included (n=3).
IFNγ and H. pylori have synergistic effects on B7-H2 downregulation
IFNγ is produced within the H. pylori infected gastric mucosa (40), and previously we showed a synergistic effect of IFNγ and H. pylori on B7-H1 upregulation on GEC (18). Thus, we examined whether IFNγ could modulate H. pylori-mediated B7-H2 downregulation on GECs. N87 cells treated with either IFNγ or H. pylori alone showed significant decreases in B7-H2 expression. However, treatment with both IFNγ and H. pylori resulted in complete abrogation of B7-H2 expression. B7-H2 expression decreased after culturing GECs with IFNγ, and decreased expression was more prominent when GEC cells were pretreated with IFNγ prior to infection with H. pylori. (Fig. 3) Blocking the IFNγ with neutralizing anti-IFNγ mAb prevented the decrease in the levels of B7-H2 expression. Our results clearly showed a synergistic effect of IFNγ and H. pylori in reduced B7-H2 expression by GECs. Interestingly, the synergism of IFNγ and H. pylori in decreasing B7-H2 expression was less obvious when cells were infected with a cagA− strain (Fig. 3). This result supports the key role of CagA in B7-H2 downregulation during H. pylori infection.
Figure 3.

IFNγ synergize H. pylori-mediated B7-H2 downregulation on gastric epithelium. N87 cells were treated with IFNγ (100 U/mL) for 48 hour. Cells treated with IFNγ were washed and cultured with medium for 24 hour before they were either infected with H. pylori 51B WT and cagA− strain in the presence or absence of IFNγ-neutralizing Ab. B7-H2 expression on N87 GEC was measured by flow cytometry after 24 hour. Graph represents the mean ± SD (n=8); P < .05.
B7-H2 downregulation involve activation of mTOR/p70 S6 kinase
Previous studies showed that H. pylori CagA protein can activate NFκB, MAPK, STAT 3 and PI3K pathways (41–44). In order to understand the underlying mechanisms regulating B7-H2 decreased expression during H. pylori infection we first globally analyzed pathways activated in H. pylori infected GEC using a Luminex cell signaling array. Our data demonstrated that, in addition to NFκB and STAT3 pathways, H. pylori infection of GEC also leads to the activation of mTOR/p70 S6 kinase within the first 5 minutes of infection (Fig. 4A, B). In contrast, no significant phosphorylation of p70 S6 kinase was observed in GEC infected with cagA− strain (Fig. 4C, D). Thus, we examined the role of these pathways in H. pylori-mediated downregulation of the B7-H2 by using specific inhibitors. We observed that downregulation of B7-H2 by the cagA+ H. pylori strain was blocked in the presence of rapamycin, a p70 S6 kinase/mTOR specific inhibitor (Fig. 4E). In contrast, inhibition of PI3K, STAT3 and NFκB pathways with pharmacological inhibitors did not affect H. pylori mediated downregulation of B7-H2 expression (Fig. 4F). These results suggest that p70 S6 kinase is a key signaling pathway in H. pylori-mediated downregulation of B7-H2 on GECs.
Figure 4.

Activation of mTOR/p70 S6 kinase involved in the H. pylori mediated downregulation of B7-H2 expression. N87 cells were incubated with H. pylori 51B WT and cagA− strain. Cell were lysed after 5 min, 15 min, 30 min, 45 min and 60 min. Cell lysates from GEC exposed to H. pylori 51B WT and cagA− strain was analyzed for (A, C) phospho and (B, D) total p70 S6 kinase using Luminex bead array respectively. (E) GECs were treated with rapamycin (100 ng/ml) an inhibitor of mTOR/p70 S6 kinase pathway, (F) or with wortmannin (100 nM), AG-490 (100 ng/mL) and CAY10512 (10 μM) inhibitor for PI3K, STAT3 and NFκB pathway respectively, for 1 hour and then were infected with H. pylori. B7-H2 expression was analyzed using flow cytometry 24 hour later. Data expressed as a percentage of positive cells. Results represents as the mean ± SD (n=8); P=N.S. (Not significant); *P < .05.
CagA+ H. pylori infection reduces induction of Th17 by human gastric epithelium
As B7-H2 has been implicated in Th17 cell differentiation, we examined whether the CagA-dependent B7-H2 downregulation impaired the capacity of GEC to induce Th17 cells differentiation from naïve CD4+ T cells. Our results showed that there is a small induction of RORγ expressing CD4+ T cells when naïve CD4+ T cells were co-cultured with N87 cells infected with H. pylori WT, but this induction was significantly increased when cells were infected with a cagA− strain (Fig. 5A). The presence of Th17 was further confirmed by measuring IL-17A in co-culture supernatants by Luminex array (Fig. 5B). Analysis of mRNA levels in parallel cultures confirmed these findings by showing increased RORγ and IL-17A mRNA levels in CD4+ T cells (Fig. 5C and D). These data demonstrate the critical role of CagA in maintaining low level Th17 responses, and this may contribute to H. pylori immune evasion.
Figure 5.

H. pylori mediated Th17 development from activated naïve T cells in co-culture with N87. N87 cells were treated with either media, with H. pylori WT, or with H. pylori cagA− strain (H. pylori:GEC=10:1) for 8 hour. After treatment, the GECs were washed and co-cultured with preactivated human CD4+ T cells (3:1 T cell: GEC) for 2 days and were analyzed for (A) RORγ expression in T cells by flow cytometry, (B) IL-17A production in supernatant by Luminex array, (C) RORγ mRNA levels, and (D) IL-17A mRNA levels in T cells by Real Time PCR. mRNA was normalized to 18S and compared to untreated cells. N=6, *P < .05.
Downregulation of B7-H2 on GEC during murine H. pylori infection depends on CagA and correlates with the decrease of Th17 responses and increase in H. pylori colonization
To understand the relevance of our observations in vitro to the H. pylori associated immunopathophysiology, we used a mouse model of H. pylori infection. Mice infected with H. pylori PMSS1, which contains a functional T4SS, had downregulated B7-H2 expression by gastric epithelial (EpCAM+) cells. Interestingly, infection of mice with H. pylori SS1 strain in which the T4SS is defective and cannot deliver CagA in GEC (45) resulted in upregulation of B7-H2 (Fig. 6A–B). No significant difference was observed in gastric mucosal inflammation in mice infected with either strain at week four. However, the H. pylori load was drastically different between the mice infected with SS1 and PMSS1 strains. Mice infected with PMSS1 strain had >100-fold higher bacterial burden when compared to those infected with SS1 strain (Fig. 6C). Further, analysis of serum cytokine profile demonstrated that in contrast to the mice infected with SS1 strain, which had significant serum levels of IL-17 and IL-21 (P<.05), mice infected with PMSS1 strain failed to upregulate pro-inflammatory Th17 cytokines (Fig. 7). Serum levels of IL-6, IL-22 and IL-23 were also increased significantly in H. pylori SS1 infected mice (Fig. 7). Taken together, our in vivo data correlate with our in vitro findings and suggest that CagA-mediated downregulation of the B7-H2 might be involved in the prevention of the Th17-mediated clearance of the H. pylori during onset of the infection.
Figure 6.

H. pylori mediated downregulation of B7-H2 on GEC in vivo involves CagA function and inversely correlate with the bacterial clearance. (A) C57BL/6 mice were challenged with H. pylori strain PMSS1, which express functional T4SS and can deliver CagA or with H. pylori SS1, which does not. Gastric mononuclear cells were isolated four weeks after H. pylori challenge using enzymatic digestion and expression of B7-H2 and epithelial cell marker EpCAM was analyzed by flow cytometry. (B) Level of the B7-H2 expressing epithelial cells (EpCam+) in the gastric mucosa from the cells was measured by flow cytometry (C) Infection rate was determined by quantification of H. pylori genome copy per half of stomach based on the analysis of H. pylori 16S gene amplification by real time PCR. Each data point represents a single mouse tested in quadruplicate. Average bar of infection rate were calculated from five mice per group and demonstrated as a mean ± SD.
Figure 7.
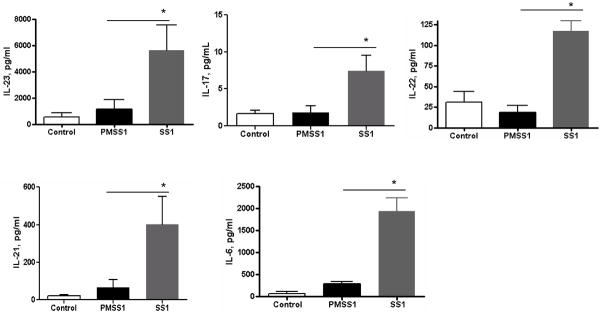
In vivo infection with H. pylori expressing functional T4SS and can deliver CagA fails to upregulate Th17 type responses. C57BL/6 mice were challenged with H. pylori strain PM-SS1, or with H. pylori SS1. Blood were collected four weeks after H. pylori challenge and cytokine profile was analyzed using luminex bead array. Data represents as mean ± SD (n=12); *P < .05.
Discussion
During H. pylori infection the host mounts an immune response, but this response is insufficient to clear the infection leading to the establishment of a persistent infection and development of chronic inflammation. Infiltration of CD4+ T cells into the gastric mucosa is among the major factors contributing to the ongoing inflammation. At the same time, these cells are required for the immunization-induced protective responses (46–48). Recent data suggested that Th17 type responses are required for the clearance of the bacteria (49). The exact mechanism(s) implicated in the H. pylori-mediated escape of host immunity to prevent clearance of H. pylori remains far from understood. Here we demonstrated that H. pylori infection of GEC leads to a decrease of B7-H2, which is a positive co-stimulatory ligand, and that this process might be important in H. pylori-mediated escape of Th17-mediated bacterial clearance.
Co-stimulatory interactions between B7 family ligands and their receptors play important roles in the growth, development and differentiation of T cells. Recent data demonstrated that interaction between B7-H2 on APCs with its putative receptor ICOS on T and B cell regulate adaptive immune responses (30, 50). Stimulation of ICOS was demonstrated to be critical for the development of human IL-17-producing CD4+ T cells (31). Further, Bauquet et. al., (29) demonstrated that ICOS, was critical for maintaining effector-memory Th17 cells. B7-H2−/− knock out (KO) mice also were noted to have lower Th17 responses to chlamydial infection than WT mice (51). The blockade of B7-H2/ICOS signaling inhibited Th1 and Th17 cells responses in chronic inflammatory conditions such as rheumatoid arthritis (30). Together these findings implicate B7-H2/ICOS signaling as an important mediator in the activation of Th17 cells in inflammation. Since expression of ICOS ligand, B7-H2, was previously reported on intestinal epithelial cells (52) and due to its importance in activation of Th17 responses, we measured the expression of B7-H2 in human biopsy samples and found that epithelial cells in gastric biopsies isolated from H. pylori infected patients had decreased levels of B7-H2 expression compared to uninfected biopsy samples. B7-H2 expression on colonic and airway epithelium was previously noted (52). However, little is known about the role of B7-H2 costimulation in the responses associated with bacterial immunopathogenesis and clearance. In the current study, we demonstrated for the first time that H. pylori significantly downregulated B7-H2 expression in gastric mucosa, particularly on GEC.
Since CagA has been shown to play an important role in H. pylori-mediated pathogenesis and immune evasion mechanisms, (53) we sought to investigate its role in the observed B7-H2 downregulation. In this study using a CagA isogenic mutant and their corresponding parental strains, we showed that CagA plays a crucial role in downregulating B7-H2 expression on GEC. Though compared to the untreated cells H. pylori cagA− strains always showed some downregulation of B7-H2, suggesting that other components of H. pylori might also have an influence in downregulating B7-H2 expression, but it is less effective than the wild type CagA+ strains. Our in vivo data using C57BL/6 mice also showed that H. pylori mediated transfer of CagA via a T4SS significantly downregulates B7-H2 expression in the GEC in the murine gastric mucosa. These in vitro and in vivo data reveal a novel mechanism whereby H. pylori uses one of its important virulence factors, CagA, to create a favorable environment for its persistence via suppression of the positive costimulators required for an efficient effector T cell response.
As cytokines play an important role in regulating immune function and IFNγ has been detected in H. pylori-infected gastric tissues in both humans and mice, (54) we also investigated whether IFNγ has any role in B7-H2 expression. Our results showed that IFNγ synergizes with the effect of H. pylori cagA+ strains in downregulating B7-H2 expression by GEC. Several studies showed induction of B7–2, B7-H1, and B7-DC in different classical APCs by IFNγ (55, 56). Stanciu et. al., (57) showed a synergistic effect of Respiratory Syncytial Virus (RSV) and IFNγ in the upregulation of B7-H1 and B7-DC in respiratory epithelial cells. In addition, their study showed that treatment of RSV infected cells with IFNγ causes downregulation of B7-H2 and B7-H3 expression. Previous findings from our group showed IFNγ and H. pylori synergize in B7-H1 upregulation (18). Thus, the synergistic effect of IFNγ and H. pylori in B7-H2 downregulation could result from IFNγ mediated increase in expression by GEC receptors that are used by H. pylori (32).
CagA interacts with several intracellular components of signal transduction and activates NFκB, MAPK, STAT3 and PI3K/Akt pathways (41–44). Previous reports have highlighted the fact that H. pylori activates STAT3 to modulate host immune responses (43). Though our results showed activation of these pathways by CagA expressing H. pylori strains, our data implies that CagA-mediated activation of NFκB, STAT3 and PI3K pathway is not required for the H. pylori-mediated downregulation of B7-H2 on GEC. Furthermore, our data for the first time demonstrate that CagA contributes to the H. pylori mediated activation of the mTOR/p70 S6 kinase pathway. Serine/threonine protein kinase mTOR acts in a signaling pathway downstream from PI3K/Akt and regulates the activation of the p70 S6 kinase, which is required for translational regulation of ribosomal proteins (58). Using specific cell signaling inhibitors we showed that H. pylori use CagA to manipulate B7-H2 expression by activating p70 S6 kinase pathway to prevent GEC from providing positive costimulation needed for protective Th17 cells.
Previously our group showed that H. pylori uses its CagA and VacA proteins to induce TGF-β production from GEC which causes inhibition of CD4+ effector T cell proliferation and induction of Treg cells (17). Herein using in vitro GEC-T cell co-cultures we showed that H. pylori uses CagA cytotoxin to downregulate Th17 cell type responses. A significant downregulation of Th17 cell transcription factor RORγτ and IL-17A was observed when GEC were exposed to the H. pylori strains expressing CagA, but not in presence of the cagA− mutant. Our in vivo data with the H. pylori mouse model support this in vitro observation. We have further shown that H. pylori CagA-mediated B7-H2 downregulation correlates with a decrease in Th17 type responses detected in murine serum and an increase in H. pylori colonization of the gastric mucosa. Though our data showed increased H. pylori bacterial load and decreased Th17 type of response in PMSS1 infected mice compared with those infected with the SS1 strain, we did not observe severe inflammatory changes in any of the H. pylori infected mice. This might be because H. pylori infection in mouse model results mostly in lymphocytic gastritis which does not progress to severe inflammation. An optimum induction of chronic gastritis can be achieved using H. felis as a model (59). Th17 cells have been suggested to have dual roles in both infection control on one hand and preneoplastic changes on the other hand. Several studies showed that protective immunity against H. pylori infection requires strong Th17 response (60). Our study showed reduced Th17 cell cytokines and increased bacterial load in the PMSS1 infected mice correlates with results from another recent study (49). Horvath et. al., showed that mice lacking IL-23 when infected with H. pylori showed reduced IL-17 production and increased bacterial load in their stomachs. Another study suggested that ICOS-induced signaling is essential for IL-21 mediated regulation of IL-23R expression in differentiated Th17 cells and for IL-23 driven expansion of Th17 cell (29). Our study also supports the importance of B7-H2/ICOS signaling in Th17 cell development since the downregulation of B7-H2 expression in the gastric mucosa of PMSS1 H. pylori-infected mice correlates with decreased Th17 type responses.
Our data suggest a novel CagA-dependent mechanism, which involves downregulation of B7-H2 on GEC, a primary target for H. pylori infection, used by the bacteria to avoid a Th17 cell-mediated clearance. Thus, our in vitro and in vivo studies suggest that H. pylori use the T4SS to downregulate Th17 cell responses, which are critical for clearing the pathogen. Therefore, H. pylori CagA delivery system may be an important target in vaccine development for achieving acceptable levels of immune protection and to design a therapeutic strategy to treat patients infected with this prevalent and deadly human pathogen.
Acknowledgments
Taslima T. Lina is a recipient of Sealy Centre for Vaccine Development Pre-doctoral fellowship, UTMB. We thank our technical assistant Ms. Yu Lin for her excellent assistance in performing the animal work.
This work was supported by National Institutes of Health grants AI68712, DK090090-01 and CA127022; American Gastroenterological Association Research Scholar Award, NIH 1U54RR02614; The University of Texas Medical Branch Clinical and Translational Sciences Award; UTMB Sealy Centre for Vaccine Development Pre-doctoral Fellowship;. The American Cancer Society RSG-10-159-01-LIB, NIH 8UL1TR000041, The University of New Mexico clinical and Translational Science Center.
Abbreviations used in this paper
- APC
antigen presenting cells
- CagA
cytotoxin associated gene A
- CFU
colony forming unit
- GEC
Gastric epithelial cells
- H. pylori
Helicobacter pylori
- IL
interleukin
- mRNA
messenger RNA
- Th17
T helper 17 cells
- Treg
T regulatory cells
References
- 1.Marshall BJ. Helicobacter pylori. The American journal of gastroenterology. 1994;89:S116–128. [PubMed] [Google Scholar]
- 2.Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, Sibley RK. Helicobacter pylori infection and the risk of gastric carcinoma. The New England journal of medicine. 1991;325:1127–1131. doi: 10.1056/NEJM199110173251603. [DOI] [PubMed] [Google Scholar]
- 3.Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology. 1998;114:1169–1179. doi: 10.1016/s0016-5085(98)70422-6. [DOI] [PubMed] [Google Scholar]
- 4.Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N, Schlemper RJ. Helicobacter pylori infection and the development of gastric cancer. The New England journal of medicine. 2001;345:784–789. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- 5.Parsonnet J, Hansen S, Rodriguez L, Gelb AB, Warnke RA, Jellum E, Orentreich N, Vogelman JH, Friedman GD. Helicobacter pylori infection and gastric lymphoma. The New England journal of medicine. 1994;330:1267–1271. doi: 10.1056/NEJM199405053301803. [DOI] [PubMed] [Google Scholar]
- 6.Rauws EA, Tytgat GN. Cure of duodenal ulcer associated with eradication of Helicobacter pylori. Lancet. 1990;335:1233–1235. doi: 10.1016/0140-6736(90)91301-p. [DOI] [PubMed] [Google Scholar]
- 7.Wu XC, Andrews P, Chen VW, Groves FD. Incidence of extranodal non-Hodgkin lymphomas among whites, blacks, and Asians/Pacific Islanders in the United States: anatomic site and histology differences. Cancer epidemiology. 2009;33:337–346. doi: 10.1016/j.canep.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Coghlan JG, Gilligan D, Humphries H, McKenna D, Dooley C, Sweeney E, Keane C, O’Morain C. Campylobacter pylori and recurrence of duodenal ulcers--a 12-month follow-up study. Lancet. 1987;2:1109–1111. doi: 10.1016/s0140-6736(87)91545-5. [DOI] [PubMed] [Google Scholar]
- 9.Blaser MJ, Crabtree JE. CagA and the outcome of Helicobacter pylori infection. American journal of clinical pathology. 1996;106:565–567. doi: 10.1093/ajcp/106.5.565. [DOI] [PubMed] [Google Scholar]
- 10.Asahi M, Azuma T, Ito S, Ito Y, Suto H, Nagai Y, Tsubokawa M, Tohyama Y, Maeda S, Omata M, Suzuki T, Sasakawa C. Helicobacter pylori CagA protein can be tyrosine phosphorylated in gastric epithelial cells. The Journal of experimental medicine. 2000;191:593–602. doi: 10.1084/jem.191.4.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bourzac KM, Guillemin K. Helicobacter pylori-host cell interactions mediated by type IV secretion. Cellular microbiology. 2005;7:911–919. doi: 10.1111/j.1462-5822.2005.00541.x. [DOI] [PubMed] [Google Scholar]
- 12.Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nature reviews Cancer. 2004;4:688–694. doi: 10.1038/nrc1433. [DOI] [PubMed] [Google Scholar]
- 13.Beswick EJ, Bland DA, Suarez G, Barrera CA, Fan X, Reyes VE. Helicobacter pylori binds to CD74 on gastric epithelial cells and stimulates interleukin-8 production. Infection and immunity. 2005;73:2736–2743. doi: 10.1128/IAI.73.5.2736-2743.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muller A, Oertli M, Arnold IC. H. pylori exploits and manipulates innate and adaptive immune cell signaling pathways to establish persistent infection. Cell communication and signaling: CCS. 2011;9:25. doi: 10.1186/1478-811X-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris PR, Wright SW, Serrano C, Riera F, Duarte I, Torres J, Pena A, Rollan A, Viviani P, Guiraldes E, Schmitz JM, Lorenz RG, Novak L, Smythies LE, Smith PD. Helicobacter pylori gastritis in children is associated with a regulatory T-cell response. Gastroenterology. 2008;134:491–499. doi: 10.1053/j.gastro.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 16.Mitchell P, Germain C, Fiori PL, Khamri W, Foster GR, Ghosh S, Lechler RI, Bamford KB, Lombardi G. Chronic exposure to Helicobacter pylori impairs dendritic cell function and inhibits Th1 development. Infection and immunity. 2007;75:810–819. doi: 10.1128/IAI.00228-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beswick EJ, I, Pinchuk V, Earley RB, Schmitt DA, Reyes VE. Role of gastric epithelial cell-derived transforming growth factor beta in reduced CD4+ T cell proliferation and development of regulatory T cells during Helicobacter pylori infection. Infection and immunity. 2011;79:2737–2745. doi: 10.1128/IAI.01146-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Das S, Suarez G, Beswick EJ, Sierra JC, Graham DY, Reyes VE. Expression of B7-H1 on gastric epithelial cells: its potential role in regulating T cells during Helicobacter pylori infection. J Immunol. 2006;176:3000–3009. doi: 10.4049/jimmunol.176.5.3000. [DOI] [PubMed] [Google Scholar]
- 19.Kabir S. The role of interleukin-17 in the Helicobacter pylori induced infection and immunity. Helicobacter. 2011;16:1–8. doi: 10.1111/j.1523-5378.2010.00812.x. [DOI] [PubMed] [Google Scholar]
- 20.Luzza F, Parrello T, Monteleone G, Sebkova L, Romano M, Zarrilli R, Imeneo M, Pallone F. Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J Immunol. 2000;165:5332–5337. doi: 10.4049/jimmunol.165.9.5332. [DOI] [PubMed] [Google Scholar]
- 21.Resende C, Thiel A, Machado JC, Ristimaki A. Gastric cancer: basic aspects. Helicobacter. 2011;16(Suppl 1):38–44. doi: 10.1111/j.1523-5378.2011.00879.x. [DOI] [PubMed] [Google Scholar]
- 22.DeLyria ES, Redline RW, Blanchard TG. Vaccination of mice against H. pylori induces a strong Th-17 response and immunity that is neutrophil dependent. Gastroenterology. 2009;136:247–256. doi: 10.1053/j.gastro.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kao JY, Zhang M, Miller MJ, Mills JC, Wang B, Liu M, Eaton KA, Zou W, Berndt BE, Cole TS, Takeuchi T, Owyang SY, Luther J. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology. 2010;138:1046–1054. doi: 10.1053/j.gastro.2009.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishii N, Chiba M, Iizuka M, Watanabe H, Ishioka T, Masamune O. Expression of MHC class II antigens (HLA-DR, -DP, and -DQ) on human gastric epithelium. Gastroenterologia Japonica. 1992;27:23–28. doi: 10.1007/BF02775060. [DOI] [PubMed] [Google Scholar]
- 25.Ye G, Barrera C, Fan X, Gourley WK, Crowe SE, Ernst PB, Reyes VE. Expression of B7-1 and B7-2 costimulatory molecules by human gastric epithelial cells: potential role in CD4+ T cell activation during Helicobacter pylori infection. The Journal of clinical investigation. 1997;99:1628–1636. doi: 10.1172/JCI119325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annual review of immunology. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 27.Beswick EJ, I, Pinchuk V, Das S, Powell DW, Reyes VE. Expression of the programmed death ligand 1, B7-H1, on gastric epithelial cells after Helicobacter pylori exposure promotes development of CD4+ CD25+ FoxP3+ regulatory T cells. Infection and immunity. 2007;75:4334–4341. doi: 10.1128/IAI.00553-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aicher A, Hayden-Ledbetter M, Brady WA, Pezzutto A, Richter G, Magaletti D, Buckwalter S, Ledbetter JA, Clark EA. Characterization of human inducible costimulator ligand expression and function. J Immunol. 2000;164:4689–4696. doi: 10.4049/jimmunol.164.9.4689. [DOI] [PubMed] [Google Scholar]
- 29.Bauquet AT, Jin H, Paterson AM, Mitsdoerffer M, Ho IC, Sharpe AH, Kuchroo VK. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nature immunology. 2009;10:167–175. doi: 10.1038/ni.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frey O, Meisel J, Hutloff A, Bonhagen K, Bruns L, Kroczek RA, Morawietz L, Kamradt T. Inducible costimulator (ICOS) blockade inhibits accumulation of polyfunctional T helper 1/T helper 17 cells and mitigates autoimmune arthritis. Annals of the rheumatic diseases. 2010;69:1495–1501. doi: 10.1136/ard.2009.119164. [DOI] [PubMed] [Google Scholar]
- 31.Paulos CM, Carpenito C, Plesa G, Suhoski MM, Varela-Rohena A, Golovina TN, Carroll RG, Riley JL, June CH. The inducible costimulator (ICOS) is critical for the development of human T(H)17 cells. Science translational medicine. 2010;2:55ra78. doi: 10.1126/scitranslmed.3000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan X, Crowe SE, Behar S, Gunasena H, Ye G, Haeberle H, Van Houten N, Gourley WK, Ernst PB, Reyes VE. The effect of class II major histocompatibility complex expression on adherence of Helicobacter pylori and induction of apoptosis in gastric epithelial cells: a mechanism for T helper cell type 1-mediated damage. The Journal of experimental medicine. 1998;187:1659–1669. doi: 10.1084/jem.187.10.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crowe SE, Alvarez L, Dytoc M, Hunt RH, Muller M, Sherman P, Patel J, Jin Y, Ernst PB. Expression of interleukin 8 and CD54 by human gastric epithelium after Helicobacter pylori infection in vitro. Gastroenterology. 1995;108:65–74. doi: 10.1016/0016-5085(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 34.Lu H, Wu JY, Beswick EJ, Ohno T, Odenbreit S, Haas R, Reyes VE, Kita M, Graham DY, Yamaoka Y. Functional and intracellular signaling differences associated with the Helicobacter pylori AlpAB adhesin from Western and East Asian strains. The Journal of biological chemistry. 2007;282:6242–6254. doi: 10.1074/jbc.M611178200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arnold IC, Lee JY, Amieva MR, Roers A, Flavell RA, Sparwasser T, Muller A. Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology. 2011;140:199–209. doi: 10.1053/j.gastro.2010.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heuermann D, Haas R. A stable shuttle vector system for efficient genetic complementation of Helicobacter pylori strains by transformation and conjugation. Molecular & general genetics: MGG. 1998;257:519–528. doi: 10.1007/s004380050677. [DOI] [PubMed] [Google Scholar]
- 37.Saada JI, I, Pinchuk V, Barrera CA, Adegboyega PA, Suarez G, Mifflin RC, Di Mari JF, Reyes VE, Powell DW. Subepithelial myofibroblasts are novel nonprofessional APCs in the human colonic mucosa. J Immunol. 2006;177:5968–5979. doi: 10.4049/jimmunol.177.9.5968. [DOI] [PubMed] [Google Scholar]
- 38.Roussel Y, Harris A, Lee MH, Wilks M. Novel methods of quantitative real-time PCR data analysis in a murine Helicobacter pylori vaccine model. Vaccine. 2007;25:2919–2929. doi: 10.1016/j.vaccine.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 39.Backert S, Selbach M. Role of type IV secretion in Helicobacter pylori pathogenesis. Cellular microbiology. 2008;10:1573–1581. doi: 10.1111/j.1462-5822.2008.01156.x. [DOI] [PubMed] [Google Scholar]
- 40.Shimada M, Ando T, Peek RM, Watanabe O, Ishiguro K, Maeda O, Ishikawa D, Hasegawa M, Ina K, Ohmiya N, Niwa Y, Goto H. Helicobacter pylori infection upregulates interleukin-18 production from gastric epithelial cells. European journal of gastroenterology & hepatology. 2008;20:1144–1150. doi: 10.1097/MEG.0b013e32830edb15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Churin Y, Al-Ghoul L, Kepp O, Meyer TF, Birchmeier W, Naumann M. Helicobacter pylori CagA protein targets the c-Met receptor and enhances the motogenic response. The Journal of cell biology. 2003;161:249–255. doi: 10.1083/jcb.200208039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Higashi H, Nakaya A, Tsutsumi R, Yokoyama K, Fujii Y, Ishikawa S, Higuchi M, Takahashi A, Kurashima Y, Teishikata Y, Tanaka S, Azuma T, Hatakeyama M. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. The Journal of biological chemistry. 2004;279:17205–17216. doi: 10.1074/jbc.M309964200. [DOI] [PubMed] [Google Scholar]
- 43.Lee KS, Kalantzis A, Jackson CB, O’Connor L, Murata-Kamiya N, Hatakeyama M, Judd LM, Giraud AS, Menheniott TR. Helicobacter pylori CagA triggers expression of the bactericidal lectin REG3γ via gastric STAT3 activation. PloS one. 2012;7:e30786. doi: 10.1371/journal.pone.0030786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li SP, Chen XJ, Sun AH, Zhao JF, Yan J. CagA(+) Helicobater pylori induces Akt1 phosphorylation and inhibits transcription of p21(WAF1/CIP1) and p27(KIP1) via PI3K/Akt1 pathway. Biomedical and environmental sciences: BES. 2010;23:273–278. doi: 10.1016/S0895-3988(10)60063-3. [DOI] [PubMed] [Google Scholar]
- 45.Crabtree JE, Ferrero RL, Kusters JG. The mouse colonizing Helicobacter pylori strain SS1 may lack a functional cag pathogenicity island. Helicobacter. 2002;7:139–140. doi: 10.1046/j.1083-4389.2002.00071.x. author reply 140–131. [DOI] [PubMed] [Google Scholar]
- 46.Smythies LE, Waites KB, Lindsey JR, Harris PR, Ghiara P, Smith PD. Helicobacter pylori-induced mucosal inflammation is Th1 mediated and exacerbated in IL-4, but not IFN-gamma, gene-deficient mice. J Immunol. 2000;165:1022–1029. doi: 10.4049/jimmunol.165.2.1022. [DOI] [PubMed] [Google Scholar]
- 47.Eaton KA, Mefford M, Thevenot T. The role of T cell subsets and cytokines in the pathogenesis of Helicobacter pylori gastritis in mice. J Immunol. 2001;166:7456–7461. doi: 10.4049/jimmunol.166.12.7456. [DOI] [PubMed] [Google Scholar]
- 48.Mohammadi M, Nedrud J, Redline R, Lycke N, Czinn SJ. Murine CD4 T-cell response to Helicobacter infection: TH1 cells enhance gastritis and TH2 cells reduce bacterial load. Gastroenterology. 1997;113:1848–1857. doi: 10.1016/s0016-5085(97)70004-0. [DOI] [PubMed] [Google Scholar]
- 49.Horvath DJ, Jr, Washington MK, Cope VA, Algood HM. IL-23 contributes to control of chronic Helicobacter pylori infection and the development of T helper responses in a mouse model. Frontiers in immunology. 2012;3:56. doi: 10.3389/fimmu.2012.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burmeister Y, Lischke T, Dahler AC, Mages HW, Lam KP, Coyle AJ, Kroczek RA, Hutloff A. ICOS controls the pool size of effector-memory and regulatory T cells. J Immunol. 2008;180:774–782. doi: 10.4049/jimmunol.180.2.774. [DOI] [PubMed] [Google Scholar]
- 51.Kadkhoda K, Wang S, Joyee AG, Fan Y, Yang J, Yang X. Th1 cytokine responses fail to effectively control Chlamydia lung infection in ICOS ligand knockout mice. J Immunol. 2010;184:3780–3788. doi: 10.4049/jimmunol.0901384. [DOI] [PubMed] [Google Scholar]
- 52.Nakazawa A, Dotan I, Brimnes J, Allez M, Shao L, Tsushima F, Azuma M, Mayer L. The expression and function of costimulatory molecules B7-H and B7-H1 on colonic epithelial cells. Gastroenterology. 2004;126:1347–1357. doi: 10.1053/j.gastro.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 53.Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clinical microbiology reviews. 2006;19:449–490. doi: 10.1128/CMR.00054-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Karttunen R, Karttunen T, Ekre HP, MacDonald TT. Interferon gamma and interleukin 4 secreting cells in the gastric antrum in Helicobacter pylori positive and negative gastritis. Gut. 1995;36:341–345. doi: 10.1136/gut.36.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim J, Myers AC, Chen L, Pardoll DM, Truong-Tran QA, Lane AP, McDyer JF, Fortuno L, Schleimer RP. Constitutive and inducible expression of B7 family of ligands by human airway epithelial cells. American journal of respiratory cell and molecular biology. 2005;33:280–289. doi: 10.1165/rcmb.2004-0129OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morgado P, Ong YC, Boothroyd JC, Lodoen MB. Toxoplasma gondii induces B7-2 expression through activation of JNK signal transduction. Infection and immunity. 2011;79:4401–4412. doi: 10.1128/IAI.05562-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stanciu LA, Bellettato CM, Laza-Stanca V, Coyle AJ, Papi A, Johnston SL. Expression of programmed death-1 ligand (PD-L) 1, PD-L2, B7-H3, and inducible costimulator ligand on human respiratory tract epithelial cells and regulation by respiratory syncytial virus and type 1 and 2 cytokines. The Journal of infectious diseases. 2006;193:404–412. doi: 10.1086/499275. [DOI] [PubMed] [Google Scholar]
- 58.Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Molecular and cellular biology. 2004;24:200–216. doi: 10.1128/MCB.24.1.200-216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O’Rourke JL, Lee A. Animal models of Helicobacter pylori infection and disease. Microbes and infection / Institut Pasteur. 2003;5:741–748. doi: 10.1016/s1286-4579(03)00123-0. [DOI] [PubMed] [Google Scholar]
- 60.Czinn SJ, Blanchard T. Vaccinating against Helicobacter pylori infection. Nature reviews Gastroenterology & hepatology. 2011;8:133–140. doi: 10.1038/nrgastro.2011.1. [DOI] [PubMed] [Google Scholar]