

Abstract
In the pathogenesis of diabetic retinopathy, H-Ras (a small molecular weight G-protein) and matrix metalloproteinase-9 (MMP9) act as pro-apoptotic, accelerating the apoptosis of retinal capillary cells, a phenomenon that predicts its development and the activation of MMP9 is under the control of H-Ras. The goal of this study is to elucidate the cellular mechanism by which H-Ras activates MMP9 culminating in the development of diabetic retinopathy. Using isolated retinal endothelial cells, the effect of regulation of H-Ras downstream signaling cascade, Raf-1, MEK, and ERK, was investigated on glucose-induced activation of MMP9. In vitro results were confirmed in the retina obtained from diabetic mice manipulated for MMP9 gene, and also in the retinal microvasculature obtained from human donors with diabetic retinopathy. Regulation of Raf-1/MEK/ERK by their specific siRNAs and pharmacologic inhibitors prevented glucose-induced activation of MMP9 in retinal endothelial cells. In MMP9-KO mice, diabetes had no effect on retinal MMP9 activation, and H-Ras/Raf-1/MEK signaling cascade remained normal. Similarly, donors with diabetic retinopathy had increased MMP9 activity in their retinal microvessels, the site of histopathology associated with diabetic retinopathy, and this was accompanied by activated H-Ras signaling pathway (Raf-1/ERK). Collectively, these results suggest that Ras/Raf-1/MEK/ERK cascade has an important role in the activation of retinal MMP9 resulting in the apoptosis of its capillary cells. Understanding the upstream mechanism responsible for the activation of MMP9 should help identify novel molecular targets for future pharmacological interventions to inhibit the development/progression of diabetic retinopathy.
Retinopathy is one of the most common microvascular complications of diabetes. In the development of diabetic retinopathy the microvasculature of the retina is damaged, and its permeability is increased. Considerable experimental evidence generated from isolated retinal cell and animal models indicates that in the development of diabetic retinopathy, retinal cells (capillary and non-capillary), undergo accelerated apoptosis before the histopathology appear, and apoptosis of retinal capillary cells culminates in the formation of degenerative capillaries and pericyte ghosts in retinal vasculature (Mizutani et al., 1996; Kern et al., 2000; Kowluru et al., 2001).
Diabetic retinopathy is considered a multi-factorial disease, and the molecular mechanism involved in its development is complex which requires proper cellular signal coordination and interactions of various growth factors, cytokines, and enzymes produced by the retinal cells (Frank, 2004; Kowluru and Odenbach, 2004; Huang and Sheibani, 2008; Geraldes and King, 2010). Over the last several years multiple mechanisms, including oxidative stress, activation of protein kinase C and mitogen-activated protein kinases, advanced glycation end products formation, have been implicated in the accelerated apoptosis of retinal capillaries cells in diabetes (Stitt, 2003; Kowluru et al., 2004; Khan and Chakrabarti, 2007; Huang and Sheibani, 2008), but the exact signaling cascade leading to capillary cell apoptosis remains elusive.
Diabetic environment activates several matrix metalloproteinases (MMPs) that are considered to participate in many of its complications, including retinopathy, nephropathy, and cardiomyopathy (Tyagi et al., 2005; Thrailkill et al., 2009; Kowluru, 2010; Mohammad and Kowluru, 2010). MMPs are calcium or zinc-dependent extracellular proteolytic enzymes that play pivotal roles in degrading and remodeling of extracellular matrix in various physiological and pathologic conditions, including ovarian cancer, kidney disease, and atherosclerosis (Malemud, 2006; Schäfers et al., 2010). Diabetes-induced activation of MMP9 in the retina and its capillary cells is suggested to play a role in the pathogenesis of diabetic retinopathy (Das et al., 1999; Giebel et al., 2005; Bhatt and Addepalli, 2010; Kowluru, 2010), but the mechanism responsible for its activation is not clear.
We have shown that the activation of MMP9 in retinal capillary cells in hyperglycemic conditions is downstream of H-Ras, a small molecular weight guanine nucleotide-binding protein (Kowluru, 2010). H-Ras is normally activated in response to the binding of extracellular signals and transduces signals from cell surface receptors into the nucleus via activation of the Raf-1/mitogen activated kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) (Cox and Der, 2002; Schubbert et al., 2007; Omerovic and Prior, 2009). Raf-1/MEK/ERK pathway is activated in the retina and its endothelial cells in diabetes, and this cascade acts as a pro-apoptotic stimulus in the pathogenesis of diabetic retinopathy (Kowluru et al., 2004; Kanwar and Kowluru, 2008; Kowluru and Kanwar, 2009b). Activated H-Ras, however, has potential to interact with various effector proteins and can stimulate other signaling cascades, including PI3K-Akt pathway (Serban et al., 2008), and the cellular mechanism by which it regulates MMP9 in diabetes is not fully understood. The aim of the present study is to elucidate the signaling mechanism by which activation of H-Ras in diabetes results in MMP9 activation, inducing apoptosis of retinal capillary cells, and ultimately in the development of retinopathy. We have investigated H-Ras mediated signaling cascade leading to the activation of MMP9 in the isolated retinal endothelial cells with the target-specific gene modified. In vitro results are confirmed in the retina obtained from diabetic mice manipulated for MMP9 gene, and in the retinal microvasculature prepared from donors with diabetic retinopathy.
Methods
Retinal endothelial cells
Endothelial cells, isolated from bovine retina, were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 15% inactivated fetal bovine serum, 5% replacement serum (Nu-serum; BD Bioscience, San Jose, CA), heparin (50 μg/ml), and endothelial cell growth supplement (25 μg/ml). Confluent cells from 3rd to 5th passages were incubated in 5 mM glucose (normal) or 20 mM glucose (high) for 4 days in the presence or absence of either farnesylation inhibitor (25 μM FTI-277) or Raf-1 kinase inhibitor, GW5074 (10 μM from Biomol International, Plymouth Meeting, PA) or MEK inhibitor, PD098059 (30 μM) or U-0126 (10 μM, EMD-Calbiochem, San Diego, CA) (Kowluru et al., 2004; Kowluru, 2010).
Transfection of endothelial cells
BRECs from 3rd to 4th passage were transfected with small interfering RNA (siRNA) of H-Ras, Raf-1, MEK, or MMP9 using transfection reagents and siRNA duplex from Santa Cruz Biotechnology (Santa Cruz, CA). The transfection complex was prepared by adding siRNA duplex (0.25–1 μg) and siRNA transfection reagent. The mixture was incubated for 30 min at room temperature, and the cells were washed with the transfection medium and incubated with the transfection complex for 8 h at 37°C. Incubations carried out using cells transfected with non-targeting scramble RNA served as transfection control. The medium was then replaced with the 5 or 20 mM glucose media and the cells were incubated for 4 additional days. These procedures are routinely used in our laboratory (Kowluru, 2010; Mohammad and Kowluru, 2010), and as shown in Figure 1a, transfection of BRECs with siRNA or scramble RNA did not affect their survival or proliferation. (Fig. 1b–e shows the transfection efficiencies of HRas, Raf-1, MEK, and MMP9, respectively, by their specific siRNAs.) Parallel incubations were run using non-transfected cells. Control incubations containing 20 mM mannitol, instead of 20 mM glucose, were used to rule out the effect of osmolarity. The cells were rinsed with phosphate-buffered saline (PBS) and homogenized in RIPA buffer (20 mM Tris–HCl pH 7.4 containing Nonidet P40, 150 mM NaCl, 1 mM Na3VO4, 5 mM EDTA, 0.1 mg/ml aprotinin, 1 mM phenylmethionylsulfonyl fluoride, pH 7.4). The homogenate was centrifuged at 750g for 5 min to remove the cell debris, and protein was quantified by bicinchoninic acid assay (Sigma-Aldrich, St. Louis, MO). Each experiment was repeated with three to four different cell preparations.
Fig. 1.

Effect of siRNA transfection on cellular morphology, and transfection efficiency of various siRNAs: (a) retinal endothelial cells untransfected, or transfected with H-Ras siRNA or scramble RNA were visualized under an Olympus BX50 microscope and imaged at 20× magnification. Gene expressions of H-Ras, Raf-1, MEK, and MMP9, respectively, were quantified by semi-quantitative PCR, in the retinal endothelial cells transfected with (b) H-Ras siRNA, H-si; (c) Raf-1 siRNA, R-si; (d) MEK siRNA (M-si); or (e) MMP9 siRNA, MMP-si. β-actin was used as loading standard. Untrans = untransfeted cells.
Mice
Male, wild-type (WT) C57BL/6J or MMP-9 gene-knockout (MMP9-KO, B6.FVB (Cg)-Mmp9tm1Tvu/J) were obtained from Jackson Laboratory (Bar Harbor, ME). A group of mice from each strain were made diabetic by streptozotocin injection (55 mg/kg BW) for 5 consecutive days. The mice presenting blood glucose 250 mg/dl or higher, 3 days after the last injection of streptozotocin, were considered as diabetic. Mice were sacrificed approximately 2 months after the induction of diabetes by an overdose of pentobarbital (120 mg/kg BW); one eye was used to isolate the retina and frozen immediately in liquid nitrogen for biochemical analysis, and the other eye was incubated in 10% formaldehyde for 30 min, washed with PBS, fixed in OCT, and frozen in liquid nitrogen for sectioning and immunostaining (Mohammad and Kowluru, 2010). The treatment of the animals conformed to the Association for Research in Vision and Ophthalmology Resolution on the Use of Animals in Research, and Institutional Guidelines.
Preparation of microvessels from human retina
Human post-mortem eyes were obtained from Midwest Eye. Donors with diabetes (10–12 years) were 54–75 years of age, and non-diabetic donors were of 44–72 years of age (Table I). The retina was isolated, and to prepare microvessels, a portion of the retina was incubated in distilled water for 1 h at 37°C, followed by incubation with DNase to remove neural and glial cells (Kowluru et al., 1998; Kowluru and Kanwar, 2009a). The retinal vasculature was cleaned of debris under a microscope by repetitive inspiration and ejection through Pasteur pipette.
TABLE I.
Patient Characteristics
| Case number | Age | Type of diabetes | Duration of diabetes (years) | Retinopathy status | Cause of death | Death-enucleation time (h) |
|---|---|---|---|---|---|---|
| Diabetic patients | ||||||
| 1 | 75 | T2DM | 12 | PDR | Pulmonary edema | 9 |
| 2 | 54 | T2DM | 10 | PDR | Congestive heart failure | 6 |
| 3 | 69 | T2DM | 10 | PDR | Respiratory failure | 6 |
| 4 | 70 | T2DM | Not available | Unknown | Stroke | 10 |
| Control patients | ||||||
| 5 | 44 | — | Not applicable | — | Intracranial hemorrhage | 11 |
| 6 | 70 | — | Not applicable | — | Stroke | 6 |
| 7 | 72 | — | Not applicable | — | Unknown | 7 |
| 8 | 55 | — | Not applicable | — | Subarachnoid hemorrhage | 10 |
T2DM, type II diabetes mellitus.
MMP9 activity
Gelatinolytic activity of MMP9 was quantified in the media and human retinal microvessels by zymography (Kowluru and Kanwar, 2009a; Kowluru, 2010; Mohammad and Kowluru, 2010). To obtain better resolution of active and pro-MMP9, the method was slightly modified. Protein (10–20 μg) was applied onto non-reduced 8% SDS–PAGE containing 0.1% gelatin; 10 ng pro-MMP9 (human recombinant, a gift from Dr. Rafael Fridman, Wayne State University) was used as a pro-MMP9 marker. For positive control, culture medium from the cells incubated in 5 mM glucose was treated with 1 mM p-aminophenylmercuric acetate (APMA, Amersham, Buckinghamshire, UK) for 3 h at 37°C. After electrophoresis, the gels were incubated in 2.5% Triton X-100 for 1 h, and after washing with distilled water, they were incubated overnight at 37°C in substrate buffer containing 50 mM Tris–HCl, pH 7.5, 5 mM CaCl2, 200 mM NaCl2, and 0.02% Brij 35. The gels were stained with 0.5% Coomassie Brilliant Blue R-250 (Sigma Chemicals, St. Louis, MO), followed by de-staining with 10% acetic acid and 30% methanol. The intensity of the active band of MMP9 (~80 kDa) was quantified using Un-Scan-It Gel digitizing software.
Activation of MMP9 was also confirmed by ELISA kit (Amersham) according to manufactures’ instruction. The assay, specific for MMP9, measures latent or active form of MMP9 depending on the treatment of immunocaptured MMP9 with APMA. Briefly, standards 0.25–5 ng/ml or 10–20 μg protein was incubated over night at 4°C in microtiter plates coated with anti-MMP9 antibody, and the cleavage of the chromogenic peptide was quantified spectrophotometrically at 405 nm. The values obtained from the immunocaptured MMP9 that was not treated with APMA was considered as the active form of MMP9 induced by high glucose.
Activation of Ras by Raf-1 binding
The relative abundance of GTP-bound active H-Ras was quantified in isolated retinal endothelial cells, and in retina and its microvasculature using a Raf-1 binding assay kit (Pierce, Rockford, IL) as reported by us previously (Kowluru and Kowluru, 2007). Protein (100–150 μg) was incubated with Raf-RBD-GTP, and the resin was centrifuged for 10–30 sec, washed, and the beads were suspended in the reducing sample buffer containing β-mercaptoethanol and 2× sample buffer. These proteins were then analyzed by Western blot analysis.
Quantification of gene expression
Total RNA, was isolated from endothelial cells, mouse retina, or human microvessels with TRIZOL reagent (Invitrogen, Carlsbad, CA) and converted to cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). For quantitative real-time RT-PCR, gene-specific TaqMan assays (Applied Biosystems) and 90 ng cDNA were used to assess mRNA abundance of Raf-1 and MEK 1/2; β-actin served as an internal control. Fold-change in mRNA abundance was calculated with the ddCt method as routinely used by us (Kowluru and Kanwar, 2009a; Kowluru, 2010; Mohammad and Kowluru, 2010). For semi-quantitative RT-PCR, the PCR primers for bovine cells were: MMP-9 forward 5′-CGCACGACATCTTTCAGTACCA-3′ and reverse 5′-GGAACTCACGCGCCAGTA-3′; H-Ras forward 5′-CAGAGCTCCTGGTTTGGCAG-3′ and reverse 5′-TCTATAGAGGGATCATACTC-3′; Raf-1 forward 5′-TGTCCTCCGCAAAACCCGGC-3′ and reverse 5′-TAGAGGCTGCTGCCCTCGCA-3′; MEK forward 5′-CTTCGGTTCCCTGTGCACTGAGC-3′ and reverse 5′-TTGGACCCAGGAAAGGCGCT-3′. The primers used for mouse retina included MMP9 forward 5′-GCTCCTGGCTCTCCTGGCTT-3′ and reverse 5′-GTCCCACTTGAGGCCTTTGA-3′, and for human retina MMP9 forward 5′-CACTGTCCACCCCTCAGAGC-3′ and reverse 5′-GCCACTTGTCGGCGATAAGG-3′ and β-actin forward 5′-CCT CTA TGC CAA CAC AGT GC-3′ and reverse 5′-CAT CGT ACT CCT GCT TGC TG-3′. cDNA (1 μl) was amplified with GoTaq DNA polymerase (Promega, Madison, WI) and PCR was conducted at 94°C for 45 sec, 50–55°C for 45–70 sec, and 72°C for 45 sec for 30–38 cycles. The DNA was run on a 1.2% agarose gel, with a 100 bp DNA ladder (standard) run simultaneously on each gel. Relative amplification was quantified by normalizing the gene-specific band intensity to that of β-actin. Quantitative analysis was achieved by measuring the integrated density value of PCR products in gel photographs using Un-Scan-It Gel digitizing software (Kowluru and Kanwar, 2009a; Kowluru, 2010; Mohammad and Kowluru, 2010).
Protein expression
Protein (30–50 μg) was separated on a 4–20% denaturing polyacrylamide gel and transferred to nitrocellulose membranes. The membranes were blocked with 5% milk, followed by the incubation with the antibody against the target protein (H-Ras, Raf-1, MEK, and ERK; Cell Signaling, Beverly, MA). After incubation with horseradish peroxidase-conjugated secondary antibody, the membranes were developed using ECL-Plus Western blotting detection kit (Pierce). To ensure equal loading among the lanes, membranes were re-probed with the housekeeping protein, β-actin. The band intensities were quantified by Un-Scan-It software as reported previously (Kowluru and Kanwar, 2009a; Kowluru, 2010; Mohammad and Kowluru, 2010).
Apoptosis of retinal endothelial cells
Apoptosis was determined by ELISA using a Cell Death Detection ELISAPLUS kit (Roche Diagnostics, Indianapolis, IN), which uses monoclonal antibodies directed against DNA and histones to quantify the relative amounts of mono- and oligonucleosomes generated from apoptotic cells, as routinely performed in our laboratory (Kowluru et al., 2004, 2007).
Immunohistochemical analysis
Cryosection (10 μm) prepared from mouse or human retina were fixed in ice-cold acetone for 10 min at room temperature and blocked with 10% normal goat serum. For single immunostaning the sections were rinsed with PBS, permeabilized with 0.1% Triton X-100, and incubated over night with anti-MMP9 (rabbit polyclonal, Santa Cruz Biotechnology). After rinsing the slides with PBS, they were incubated with the secondary antibody (anti-rabbit-FITC conjugated) for 1 h.
For double staining, the slides were incubated overnight with two antibodies obtained from two different species (H-Ras mouse monoclonal and Von Willebrand Factor, VWF, rabbit polyclonal from Dako Japan Ltd (Carpinteria, CA), or MMP9 rabbit polyclonal). After treating the sections with 0.1% Tween-20, they were incubated with a mixture of two secondary antibodies (anti-rabbit-FITC conjugated and anti mouse-Texas red, Vector Laboratories, Burlington, CA) for 1 h. The slides were rinsed with PBS, mounted with DAPI containing mounting media (Vector Laboratories), and imaged with Olympus BX50 fluorescent microscope (20× magnification). Control slides included staining of the samples that were incubated under identical conditions except the primary antibody (Mohammad and Kowluru, 2010).
Results
Endothelial cells
High glucose activates MMP9, and H-Ras and its signaling cascade in retinal endothelial cells
Consistent with our previous reports (Kowluru, 2010), exposure of retinal endothelial cells to high glucose activated MMP9, as evidenced by its gelatinase activity and by ELISA (Fig. 2a,b). The transcripts of MMP9 were increased by about 70% (Fig. 2c) compared to the cells incubated in normal glucose conditions. In the same cells, H-Ras was activated by 30–40% (Fig. 3a), and its downstream signaling molecules, Raf-1, MEK1/2, and ERK1/2 were activated by 30–50% (Fig. 3b). Regulation of H-Ras by its siRNA ameliorated glucose-induced activation of MMP9 (Fig. 2a–c), and prevented activation of its downstream signaling, Raf-1/MEK/ERK cascade (Fig. 3b).
Fig. 2.

High glucose activates MMP9 via H-Ras/Raf-1/MEK pathway: retinal endothelial cells transfected with H-Ras siRNA, Raf-1 siRNA, MEK siRNA, or scramble RNA (SC) were incubated in 20 mM glucose for 4 days. (a) Gelatinase activity of MMP9 was measured in the incubation medium by in situ zymography using pro-MMP9 (human recombinant) as a pro-MMP9 marker. For positive control, culture medium from the cells incubated in 5 mM glucose was incubated with APMA. The active form of MMP9 (~80 kDa) was quantified, and presented in the accompanying histogram. Cells were harvested and (b) the activity of MMP9 was quantified using an ELISA kit, and (c) gene expression of MMP9 was measured by semi-quantitative PCR. Each measurement was performed in three to four different cell preparations. The values obtained from the untransfected cells incubated in 5 mM glucose are considered as 100% (control). Five and 20 = untransfected cells incubated in 5 or 20 mM glucose for 4 days, 1 = recombinant pro-MMP9, 2 = untreated medium, 3 = APMA-treated medium, 4 = recombinant pro-MMP9, 5 = 5 mM glucose, 6 = 20 mM glucose, 7 = H-Ras siRNA, 8 = Raf-1 siRNA, 9 = MEK siRNA, 10 = scrambled RNA. *P < 0.05 compared to the values obtained from the cells incubated in 5 mM glucose and #P < 0.05 scramble RNA transfected or untransfected cells, exposed to 20 mM glucose for 4 days.
Fig. 3.

H-Ras siRNA inhibits glucose-induced activation of Raf-1/MEK/ERK pathway: retinal endothelial cells, either transfected with H-Ras siRNA or untransfected, were incubated in 5 or 20 mM glucose for 4 days. Activation of (a) H-Ras and (b) Raf-1, MEK and ERK1/2 was determined by Western blot technique using specific antibodies (Cell Signaling). Effect of high glucose on the activation of (c) Raf-1 was quantified in the cells transfected with Raf-1 siRNA, and that of (d) MEK in the cells transfected with MEK siRNA by Western blot technique. The data are mean ± SD obtained from three to four different experiments. The values obtained from cells incubated in 5 mM glucose are considered as 100% (control). Five = 5 mM glucose, 20 = 20 mM glucose, H-si = H-Ras siRNA, and SC = scramble RNA. *P < 0.05 compared with the values obtained from cells incubated in 5 mM glucose, or #P < 0.05 compared with values from cells transfected with scramble RNA and incubated in 20 mM glucose for 4 days.
H-Ras activates MMP9 via Raf-1/MEK/ERK pathways
To investigate the mechanism via which H-Ras activates MMP9, we used two independent approaches, genetic manipulations and pharmacologic inhibitors. Regulation of Raf-1, the principal effector protein of H-Ras, by Raf-1 siRNA ameliorated activation of MMP9, as confirmed by its activity and gene expression (Fig. 2a–c). Similarly, inhibition of downstream signaling steps of H-Ras/Raf-1 by MEK siRNA prevented glucose-induced activation of MMP9 compared to the cells transfected with scramble RNA (Fig. 2a–c). Transfection of cells with Raf-1 siRNA or MEK siRNA also ameliorated glucose-induced activation of Raf-1 and pMEK1/2, respectively (Fig. 3c,d).
The role of Raf-1/MEK/ERK in the activation of MMP9 was further confirmed by the use of their specific inhibitors, FTI-277, GW5074, PD098059, and U-0126, respectively. Glucose-induced MMP9 activation was prevented when high glucose exposure was supplemented with these specific inhibitors (Fig. 4).
Fig. 4.
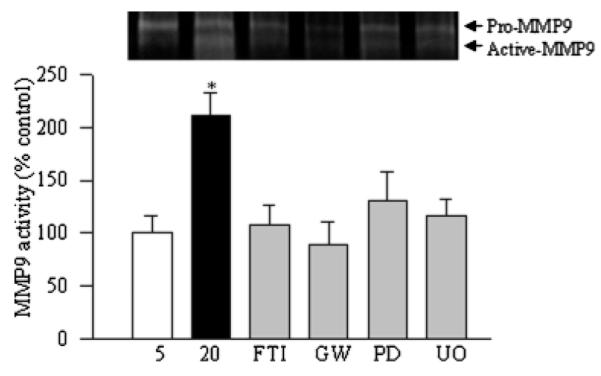
Inhibitors of Ras-Raf-1-MEK pathway attenuate high glucose-induced MMP9 in retinal endothelial cells: cells were incubated in 5 mM glucose or 20 mM glucose for 4 days in the presence or absence of specific inhibitors of H-Ras (FTI), Raf-1 (GW-5074), or MEK/ERK (PD098059 or U-0126). Gelatinase activity of MMP9 was quantified in the incubation media. Each measurement was performed in three to four different cell preparations. The values obtained from the cells incubated in 5 mM glucose are considered as 100% (control). Five = 5 mM glucose, 20 = 20 mM glucose, FTI = 20 mM glucose + FTI-277, PD = 20 mM glucose + PD098059, GW = 20 mM glucose + GW-5074, U0 = 20 mM glucose + U-0126. *P < 0.05 compared to the values obtained from the cells incubated in 5 mM glucose.
Our previous work has demonstrated that diabetes activates both, H-Ras and MMP9 in retinal microvasculature (Kanwar and Kowluru, 2008; Kowluru, 2010). In order to investigate if activated MMP9 can regulate H-Ras, the cells transfected with MMP9 siRNA were used. Inhibition of MMP9 by its siRNA failed to prevent glucose-induced activation of H-Ras (determined by the pull-down assay), and the protein expression of H-Ras remained elevated compared to the cells transfected with scramble RNA (Fig. 5a,b).
Fig. 5.
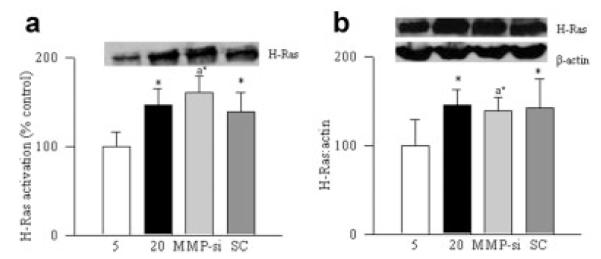
Regulation of MMP9 activation in retinal endothelial cells does not protect H-Ras activation. Cells transfected with MMP9 siRNA (MMP-si) or scramble RNA (SC) were incubated in 5 mM or 20 mM glucose for 4 days. The effect of MMP9 regulation on H-Ras activation was determined by (a) pull-down assay, and (b) by quantifying its protein expression. *P < 0.05 compared with values obtained from cells incubated in 5 mM glucose.
Effect of regulation of H-Ras or MMP9 on apoptosis of retinal endothelial cells
As expected, high glucose exposure of retinal endothelial cells increased the apoptosis of retinal endothelial cells by 70%, and regulation of H-Ras or MMP9 by their specific siRNAs, ameliorated glucose-induced increase in endothelial cell apoptosis (Fig. 6).
Fig. 6.
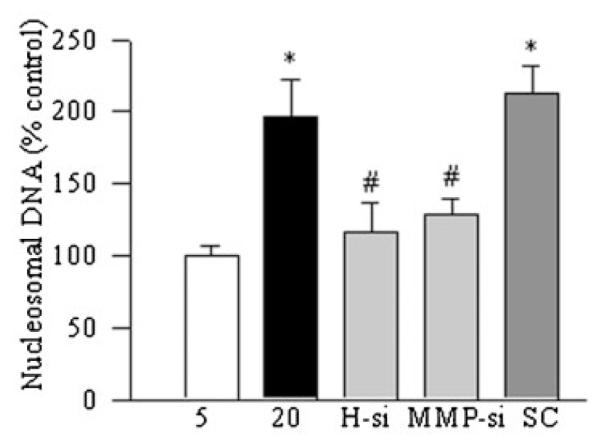
Effect of regulation H-Ras siRNA or MMP9 siRNA on glucose-induced apoptosis of retinal endothelial cells: apoptosis was measured by performing ELISA for cytoplasmic histone-associated DNA fragments using a kit from Roche Diagnostics. The graph shows the values obtained from the transfected cells incubated in 5 or 20 mM glucose for 4 days, and these values were adjusted to the total DNA in each sample. *P < 0.05 compared to the values obtained from the cells incubated in 5 mM glucose, and #P < 0.05 compared to the values obtained from cells transfected with scramble RNA and incubated in 20 mM glucose for 4 days.
Mouse retina
Diabetic mice had blood glucose values ~4 times higher compared to their age-matched non-diabetic mice. However, the values among the two diabetic groups (WT and MMP9-KO) were not different from each other (477 ± 64 and 494 ± 50 mg/dl, respectively), suggesting that the severity of hyperglycemia was similar in both MMP9-KO diabetic and WT diabetic mice.
Gene expression of MMP9 was significantly decreased in the retina of MMP9-KO mice (Fig. 7a). This was accompanied by decreased expression of its active form as measured by the ELISA (Fig. 7b). In the same mice, MMP9 immunostaining (detected by green FITC fluorescent staining) was also significantly lower (Fig. 7c), suggesting that MMP9 was downregulated in the retina of these MMP9-KO mice.
Fig. 7.

Diabetes does not activate retinal MMP9 in the mouse lacking MMP9. (a) Gene expression of MMP9 was quantified in the retina of wild-type (WT) mice and MMP9-KO (KO) mice diabetic for 2 months by semi-quantitative PCR. (b) Activity of MMP-9 was quantified by ELISA. (c) Immunostaining of MMP9 was performed in the retinal using MMP9 antibodies (green) and DAPI (blue) was used to stain the nuclei. The sections were imaged at 20× magnification using Olympus BX50 fluorescent microscope. The images presented here are representative of three or more mice in each group. The effect of MMP9 inhibition on retinal (d) H-Ras, (e) Raf-1, and (f) phosphorylated MEK was determined by Western blot using their respective antibodies, and their mRNA levels were quantified by real-time quantitative PCR using Taqman primers. The protein and mRNA levels of the targeted proteins were adjusted to the levels of β-actin in each sample. The values obtained from WT non-diabetic mice are considered as 100%. Results are expressed as mean ± SD of at least five to six mice in each group. *P < 0.05 compared with WT non-diabetic mice and #P < 0.05 compared to WT diabetic mouse. WT-N and WT-D = wild-type non-diabetic and diabetic, respectively, and KO-N and KO-D = MMP9-KO mice non-diabetic and diabetic, respectively.
Diabetes in WT mice, as expected, increased the expressions and the activity of MMP9 in the retina by 30–40% compared to the WT-normal mice. In contrast, in MMP9-KO mice diabetes had no effect on retinal MMP9 activity and expression, the values obtained from non-diabetic and diabetic MMP9-KO mice retina were similar to each other, and were significantly lower compared to those obtained from WT-diabetic mice (Fig. 7a,b). Similar results were obtained from immunofluorescence; the intensity of MMP9 staining was increased in the retinal vessels and in several areas surrounding them in WT-diabetic mice compared to age-matched WT-normal mice (Fig. 7c), however, MMP9 staining in retinal sections from MMP9-KO-diabetic mice was significantly lower compared to WT-diabetic mice.
Conversely, retina from the MMP9-KO mice was also protected from diabetes-induced activation of H-Ras, Raf-1, and MEK1/2 (Fig. 7d–f), and the values obtained from diabetic and non-diabetic MMP9-KO mice retina were not different from those obtained from WT-normal mice (P < 0.05).
Human retina
To confirm our results from isolated retinal endothelial cells and mouse retina, the activation of MMP9 and H-Ras was also investigated in the retinal microvessels (the site of histopathology characteristic of diabetic retinopathy) from non-diabetic donors and diabetic donors with retinopathy. Figure 8a represents the integrity of the retinal microvessels prepared by the osmotic-shock method from a donor with diabetic retinopathy; the microvessel preparations showed a normal complement of nuclei and were generally devoid of nonvascular contamination. As in the isolated retinal endothelial cells and retina from diabetic rodents, the active form of MMP9 and its mRNA levels were elevated significantly in the retinal microvessels prepared from the donors with diabetic retinopathy compared to the values obtained from non-diabetic donors (Fig. 8b). In the same microvessels, H-Ras, Raf-1, and ERK1/2 were activated by 60–80% (Fig. 8c).
Fig. 8.

MMP9, H-Ras, and ERK are activated in the retinal microvasculature of donors with diabetic retinopathy. Microvessels were isolated from the retina of donors with diabetic retinopathy and age-matched non-diabetic subjects by the osmotic shock method, and stained with periodic acid and hematoxylin, and (a) represents microvessels obtained from a donor with diabetes. (b) The activity of MMP-9 was quantified in the microvasculature by ELISA and by in situ zymography, and its gene expression was measured by semi-quantitative PCR. (c) Activation of H-Ras was quantified by pull-down assay, and that of Raf-1 and ERK by Western blot technique. Each measurement was performed in duplicate in the retinal microvessels obtained from four each, non-diabetic and diabetic donors. *P < 0.05 compared with the values obtained from non-diabetic donors.
The localization of H-Ras and MMP9 in retinal microvessels was also performed by immunostaining of retinal cryosections for H-Ras or MMP9; Figure 9a shows the morphology of a cryosection and the box represents the region imaged for immunostaining. In the retinal sections from donors with diabetic retinopathy compared to the sections from non-diabetic donors, immunostaining of H-Ras (red) was significantly higher in the lumen of the capillaries with positive green staining for VWF (Fig. 9b). Figure 9c shows the portion of the section representing capillaries imaged at 20× magnification. To co-localize H-Ras and MMP9 in the retinal vasculature, the sections were stained with H-Ras (red) and MMP9 (green), and Figure 9d shows significant co-localization of H-Ras and MMP9 in the retinal vessels from a donor with diabetic retinopathy.
Fig. 9.

Immunofluorescence staining of H-Ras in human retina. Retinal cryosections (10 μm) from non-diabetic donors and diabetic donors with retinopathy were immunostained for H-Ras. (a) The upper part shows the morphology of a cryosection and the box represents a region used for immunostaining. (b) H-Ras (red) and VWF (green) DAPI (blue) staining in a non-diabetic donor (left part) and a diabetic donor (right part). Portions of the section representing capillaries were imaged at 20× magnification using Olympus BX50 fluorescent microscope and (c) shows the co-localization of H-Ras and VWF, and (d) a section was stained with H-Ras (red) and MMP9 (green) to show their co-localization. These images are representative of retina samples obtained from three or more non-diabetic and diabetic donors. GCL, ganglion cell layer; ONL, outer nuclear layer.
Discussion
Our studies have shown that diabetes activates H-Ras and MMP9 in the retina and its capillary cells, and the activation is associated with the accelerated capillary cell apoptosis (Kowluru et al., 2004, 2007; Kowluru, 2010). Here we provide the signaling cascade via which activation of H-Ras activates MMP9 in diabetic milieu culminating in the loss of retinal capillary cells and the development of retinopathy. Our exciting data show that H-Ras mediated activation of MMP9 in diabetes is via activation of its effector protein Raf-1, which then activates MEK/ERK. These results are confirmed in both, isolated retinal endothelial cells in culture and retina from a rodent model of diabetic retinopathy. Genetic manipulation of MMP9 in endothelial cells prevents glucose-mediated apoptosis, but fails to prevent activation of H-Ras/Raf-1 cascade suggesting that this is upstream of MMP9. In MMP9-KO mice, diabetes fails to activate retinal MMP9, but the retina is also protected from the activation of H-Ras/Raf-1/MEK, suggesting a compensatory mechanism. Furthermore, we provide novel data from donors with diabetic retinopathy showing the activation of MMP9 and H-Ras/Raf-1/MEK cascade in the retinal microvessels (the site of histopathology), and the co-localization of H-Ras and MMP9 in the retinal capillaries.
H-Ras functions as a “molecular switch” converting signals from the cell membrane to the nucleus; the switch is turned on and off by binding to GTP, and the enzyme becomes active. While transient activation results in cell differentiation, sustained activation is associated with cell growth, and activation in selective compartments is considered to have distinct physiological outcomes (Klein et al., 2006). Active H-Ras stimulates a group of kinases involved in various signaling pathways including, Raf-1/MEK cascade, phosphatidyl inositol-3 kinase/Akt pathway, and guanine nucleotide-exchange factors for other GTPases (Karasarides et al., 2006; Serban et al., 2008). These pathways regulate a wide range of cell functions including cell proliferation, differentiation, migration and apoptosis. However, the Ras/Raf-1/MEK-ERK pathway is considered to be a major route for the transmission of signals from Ras to the transcription factor in the nucleus (Cox and Der, 2002; Schubbert et al., 2007; Omerovic and Prior, 2009). In the pathogenesis of diabetic retinopathy, activation of H-Ras in the retina and its vasculature is under the control of superoxide, which can be prevented by inhibiting superoxide accumulation, and regulation of Raf-1 kinase is associated with the apoptosis of retinal endothelial cells in high glucose conditions. The therapies that prevent the accelerated apoptosis and the development of diabetic retinopathy in rodents also inhibit activation of Ras-Raf-1 in the retina (Kowluru et al., 2004, 2007; Kowluru and Kowluru, 2007; Kanwar and Kowluru, 2008; Rayappa and Kowluru, 2008; Kowluru and Kanwar, 2009a). Here we provide exciting data showing that the inhibition of Raf-1 by Raf-1 siRNA or pharmacologic inhibitor prevents glucose-induced increase in active MMP9. These data suggest that the regulation of MMP9 by H-Ras is via H-Ras/Raf-1 pathway.
Activated Raf-1 subsequently phosphorylates MEK1/2, and MEK1/2 then phosphorylate their two known substrates, ERK1/2 (Cheng et al., 2001; Schubbert et al., 2007). MEK pathway is implicated in the regulation of vascular permeability of the retina, a functional abnormalities observed before the pathology of diabetic retinopathy (Miyamoto et al., 2007), and inhibition of p38 MAP kinae prevents the development of early stages of diabetic retinopathy (Du et al., 2010). Also, Raf-1 activation regulates MAP kinase, and glucose-induced accelerated apoptosis of retinal capillary cells (Rayappa and Kowluru, 2008). Here, we show that in high glucose, activate MEK regulates MMP9 activation, and this is attenuated by inhibition of MEK activation. In support, others have shown that MEK inhibitors block MMP9 in malignant astrocytoma cells (Chattopadhyay and Shubayev, 2009). Activation of ERK induces apoptosis of renal epithelial cells (Zhuang and Schnellmann, 2006), and in the early stages of diabetes MMP9 acts as a pro-apoptotic factor (Kowluru, 2010). The results presented here are in agreement with the reports showing that the constitutive activation of the Ras/ERK pathway modulates MMP9 (Geraldes and King, 2010), and TNF-stimulated MMP9 activation is via MAPK signaling pathway (Li et al., 2010). In addition, H-Ras mediated activation of MEK regulates the nuclear transcriptional factor, NF-κB (Lee et al., 2008), and this pivotal transcriptional factor is considered to play an important role in the development of diabetic retinopathy (Romeo et al., 2002; Kowluru et al., 2003; Zheng et al., 2004). Transfection of retinal endothelial cells with MMP9 siRNA fails to provide any benefit to glucose-induced activation of H-Ras, clearly suggesting that the activation of MMP9 is downstream of H-Ras mediated Raf-1-MEK cascade. However, it should be acknowledged that in diabetes several retinal proteins/enzymes are modified via post-translational modifications (Kanwar and Kowluru, 2009; Stitt, 2010; Zhong and Kowluru, 2010), and the role of such modifications in activating retinal MMP9 cannot be ruled out.
The activity of MMP9 in the retina of diabetic mice with MMP9 gene manipulated remains normal, and this is observed despite similar severity of hyperglycemia in WT-diabetic and MMP9-KO diabetic mice. However, to our surprise, in the same MMP9-KO diabetic mice, diabetes-induced activation of retinal H-Ras and its downstream signaling cascade-Raf-1/MEK is inhibited. This is in discordance with our in vitro results using specific siRNAs. The possibility that in a global knockout mouse model, the ablation of a gene in an in vivo system could be confounded by inherent variables, such as unknown compensatory changes in the expression of other genes to counterbalance activation/inhibition of other pathways, which may not exist in a cell culture setting, cannot be ruled out. Furthermore, MMPs can act as anti-malignant or pro- or anti-angiogenic factors depending on the stage of a particular cancer development (Deryugina and Quigley, 2010), and receptor tyrosine kinases are shown to regulate the hypoxia-inducible factors in normoxia and hypoxia via compensatory pathways (Arch, 2002; Nilsson et al., 2010).
The role of MMP9 in diabetic retinopathy was confirmed in the retinal microvessels from diabetic donors with documented retinopathy. Co-localization of H-Ras and MMP9 in the lumen of the capillaries, and similar activation of H-Ras/Raf-1/MEK-ERK cascade and MMP9 in the retinal microvessels from donors with diabetic retinopathy further supports the importance of H-Ras/Raf-1/ERK mediated regulation of MMP9 in development of diabetic retinopathy.
Thus, in summary, we have elucidated a signaling cascade responsible for H-Ras-mediated activation of MMP9 in the retina, which increases the apoptosis of capillary cells, and ultimately results in the development of diabetic retinopathy. Understanding the upstream mechanism responsible for the activation of MMP9 should help identify novel molecular targets for future pharmacological interventions to inhibit the development/progression of diabetic retinopathy.
Acknowledgments
Technical assistance of Yakov Shamailov and Doug Putt, and initial help by Dr. Gaurav Dave is sincerely appreciated. This study was supported in parts by grants from the National Institutes of Health (nos. EY014370; EY017313), Juvenile Diabetes Research Foundation (no. 1-2008-764), Thomas Foundation, and Research to Prevent Blindness.
Footnotes
The authors confirm that there are no conflicts of interest.
Literature Cited
- Arch JR. Lessons in obesity from transgenic animals. J Endocrinol Invest. 2002;25:867–875. doi: 10.1007/BF03344050. [DOI] [PubMed] [Google Scholar]
- Bhatt LK, Addepalli V. Attenuation of diabetic retinopathy by enhanced inhibition of MMP-2 and MMP-9 using aspirin and minocycline in streptozotocin-diabetic rats. Am J Transl Res. 2010;12:181–189. [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S, Shubayev VI. MMP-9 controls Schwann cell proliferation and phenotypic remodeling via IGF-1 and ErbB receptor-mediated activation of MEK/ERK pathway. Glia. 2009;57:1316–1325. doi: 10.1002/glia.20851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng TH, Shih NL, Chen SY, Loh SH, Cheng PY, Tsai CS, Liu SH, Wang DL, Chen JJ. Reactive oxygen species mediate cyclic strain-induced endothelin-1 gene expression via Ras/Raf/extracellular signal-regulated kinase pathway in endothelial cells. J Mol Cell Cardiol. 2001;42:1802–1814. doi: 10.1006/jmcc.2001.1444. [DOI] [PubMed] [Google Scholar]
- Cox AD, Der CJ. Ras family signaling: Therapeutic targeting. Cancer Biol Ther. 2002;1:599–606. doi: 10.4161/cbt.306. [DOI] [PubMed] [Google Scholar]
- Das A, McLamore A, Song W, McGuire PG. Retinal neovascularization is suppressed with a matrix metalloproteinase inhibitor. Arch Ophthalmol. 1999;117:498–503. doi: 10.1001/archopht.117.4.498. [DOI] [PubMed] [Google Scholar]
- Deryugina EI, Quigley JP. Pleiotropic roles of matrix metalloproteinases in tumor angiogenesis: Contrasting, overlapping and compensatory functions. Biochim Biophys Acta. 2010;1803:103–120. doi: 10.1016/j.bbamcr.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Tang J, Li G, Berti-Mattera L, Lee CA, Bartkowski D, Gale D, Monahan J, Niesman MR, Alton G, Kern TS. Effects of p38 MAPK inhibition on early stages of diabetic retinopathy and sensory nerve function. Invest Ophthalmol Vis Sci. 2010;51:2158–2164. doi: 10.1167/iovs.09-3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank RN. Diabetic retinopathy. N Engl J Med. 2004;350:48–58. doi: 10.1056/NEJMra021678. [DOI] [PubMed] [Google Scholar]
- Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. 2010;106:1319–1331. doi: 10.1161/CIRCRESAHA.110.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giebel SJ, Menicucci G, McGuire PG, Das A. Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood-retinal barrier. Lab Invest. 2005;85:567–607. doi: 10.1038/labinvest.3700251. [DOI] [PubMed] [Google Scholar]
- Huang Q, Sheibani N. High glucose promotes retinal endothelial cell migration through activation of Src, PI3K/Akt1/eNOS, and ERKs. Am J Physiol Cell Physiol. 2008;295:C1647–C1657. doi: 10.1152/ajpcell.00322.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanwar M, Kowluru RA. Diabetes regulates small molecular weight G-protein, H-Ras, in the microvasculature of the retina: Implication in the development of retinopathy. Microvasc Res. 2008;76:189–193. doi: 10.1016/j.mvr.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanwar M, Kowluru RA. Role of glyceraldehyde 3-phosphate dehydrogenase in the development and progression of diabetic retinopathy. Diabetes. 2009;58:227–234. doi: 10.2337/db08-1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasarides M, Dee K, Schulman D, Wolfman A, Weyman CM. Active Ras-induced effects on skeletal myoblast differentiation and apoptosis are independent of constitutive PI3-kinase activity. Cell Biol Int. 2006;30:308–318. doi: 10.1016/j.cellbi.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Kern TS, Tang J, Mizutani M, Kowluru R, Nagraj R, Lorenzi M. Response of capillary cell death to aminoguanidine predicts the development of retinopathy: Comparison of diabetes and galactosemia. Invest Ophthalmol Vis Sci. 2000;41:3972–3978. [PubMed] [Google Scholar]
- Khan ZA, Chakrabarti S. Cellular signaling and potential new treatment targets in diabetic retinopathy. Exp Diabetes Res. 2007;2007:31867. doi: 10.1155/2007/31867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Creach K, Irintcheva V, Hughes KJ, Blackwell PL, Corbett JA, Baldassare JJ. Zinc induces ERK-dependent cell death through a specific Ras isoform. Apoptosis. 2006;11:1933–11944. doi: 10.1007/s10495-006-0089-6. [DOI] [PubMed] [Google Scholar]
- Kowluru RA. Role of matrix metalloproteinase-9 in the development of diabetic retinopathy and its regulation by H-Ras. Invest Ophthalmol Vis Sci. 2010;51:4320–4326. doi: 10.1167/iovs.09-4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru RA, Kanwar M. Oxidative stress and the development of diabetic retinopathy: Contributory role of matrix metalloproteinase-2. Free Radic Biol Med. 2009a;46:1677–1685. doi: 10.1016/j.freeradbiomed.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru RA, Kanwar M. Translocation of H-Ras and its implications in the development of diabetic retinopathy. Biochem Biophys Res Commun. 2009b;387:461–466. doi: 10.1016/j.bbrc.2009.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru V, Kowluru RA. Increased oxidative stress in diabetes regulates activation of a small molecular weight G-protein, H-Ras, in the retina. Mol Vis. 2007;13:602–610. [PMC free article] [PubMed] [Google Scholar]
- Kowluru RA, Odenbach S. Role of interleukin-1beta in the pathogenesis of diabetic retinopathy. Br J Ophthalmol. 2004;88:1343–1347. doi: 10.1136/bjo.2003.038133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowluru RA, Jirousek MR, Stramm LE, Farid NA, Engerman RL, Kern TS. Abnormalities of retinal metabolism in diabetes or experimental galactosemia. V. Relationship between protein kinase C and ATPases. Diabetes. 1998;47:464–469. doi: 10.2337/diabetes.47.3.464. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Tang J, Kern TS. Abnormalities of retinal metabolism in diabetes and experimental galactosemia. VII. Effect of long-term administration of antioxidants on the development of retinopathy. Diabetes. 2001;50:1938–1942. doi: 10.2337/diabetes.50.8.1938. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Koppolu P, Chakrabarti S, Chen S. Diabetes-induced activation of nuclear transcriptional factor in the retina, and its inhibition by antioxidants. Free Radic Res. 2003;37:1169–1180. doi: 10.1080/10715760310001604189. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Kowluru A, Chakrabarti S, Khan Z. Potential contributory role of H-Ras, a small G-protein, in the development of retinopathy in diabetic rats. Diabetes. 2004;53:775–783. doi: 10.2337/diabetes.53.3.775. [DOI] [PubMed] [Google Scholar]
- Kowluru RA, Kowluru A, Kanwar M. Small molecular weight G-protein, H-Ras, and retinal endothelial cell apoptosis in diabetes. Mol Cell Biochem. 2007;296:69–76. doi: 10.1007/s11010-006-9299-z. [DOI] [PubMed] [Google Scholar]
- Lee KW, K N, Heo YS, Rogozin EA, Pugliese A, Hwang MK, Bowden GT, Bode AM, Lee HJ, Dong Z. Raf and MEK protein kinases are direct molecular targets for the chemopreventive effect of quercetin, a major flavonol in red wine. Cancer Res. 2008;68:946–955. doi: 10.1158/0008-5472.CAN-07-3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Li H, Bocking AD, Challis JR. Tumor necrosis factor stimulates matrix metalloproteinase 9 secretion from cultured human chorionic trophoblast cells through TNF receptor 1 signaling to IKBKB-NFKB and MAPK1/3 pathway. Biol Reprod. 2010;83:481–487. doi: 10.1095/biolreprod.109.082578. [DOI] [PubMed] [Google Scholar]
- Malemud CJ. Matrix metalloproteinases (MMPs) in health and disease: An overview. Front Biosci. 2006;11:1696–1701. doi: 10.2741/1915. [DOI] [PubMed] [Google Scholar]
- Miyamoto N, de Kozak Y, Jeanny JC, Glotin A, Mascarelli F, Massin P, BenEzra D, Behar-Cohen F. Placental growth factor-1 and epithelial haemato-retinal barrier breakdown: Potential implication in the pathogenesis of diabetic retinopathy. Diabetologia. 2007;50:461–470. doi: 10.1007/s00125-006-0539-2. [DOI] [PubMed] [Google Scholar]
- Mizutani M, Kern TS, Lorenzi M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest. 1996;97:2883–2890. doi: 10.1172/JCI118746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad G, Kowluru RA. Matrix metalloproteinase-2 in the development of diabetic retinopathy and mitochondrial dysfunction. Lab Invest. 2010;90:1365–1372. doi: 10.1038/labinvest.2010.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson MB, Zage PE, Zeng L, Xu L, Cascone T, Wu HK, Saigal B, Zweidler-McKay PA, Heymach JV. Multiple receptor tyrosine kinases regulate HIF-1alpha and HIF-2alpha in normoxia and hypoxia in neuroblastoma: Implications for antiangiogenic mechanisms of multikinase inhibitors. Oncogene. 2010;29:2938–2949. doi: 10.1038/onc.2010.60. [DOI] [PubMed] [Google Scholar]
- Omerovic J, Prior IA. Compartmentalized signalling: Ras proteins and signalling nanoclusters. FEBS J. 2009;276:1817–1825. doi: 10.1111/j.1742-4658.2009.06928.x. [DOI] [PubMed] [Google Scholar]
- Rayappa S, Kowluru RA. Role of Raf-1 kinase in diabetes-induced accelerated apoptosis of retinal capillary cells. Int J Biomed Sci. 2008;4:20–28. [PMC free article] [PubMed] [Google Scholar]
- Romeo G, Liu WH, Asnaghi V, Kern TS, Lorenzi M. Activation of nuclear factor-kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes. 2002;51:2241–2248. doi: 10.2337/diabetes.51.7.2241. [DOI] [PubMed] [Google Scholar]
- Schäfers M, Schober O, Hermann S. Matrix-metalloproteinases as imaging targets for inflammatory activity in atherosclerotic plaques. J Nucl Med. 2010;51:663–666. doi: 10.2967/jnumed.109.065698. [DOI] [PubMed] [Google Scholar]
- Schubbert S, Bollag G, Shannon K. Deregulated Ras signaling in developmental disorders: New tricks for an old dog. Curr Opin Genet Dev. 2007;17:15–22. doi: 10.1016/j.gde.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Serban D, Leng J, Cheresh D. H-ras regulates angiogenesis and vascular permeability by activation of distinct downstream effectors. Circ Res. 2008;102:1350–1358. doi: 10.1161/CIRCRESAHA.107.169664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitt AW. The role of advanced glycation in the pathogenesis of diabetic retinopathy. Exp Mol Pathol. 2003;75:95–108. doi: 10.1016/s0014-4800(03)00035-2. [DOI] [PubMed] [Google Scholar]
- Stitt AW. AGEs and diabetic retinopathy. Invest Ophthalmol Vis Sci. 2010;51:4867–4874. doi: 10.1167/iovs.10-5881. [DOI] [PubMed] [Google Scholar]
- Thrailkill KM, Clay Bunn R, Fowlkes JL. Matrix metalloproteinases: Their potential role in the pathogenesis of diabetic nephropathy. Endocrine. 2009;35:1–10. doi: 10.1007/s12020-008-9114-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi SC, Rodriguez WPA, Roberts AM, Falcone JC, Passmore JC, Fleming JT, Joshua IG. Hyperhomocysteinemic diabetic cardiomyopathy: Oxidative stress, remodeling, and endothelial-myocyte uncoupling. J Cardiovasc Pharmacol Ther. 2005;10:1–10. doi: 10.1177/107424840501000101. [DOI] [PubMed] [Google Scholar]
- Zheng L, Szabo C, Kern TS. Poly(ADP-ribose) polymerase is involved in the development of diabetic retinopathy via regulation of nuclear factor-kappaB. Diabetes. 2004;53:2960–2967. doi: 10.2337/diabetes.53.11.2960. [DOI] [PubMed] [Google Scholar]
- Zhong Q, Kowluru RA. Role of histone acetylation in the development of diabetic retinopathy and the metabolic memory phenomenon. J Cell Biochem. 2010;110:1306–1313. doi: 10.1002/jcb.22644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang S, Schnellmann RG. A death-promoting role for extracellular signal-regulated kinase. J Pharmacol Exp Ther. 2006;319:991–997. doi: 10.1124/jpet.106.107367. [DOI] [PubMed] [Google Scholar]