

Abstract
Bilirubin, a major end product of heme breakdown, is an important constituent of bile, responsible for its characteristic colour. Over recent decades, our understanding of bilirubin metabolism has expanded along with the processes of elimination of other endogenous and exogenous anionic substrates, mediated by the action of multiple transport systems at the sinusoidal and canalicular membrane of hepatocytes. Several inherited disorders characterised by impaired bilirubin conjugation (Crigler-Najjar syndrome type I and type II, Gilbert syndrome) or transport (Dubin-Johnson and Rotor syndrome) result in various degrees of hyperbilirubinemia of either the predominantly unconjugated or predominantly conjugated type. Moreover, disrupted regulation of hepatobiliary transport systems can explain jaundice in many acquired liver disorders. In this review, we discuss the recent data on liver bilirubin handling based on the discovery of the molecular basis of Rotor syndrome. The data show that a substantial fraction of bilirubin conjugates is primarily secreted by MRP3 at the sinusoidal membrane into the blood, from where they are subsequently reuptaken by sinusoidal membrane-bound organic anion transporting polypeptides OATP1B1 and OATP1B3. OATP1B proteins are also responsible for liver clearance of bilirubin conjugated in splanchnic organs, such as the intestine and kidney, and for a number of endogenous compounds, xenobiotics and drugs. Absence of one or both OATP1B proteins thus may have serious impact on toxicity of commonly used drugs cleared by this system such as statins, sartans, methotrexate or rifampicin. The liver-blood cycling of conjugated bilirubin is impaired in cholestatic and parenchymal liver diseases and this impairment most likely contributes to jaundice accompanying these disorders.
Keywords: Hyperbilirubinemia, Hereditary jaundice, UGT1A1, ABCC2, Organic anion transporting polypeptide 1B1, Organic anion transporting polypeptide 1B3
Core tip: Experiments with Oatp1a/1b-null mice and Oatp1a/1b; Abcc3 combination knockout mice plainly demonstrated that even under physiologic conditions a substantial portion of bilirubin glucuronides is not excreted directly into bile but is transported back to the blood by Abcc3. Oatp1a/1b activity accentuated in downstream (centrizonal) hepatocytes allows efficient reuptake of bilirubin conjugates, with a subsequent possibility being safely eliminated by excretion into bile. This and molecular findings in Rotor syndrome suggest that human transporters MRP3 and OATP1Bs form a sinusoidal liver-to-blood cycle which mediates shifting (hopping) of bilirubin and other substrates from periportal to centrizonal hepatocytes (References 18, 19, 22, 125).
INTRODUCTION
Bilirubin is the end product of heme breakdown. About 80% of bilirubin originates from degradation of erythrocyte haemoglobin in the reticuloendothelial system; the remaining 20% comes from inefficient erythropoiesis in bone marrow and degradation of other heme proteins[1-4]. Water insoluble, unconjugated bilirubin (UCB) bound to albumin is transported to the liver where it is removed from the plasma. The exact mechanism of UCB uptake is unknown; however, passive transmembrane diffusion seems to be combined with active transport mediated by several sinusoidal transporters (see below). Within the cytoplasm of hepatocytes, bilirubin is bound to ligandin and transported to endoplasmic reticulum where conjugation with glucuronic acid takes place. Conjugation is catalysed by the enzyme uridine diphosphate glycosyltransferase 1A1 (UGT1A1; EC2.4.1.17), a member of an enzyme family in the endoplasmic reticulum and nuclear envelope of hepatocytes[5-8]. In addition to the liver, UGT activity has also been detected in the small intestine and kidney[9,10]. UGT1A1 gene (ID: 54658) is a part of a complex locus encoding 13 UDP-glucuronosyltransferases[11]. The locus contains a series of thirteen unique alternate promoters and first exons, followed by four common exons No. 2-5. Theoretically, each of the unique first exons is spliced to the first of the four shared exons. The unique first exons encode different substrate binding domains whereas the other functional domains encoded by the shared exons 2-5 are the same[11-15]. In reality, only 9 of the 13 predicted UGT1As are active genes encoding functional enzymes; four are nonfunctional pseudogenes.
The excretion of conjugated bilirubin into bile is mediated by an ATP-dependent transporter identified as the multidrug resistance-associated protein MRP2/cMOAT and, to a lesser extent, also by ATP-binding cassette (ABC) efflux transporter ABCG2. MRP2 is encoded by ABCC2 and expressed under physiologic conditions at the apical (canalicular) membrane of hepatocytes and, to a much lesser extent, in the kidney, duodenum, ileum, brain and placenta[16]. Since the MRP2 mediated export represents an important step in detoxification of many endogenous and exogenous substrates, the absence of functionally active MRP2 prevents the secretion of these conjugates into bile. Absence of MRP2 mediated transport is followed by upregulation of the basolateral MRP2 homologues at the sinusoidal membrane of hepatocytes and conjugated bilirubin flow is redirected into sinusoidal blood[17]. Aside from MRP2 mediated transport of conjugated bilirubin into bile, recent studies have shown that a significant fraction of the bilirubin conjugated in the liver is, under physiologic conditions, secreted into sinusoidal blood and subsequently reuptaken by hepatocytes for final biliary excretion[18,19]. The process is mediated by sinusoidal transporters MRP3 and organic anion-transporting polypeptides OATP1B1 and OATP1B3. OATP1B transporters facilitate sodium-independent uptake of numerous endogenous and exogenous substrates[20,21]. Since expression of OATP1Bs is higher in centrilobular hepatocytes, the MRP3-OATP1B1/3 loop is likely responsible for shifting (hopping) of conjugated bilirubin and other substrates from the periportal to the centrilobular zone of the liver lobule (Figure 1). Such intralobular substrate transfer may protect periportal hepatocytes against elevated concentrations of various xenobiotics[22]. In addition, the OATP1B proteins mediate hepatic clearance of bilirubin conjugated in splanchnic organs and may represent an important alternative pathway in enterohepatic circulation[18].
Figure 1.
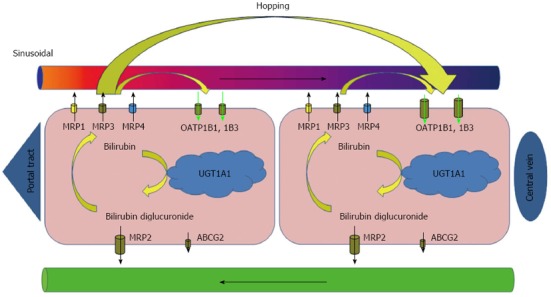
Liver cycle of conjugated bilirubin. Bilirubin conjugated in endoplasmic reticulum of hepatocytes is secreted into the bile. This process is mediated by MRP2/ABCC2 with possible minor contribution of other transporters (ABCG2) at the canalicular membrane of hepatocytes. In addition, even under physiologic conditions, a fraction of bilirubin conjugates is secreted by MRP3 across the sinusoidal membrane into the blood, from where they can be subsequently reuptaken by sinusoidal membrane-bound OATP1B1 and OATP1B3 transporters. The highest overall expression of OATP1Bs has been demonstrated at the centrilobular hepatocytes. The process of substrate shifting (hopping) from periportal to centrizonal hepatocytes may act as a protection of the periportal hepatocytes against elevated concentrations of various xenobiotics. MRP: Multidrug resistance-associated protein; OATP: Organic anion transporting polypeptide; UGT: Uridine diphosphate glucuronosyltransferase; ABC: ATP-binding cassette.
OATP1Bs may also contribute to liver uptake of UCB since complete absence of both OATP1Bs in Rotor syndrome (RS, see below) is associated with elevated levels of UCB and single nucleotide polymorphisms in genes encoding OATP1B proteins have been shown to influence serum bilirubin level[23,24]. Furthermore, results of functional studies demonstrate that OATP1B3, but not OATP1B1, may play an important role in the carrier-mediated uptake of foetal UCB by the placental trophoblast and contribute to elimination of UCB across the placental barrier[25,26].
Mild or moderately elevated serum bilirubin seems to be beneficial: Bilirubin is known as a strong antioxidant[27,28] and the protective effects of bilirubin on atherogenesis and cancerogenesis have been demonstrated in both in vitro and in vivo studies[29-33]. On the other hand, patients with profound unconjugated hyperbilirubinemia are at risk for bilirubin encephalopathy (kernicterus)[34,35]. The toxic effects of bilirubin are explained by inhibition of DNA synthesis[36]. Bilirubin may also uncouple oxidative phosphorylation and inhibit adenosine triphosphatase (ATPase) activity of brain mitochondria[37,38]. Bilirubin mediated inhibition of various enzyme systems, RNA synthesis and protein synthesis in the brain and liver, and/or alteration of carbohydrate metabolism in the brain can also contribute to its toxicity[39-43]. The accumulation of bilirubin in plasma and tissues results in characteristic yellow discoloration of tissues known as icterus or jaundice.
Inherited disorders of bilirubin excretory pathway played the key role in understanding the individual steps of the bilirubin excretory pathway. Disrupted regulation of hepatobiliary transport systems explained jaundice in many acquired liver disorders[44-48]. Additional information was obtained from a number of animal models of hereditary jaundice. These include the Gunn rat and Ugt1(-/-) mouse mimicking the Crigler-Najjar syndrome type I[49-51], the Bolivian population of squirrel monkeys mimicking Gilbert syndrome (GS)[52,53] and mutant TR or GY (Groningen yellow) rats with organic anion excretion defect (TR -/-), Eizai hyperbilirubinuria rats (EHBR), mutant Corriedale sheep, and Mrp2(-/-) mice, all modelling the Dubin-Johnson syndrome (DJS)[54-58].
HEREDITARY PREDOMINANTLY UNCONJUGATED HYPERBILIRUBINEMIA
Conjugation of bilirubin in endoplasmic reticulum is catalysed by the enzyme UGT1A1. Mutations in UGT1A1 can lead to decreased expression or partial or even complete inactivation of the enzyme[59]. By contrast, expression of UGT1A1 can be increased by phenobarbital (PB) administration. PB response activity is delineated to a 290-bp distal enhancer module sequence (-3483/-3194) glucuronosyltransferase phenobarbital response enhancing motif (gtBPREM) of the human UGT1A1[59,60]. gtBPREM is activated by the nuclear orphan receptor, human constitutive active receptor (hCAR). CAR is a cytoplasmic receptor which, after treatment with activators such as PB, translocates into the nucleus, forms a heterodimer with the retinoid X receptor and activates the PB response enhancer element.
Three types of inherited, predominantly unconjugated hyperbilirubinemia with different levels of UGT1A1 activity are recognised: Crigler-Najjar syndrome type I (CN1), type II (CN2) and GS.
CN1 (MIM#218800), the most deleterious form, described in 1952 by Crigler and Najjar[61], is characterised by complete or almost complete absence of UGT1A1 enzyme activity with severe jaundice[62]. Icterus occurring shortly after birth is complicated by bilirubin encephalopathy (kernicterus). Until the introduction of phototherapy and plasmapheresis, kernicterus was fatal in almost all cases during the first two years of life or caused serious brain damage with permanent neurologic sequelae. Intermittent phototherapy is lifelong and it results in a thorough elimination of water-soluble photoisomers of unconjugated bilirubin via bile. The effectiveness of phototherapy may decrease gradually with age and patients are at higher risk of sudden brain damage[63].
Although new treatment modalities such as hepatocyte or hepatic progenitor cell transplantation have already been used to treat CN1 patients, liver transplantation is still considered to be the only definitive treatment for CN1[63-67]. Gene therapy seems to be a promising therapeutic possibility for the patients with CN1 in the near future[68,69].
CN2 (Arias syndrome, MIM #606785), described by Arias in 1962[70], is characterised by reduced UGT1A1 enzyme activity with a moderate degree of nonhemolytic jaundice. Bilirubin levels do not exceed 350 μmol/L and CN2 is only rarely complicated by kernicterus[71]. Virtually all the mutations responsible for the syndrome are autosomal recessive, as in CN1, but several observations have also suggested the possibility of autosomal dominant pattern of inheritance[72-74].
An important clinical difference between CN type I and type II is the response to PB treatment, with no effect in type I (complete loss of the UGT1A1 enzyme activity) and a decrease of serum bilirubin levels by more than 30% in CN type II (some residual UGT1A1 activity is preserved). Moreover, bilirubin glucuronides are present in bile in CN2. However, the method of choice for the diagnosis of CN syndrome is mutation analysis of UGT1A1[75].
GS (MIM #143500), described in 1901 by Gilbert and Lereboulet[76], is characterised by fluctuating mild, unconjugated nonhemolytic hyperbilirubinemia < 85 μmol/L without overt haemolysis, usually diagnosed around puberty, and aggravated by intercurrent illness, stress, fasting or after administration of certain drugs[77,78]. Physical examination and the results of routine laboratory tests are normal apart from elevated serum bilirubin and jaundice. The clinical diagnosis of GS can be established if patients have a mild, predominantly unconjugated hyperbilirubinemia and normal activity of liver enzymes. The reduced caloric intake test and phenobarbital stimulation test have low diagnostic specificity in GS subjects[79]. Histological findings in GS are mild, with a slight centrilobular accumulation of pigment with lipofuscin-like properties[80]. Ultrastructurally, hepatocytes reveal hypertrophy of smooth endoplasmic reticulum[81,82]. Since the morphological picture of GS is completely non-specific and the disorder is benign, liver biopsy is not indicated.
GS is characterised by reduced levels of UGT1A1 activity to about 25%-30% caused by homozygous, compound heterozygous, or heterozygous mutations in the UGT1A1 with autosomal recessive transmission[80].
GS is the most frequent hereditary jaundice affecting nearly 5%-10% of the Caucasian population[83]. The genetic basis of GS was first disclosed in 1995[84] as presence of the allele UGT1A1*28, characterised by insertion of TA in the TATAA box (A[TA]7TAA) in the proximal promoter of UGT1A1. UGT1A1*28 has been identified as the most frequent mutation in Caucasian GS subjects[85]. The insertion is responsible for reduction of transcription of UGT1A1 to 20% from normal and for a decrease of hepatic glucuronidation activity by 80% in a homozygous state[86]. In Caucasians and African Americans, the frequency of UGT1A1*28 allele is about 35%-40%, but it is much lower in Asians, including Koreans (13%), Chinese (16%), and Japanese (11%)[87-89]. Moreover, in the majority of Caucasian GS subjects, expression of UGT1A1 is further decreased by the presence of the second mutation T>G in gtPBREM[59,60]. In addition to the mutations in the promoter, GS may be caused by mutations in structural regions of the UGT1A1. In Asians, other variants, such as UGT1A1*6 characterised by a missense mutation involving G to A substitution at nucleotide 211 (c.211G>A) in exon 1 (also known as p.G71R), UGT1A1*7 (p.Y486D), UGT1A1*27 (p.P229Q), and UGT1A1*62 (p.F83L) have been detected[60,87-90].
In addition to biochemical defect leading to reduced glucuronidation, other factors, such as impaired hepatic (re)uptake of bilirubin (see Rotor syndrome below for the possible mechanism) or an increased load of bilirubin, seem to be necessary for clinical manifestation of GS[86,91,92].
GS is benign and GS carriers present with no liver disease. However, the mutations in the UGT1A1 identical to those recognised in GS subjects may contribute to the development of prolonged neonatal hyperbilirubinemia in breast-fed infants[93,94].
Moreover, since the process of glucuronidation is an important step in elimination of numerous endogenous and exogenous substrates, GS subjects may be more susceptible to the adverse effects of some drugs metabolised by UGT1A1, such as indinavir, atazanavir[95-99] or irinotecan[100-102].
HEREDITARY PREDOMINANTLY CONJUGATED HYPERBILIRUBINEMIA
Two types of hereditary conjugated jaundice are known as Dubin-Johnson and Rotor syndrome. Both are characterised by the presence of mixed, predominantly conjugated hyperbilirubinemia, with conjugated bilirubin more than 50% of total bilirubin.
DJS (MIM # 237500), a benign autosomal recessive disorder described in 1954 by Dubin et al[103] and Sprinz et al[104], is characterised by fluctuating mild, predominantly conjugated hyperbilirubinemia, with typical manifestation in adolescence or young adulthood. Most patients are asymptomatic except of occasional slight abdominal pain and fatigue. Urine excretion of total coproporphyrin in 24 h is normal, but 80% are represented by coproporphyrin I. Biliary excretion of anionic dyes including bromosulfophthalein (BSP), indocyanine green and cholescintigraphy radiotracers is delayed with absent or delayed filling of the gallbladder[105]. BSP clearance in DJS subjects is normal at 45 min with the second peak at 90 min[106]. Liver histology in DJS shows an accumulation of distinctive melanin-like lysosomal pigment in an otherwise normal liver that gives the organ a characteristic dark pink or even black colour. The pigment is positive in PAS and Masson-Fontana reaction with marked autofluorescence. In contrast to melanin, DJS pigment does not reduce neutral silver ammonium solution[103,107]. The amount of pigment may vary and possible transient loss may occur in coincidence with other liver diseases[108,109]. The molecular mechanism in DJS is absence or deficiency of human canalicular multispecific organic anion transporter MRP2/cMOAT caused by homozygous or compound heterozygous mutation in ABCC2 (gene ID: 1244) on chromosome 10q24[110-114]. The ABCC2 mutation alters not only MRP2-mediated transport of conjugated bilirubin but also transport of many anionic substrates as well as a wide range of drugs, such as chemotherapeutics, uricosurics, antibiotics, leukotrienes, glutathione, toxins and heavy metals. Absence of MRP2/cMOAT may result in impaired elimination and in subsequent renal toxicity of the substrates mentioned above[115-120].
A rare type of hereditary mixed hyperbilirubinemia caused by the simultaneous presence of mutations characteristic for DJS and GS has been classified as dual hereditary jaundice[121]. Serum direct bilirubin concentrations in dual hereditary jaundice reach only 20%-50% of total bilirubin.
RS (MIM #237450), described in 1948 by Rotor et al[122], is characterised by mild, predominantly conjugated hyperbilirubinemia with delayed excretion of anionic dyes without re-increase of their concentration. Total urinary coproporphyrin excretion is significantly increased and the proportion of coproporphyrin I in urine is approximately 65% of the total in homozygotes and 43% in heterozygotes[123,124]. By histopathological examination, the liver tissue does not display any marked architectural or cytomorphological abnormalities and pigment is not present.
The presence of homozygous mutations in both SLCO1B1 and SLCO1B3 neighbouring genes located on chromosome 12 with complete and simultaneous deficiency of proteins OATP1B1 and OATP1B3 has recently been identified as the molecular mechanism of the syndrome[125]. The complete absence of both transporters OATP1B1 and OATP1B3 has been confirmed by immunohistochemistry in all studied Rotor subjects. Interestingly, the presence of a single functional allele of either SLCO1B1 or SLCO1B3 prevented the jaundice.
RS does not require any therapy but, with regard to the impact of OATP1B transporters on pharmacokinetics of a broad spectrum of commonly used drugs such as penicillins, statins, sartans, rifampicin, methotrexate and many others, it is assumed that RS subjects and also those with the deleterious mutations in either of the SLCO1B genes, even without full clinical expression of the syndrome, may be at increased risk for drug toxicity[125-129].
BILIRUBIN HANDLING PROTEINS IN CHOLESTASIS
Animal models of obstructive and intrahepatic cholestasis help us to discover and understand the main principles of acquired defects in hepatobiliary transport of bile salts and other organic anions. Up and down regulation of these mechanisms can explain impaired liver uptake and excretion of the biliary constituents resulting in the cholestasis and icterus which accompanies many common acquired liver disorders[48,130,131]. A general pattern of response to cholestatic liver injury is initiated by downregulation of the basolateral membrane bound transporters NTCP and OATP1B1. The expression of several canalicular export pumps is relatively unaffected [bile salt export pump (BSEP), multidrug resistance protein 2 (MDR2)] or even upregulated (MDR1). Decreased expression of MRP2 in sepsis or in obstructive cholestasis is followed by upregulation of several MRP homologues at basolateral membrane of hepatocytes that may extrude bile salts back to the sinusoidal blood and systemic circulation. Most of these changes are believed to help prevent an accumulation of potentially toxic bile components and other substrates in the liver.
Similar patterns of expression of the bilirubin and bile salts handling proteins and mRNA are observed in cholestatic liver diseases in humans. At the stage I and II of primary biliary cirrhosis (PBC), expression and localisation of OATP1B1, OATP1B3, NTCP, MRP2, MRP3 and MDR3 are unchanged. At stage III, immunostaining intensities of the sinusoidal uptake transporters and their mRNA levels decrease. Irregular MRP2 immunostaining suggests redistribution of MRP2 into intracellular structures in the advanced stages of PBC; however, at stage III and IV, basolateral uptake transporters NTCP and OATP1B1 are downregulated. Expression of the canalicular export pumps for bile salts (BSEP) and bilirubin (MRP2) remains unchanged and the canalicular P-glycoproteins MDR1 and MDR3 and the basolateral efflux pump MRP3 are upregulated[132-135].
At the early-stages of cholestasis in extrahepatic biliary atresia, BSEP, MDR3, MRP2, NTCP/SLC10A1, SLCO1A2 and nuclear receptor farnesoid X receptor are downregulated. At the late-stages of cholestasis, farnesoid X receptor and BSEP levels returns to normal, MDR3 and MDR1 are upregulated and MRP2 is downregulated[136].
In primary sclerosing cholangitis, the level of OATP1B1 mRNA in liver tissue has been demonstrated to represent 49% of controls and the level of MRP2 mRNA dropped to 27% of controls[137].
CONCLUSION AND PERSPECTIVES
Over the last decades, molecular basis of hyperbilirubinemia syndromes has been elucidated and mutations affecting the basolateral and apical membrane transporters responsible for accumulation of either conjugated or unconjugated bilirubin have been identified.
Except for GS, the majority of inherited hyperbilirubinemia syndromes are rare autosomal recessive disorders with a low prevalence in the general population and, apart from CN syndrome type I and some cases of CN type II in neonatal period, mostly not requiring further therapy. Nonetheless, the enzyme and transport systems involved in bilirubin metabolism may play an important role in the elimination and disposition processes of many other endogenous and exogenous substrates including hormones, drugs, toxins and heavy metals[102,138]. Dysfunction or absence of these systems, including selected ABC transporters and OATPs, may alter pharmacokinetics and pharmacodynamics of many biologically active agents, affect penetration of the substrates into various tissues and lead to their intracellular accumulation with a subsequent increase of organ toxicity[126,127,128]. In addition, the absence of the functional transport proteins involved in hepatobiliary and enterohepatic circulation may involve drug disposition, drug-drug or drug-food interactions and result in decreased effectiveness or even resistance to a diverse spectrum of chemotherapeutic agents and xenobiotics[139-141]. Individuals with mutations in the responsible gene or genes with the fully expressed phenotype of the corresponding hyperbilirubinemia syndrome, as well as subjects carrying mutations without clinical manifestation of hyperbilirubinemia under normal conditions, may be more susceptible to the adverse effects of some drugs and metabolites[142,143].
Clarifying the molecular genetic basis of hereditary hyperbilirubinemia syndromes together with the discoveries of the major systems essential for the metabolism and transport of bilirubin and other endogenous and exogenous substrates represent a substantial contribution to the current knowledge of the heme degradation pathway. Further investigation of how bilirubin transport proteins and their variations affect pharmacokinetics of drugs may be of significant clinical importance.
Footnotes
Supported by The Project (Ministry of Health, Czech Republic) for Development of Research Organization 00023001 (IKEM, Prague, Czech Republic), Institutional support
P- Reviewers Marin JJG, Ruiz-Gaspa S, Teng RJ S- Editor Wen LL L- Editor A E- Editor Zhang DN
References
- 1.Stadeler G. Uber die Farbstoffe der Galle. Anaalen d Chemie u Pharmacie. 1864;132:323–354. [Google Scholar]
- 2.London IM, West R, Shemin D, Rittenberg D. On the origin of bile pigment in normal man. J Biol Chem. 1950;184:351–358. [PubMed] [Google Scholar]
- 3.Berk PD, Howe RB, Bloomer JR, Berlin NI. Studies of bilirubin kinetics in normal adults. J Clin Invest. 1969;48:2176–2190. doi: 10.1172/JCI106184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci USA. 1968;61:748–755. doi: 10.1073/pnas.61.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmid R. The identification of direct-reacting bilirubin as bilirubin glucuronide. J Biol Chem. 1957;229:881–888. [PubMed] [Google Scholar]
- 6.Schmid R. Direct-reacting bilirubin, bilirubin glucuronide, in serum, bile and urine. Science. 1956;124:76–77. doi: 10.1126/science.124.3211.76. [DOI] [PubMed] [Google Scholar]
- 7.Gorski JP, Kasper CB. Purification and properties of microsomal UDP-glucuronosyltransferase from rat liver. J Biol Chem. 1977;252:1336–1343. [PubMed] [Google Scholar]
- 8.Burchell B. Purification of UDP-glucuronyltransferase from untreated rat liver. FEBS Lett. 1977;78:101–104. doi: 10.1016/0014-5793(77)80283-4. [DOI] [PubMed] [Google Scholar]
- 9.Fisher MB, Paine MF, Strelevitz TJ, Wrighton SA. The role of hepatic and extrahepatic UDP-glucuronosyltransferases in human drug metabolism. Drug Metab Rev. 2001;33:273–297. doi: 10.1081/dmr-120000653. [DOI] [PubMed] [Google Scholar]
- 10.Kokudo N, Takahashi S, Sugitani K, Okazaki T, Nozawa M. Supplement of liver enzyme by intestinal and kidney transplants in congenitally enzyme-deficient rat. Microsurgery. 1999;19:103–107. doi: 10.1002/(sici)1098-2752(1999)19:2<103::aid-micr12>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 11.Owens IS, Basu NK, Banerjee R. UDP-glucuronosyltransferases: gene structures of UGT1 and UGT2 families. Methods Enzymol. 2005;400:1–22. doi: 10.1016/S0076-6879(05)00001-7. [DOI] [PubMed] [Google Scholar]
- 12.Burchell B. Identification and purification of mutiple forms of UDP-glucuronosyltransferase. Rev Biochem Toxicol. 1981;3:1. [Google Scholar]
- 13.Roy Chowdhury J, Roy Chowdhury N, Falany CN, Tephly TR, Arias IM. Isolation and characterization of multiple forms of rat liver UDP-glucuronate glucuronosyltransferase. Biochem J. 1986;233:827–837. doi: 10.1042/bj2330827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burchell B, Brierley CH, Monaghan G, Clarke DJ. The structure and function of the UDP-glucuronosyltransferase gene family. Adv Pharmacol. 1998;42:335–338. doi: 10.1016/s1054-3589(08)60758-9. [DOI] [PubMed] [Google Scholar]
- 15.Tukey RH, Strassburg CP. Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annu Rev Pharmacol Toxicol. 2000;40:581–616. doi: 10.1146/annurev.pharmtox.40.1.581. [DOI] [PubMed] [Google Scholar]
- 16.Cherrington NJ, Hartley DP, Li N, Johnson DR, Klaassen CD. Organ distribution of multidrug resistance proteins 1, 2, and 3 (Mrp1, 2, and 3) mRNA and hepatic induction of Mrp3 by constitutive androstane receptor activators in rats. J Pharmacol Exp Ther. 2002;300:97–104. doi: 10.1124/jpet.300.1.97. [DOI] [PubMed] [Google Scholar]
- 17.Gartung C, Matern S. Molecular regulation of sinusoidal liver bile acid transporters during cholestasis. Yale J Biol Med. 1997;70:355–363. [PMC free article] [PubMed] [Google Scholar]
- 18.van de Steeg E, Wagenaar E, van der Kruijssen CM, Burggraaff JE, de Waart DR, Elferink RP, Kenworthy KE, Schinkel AH. Organic anion transporting polypeptide 1a/1b-knockout mice provide insights into hepatic handling of bilirubin, bile acids, and drugs. J Clin Invest. 2010;120:2942–2952. doi: 10.1172/JCI42168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iusuf D, van de Steeg E, Schinkel AH. Functions of OATP1A and 1B transporters in vivo: insights from mouse models. Trends Pharmacol Sci. 2012;33:100–108. doi: 10.1016/j.tips.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 20.König J, Cui Y, Nies AT, Keppler D. Localization and genomic organization of a new hepatocellular organic anion transporting polypeptide. J Biol Chem. 2000;275:23161–23168. doi: 10.1074/jbc.M001448200. [DOI] [PubMed] [Google Scholar]
- 21.König J, Cui Y, Nies AT, Keppler D. A novel human organic anion transporting polypeptide localized to the basolateral hepatocyte membrane. Am J Physiol Gastrointest Liver Physiol. 2000;278:G156–G164. doi: 10.1152/ajpgi.2000.278.1.G156. [DOI] [PubMed] [Google Scholar]
- 22.Iusuf D, van de Steeg E, Schinkel AH. Hepatocyte hopping of OATP1B substrates contributes to efficient hepatic detoxification. Clin Pharmacol Ther. 2012;92:559–562. doi: 10.1038/clpt.2012.143. [DOI] [PubMed] [Google Scholar]
- 23.Zhang W, He YJ, Gan Z, Fan L, Li Q, Wang A, Liu ZQ, Deng S, Huang YF, Xu LY, et al. OATP1B1 polymorphism is a major determinant of serum bilirubin level but not associated with rifampicin-mediated bilirubin elevation. Clin Exp Pharmacol Physiol. 2007;34:1240–1244. doi: 10.1111/j.1440-1681.2007.04798.x. [DOI] [PubMed] [Google Scholar]
- 24.Sanna S, Busonero F, Maschio A, McArdle PF, Usala G, Dei M, Lai S, Mulas A, Piras MG, Perseu L, et al. Common variants in the SLCO1B3 locus are associated with bilirubin levels and unconjugated hyperbilirubinemia. Hum Mol Genet. 2009;18:2711–2718. doi: 10.1093/hmg/ddp203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Briz O, Serrano MA, MacIas RI, Gonzalez-Gallego J, Marin JJ. Role of organic anion-transporting polypeptides, OATP-A, OATP-C and OATP-8, in the human placenta-maternal liver tandem excretory pathway for foetal bilirubin. Biochem J. 2003;371:897–905. doi: 10.1042/BJ20030034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macias RI, Marin JJ, Serrano MA. Excretion of biliary compounds during intrauterine life. World J Gastroenterol. 2009;15:817–828. doi: 10.3748/wjg.15.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 28.Dennery PA, McDonagh AF, Spitz DR, Rodgers PA, Stevenson DK. Hyperbilirubinemia results in reduced oxidative injury in neonatal Gunn rats exposed to hyperoxia. Free Radic Biol Med. 1995;19:395–404. doi: 10.1016/0891-5849(95)00032-s. [DOI] [PubMed] [Google Scholar]
- 29.Mayer M. Association of serum bilirubin concentration with risk of coronary artery disease. Clin Chem. 2000;46:1723–1727. [PubMed] [Google Scholar]
- 30.Ollinger R, Kogler P, Troppmair J, Hermann M, Wurm M, Drasche A, Königsrainer I, Amberger A, Weiss H, Ofner D, et al. Bilirubin inhibits tumor cell growth via activation of ERK. Cell Cycle. 2007;6:3078–3085. doi: 10.4161/cc.6.24.5022. [DOI] [PubMed] [Google Scholar]
- 31.Lacko M, Roelofs HM, Te Morsche RH, Voogd AC, Ophuis MB, Peters WH, Manni JJ. Genetic polymorphism in the conjugating enzyme UGT1A1 and the risk of head and neck cancer. Int J Cancer. 2010;127:2815–2821. doi: 10.1002/ijc.25296. [DOI] [PubMed] [Google Scholar]
- 32.Horsfall LJ, Rait G, Walters K, Swallow DM, Pereira SP, Nazareth I, Petersen I. Serum bilirubin and risk of respiratory disease and death. JAMA. 2011;305:691–697. doi: 10.1001/jama.2011.124. [DOI] [PubMed] [Google Scholar]
- 33.Keshavan P, Schwemberger SJ, Smith DL, Babcock GF, Zucker SD. Unconjugated bilirubin induces apoptosis in colon cancer cells by triggering mitochondrial depolarization. Int J Cancer. 2004;112:433–445. doi: 10.1002/ijc.20418. [DOI] [PubMed] [Google Scholar]
- 34.Hervieux J. Paris: These Med; 1847. De l’ictere des nouveau-nes. [Google Scholar]
- 35.Schmorl G. Zur Kenntnis des ikterus neonatatorum, inbesondere der dabei auftreten den gehirnveranderungen. Verh Dtsch Ges Pathol. 1903;6:109. [Google Scholar]
- 36.Schiff D, Chan G, Poznansky MJ. Bilirubin toxicity in neural cell lines N115 and NBR10A. Pediatr Res. 1985;19:908–911. doi: 10.1203/00006450-198509000-00007. [DOI] [PubMed] [Google Scholar]
- 37.Mustafa MG, Cowger ML, King TE. Effects of bilirubin on mitochondrial reactions. J Biol Chem. 1969;244:6403–6414. [PubMed] [Google Scholar]
- 38.Diamond I, Schmid R. Oxidative phosphorylation in experimental bilirubin encephalopathy. Science. 1967;155:1288–1289. doi: 10.1126/science.155.3767.1288. [DOI] [PubMed] [Google Scholar]
- 39.Strumia E. [Effect of bilirubin on some hydrolases] Boll Soc Ital Biol Sper. 1959;35:2160–2162. [PubMed] [Google Scholar]
- 40.Flitman R, Worth MH. Inhibition of hepatic alcohol dehydrogenase by bilirubin. J Biol Chem. 1966;241:669–672. [PubMed] [Google Scholar]
- 41.Katoh R, Kashiwamata S, Niwa F. Studies on cellular toxicity of bilirubin: Effect on the carbohydrate metabolism in the young rat brain. Brain Res. 1975;83:81–92. [Google Scholar]
- 42.Greenfield S, Majumdar AP. Bilirubin encephalopathy: effect on protein synthesis in the brain of the Gunn rat. J Neurol Sci. 1974;22:83–89. doi: 10.1016/0022-510x(74)90056-2. [DOI] [PubMed] [Google Scholar]
- 43.Majumdar AP. Bilirubin encephalopathy: effect on RNA polymerase activity and chromatin template activity in the brain of the Gunn rat. Neurobiology. 1974;4:425–431. [PubMed] [Google Scholar]
- 44.Geier A, Dietrich CG, Voigt S, Kim SK, Gerloff T, Kullak-Ublick GA, Lorenzen J, Matern S, Gartung C. Effects of proinflammatory cytokines on rat organic anion transporters during toxic liver injury and cholestasis. Hepatology. 2003;38:345–354. doi: 10.1053/jhep.2003.50317. [DOI] [PubMed] [Google Scholar]
- 45.Geier A, Wagner M, Dietrich CG, Trauner M. Principles of hepatic organic anion transporter regulation during cholestasis, inflammation and liver regeneration. Biochim Biophys Acta. 2007;1773:283–308. doi: 10.1016/j.bbamcr.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 46.Alrefai WA, Gill RK. Bile acid transporters: structure, function, regulation and pathophysiological implications. Pharm Res. 2007;24:1803–1823. doi: 10.1007/s11095-007-9289-1. [DOI] [PubMed] [Google Scholar]
- 47.Thompson R, Jansen PL. Genetic defects in hepatocanalicular transport. Semin Liver Dis. 2000;20:365–372. doi: 10.1055/s-2000-9384. [DOI] [PubMed] [Google Scholar]
- 48.Lee J, Boyer JL. Molecular alterations in hepatocyte transport mechanisms in acquired cholestatic liver disorders. Semin Liver Dis. 2000;20:373–384. doi: 10.1055/s-2000-9390. [DOI] [PubMed] [Google Scholar]
- 49.Gunn CH. Hereditary acholuric jaundice in a new mutant strain of rats. J Hered. 1938;29:137–139. [Google Scholar]
- 50.Roy Chowdhury J, Van ES HHG, Roy Chowdhury N. Gunn rat: An animal model of deficiency of bilirubin conjugation. In: Tavoloni N, Berk PD, editors. Hepatic Transport and Bile Secretion: Physiology and Pathophysiology. New York: Raven Press; 1992. p. 713. [Google Scholar]
- 51.Nguyen N, Bonzo JA, Chen S, Chouinard S, Kelner MJ, Hardiman G, Bélanger A, Tukey RH. Disruption of the ugt1 locus in mice resembles human Crigler-Najjar type I disease. J Biol Chem. 2008;283:7901–7911. doi: 10.1074/jbc.M709244200. [DOI] [PubMed] [Google Scholar]
- 52.Portman OW, Roy Chowdhury J, Roy Chowdhury N, Alexander M, Cornelius CE, Arias IM. A nonhuman primate model of Gilbert’s syndrome. Hepatology. 1984;4:175–179. doi: 10.1002/hep.1840040202. [DOI] [PubMed] [Google Scholar]
- 53.Portman OW, Alexander M, Cornelius CE, Chowdhury JR, Chowdhury NR, Arias IM. The effects of nutrition on unconjugated plasma bilirubin concentrations in squirrel monkeys. Hepatology. 1984;4:454–460. doi: 10.1002/hep.1840040318. [DOI] [PubMed] [Google Scholar]
- 54.Jansen PL, van Klinken JW, van Gelder M, Ottenhoff R, Elferink RP. Preserved organic anion transport in mutant TR- rats with a hepatobiliary secretion defect. Am J Physiol. 1993;265:G445–G452. doi: 10.1152/ajpgi.1993.265.3.G445. [DOI] [PubMed] [Google Scholar]
- 55.Kawaguchi A, Nozaki Y, Hosokawa S, Tagaya O, Mikami T, Wakabayashi T. [Establishment of hyperbilirubinuria rat mutant--a new animal model for jaundice] Jikken Dobutsu. 1994;43:37–44. doi: 10.1538/expanim1978.43.1_37. [DOI] [PubMed] [Google Scholar]
- 56.Yamazaki K, Mikami T, Hosokawa S, Tagaya O, Nozaki Y, Kawaguchi A, Funami H, Katoh H, Yamamoto N, Wakabayashi T. A new mutant rat with hyperbilirubinuria (hyb) J Hered. 1995;86:314–317. doi: 10.1093/oxfordjournals.jhered.a111592. [DOI] [PubMed] [Google Scholar]
- 57.Cornelius CE, Arias IM, Osburn BI. Hepatic pigmentation with photosensitivity: A Syndrome in corriedale sheep resembling dubin-johnson syndrome in man. J Am Vet Med Assoc. 1965;146:709–713. [PubMed] [Google Scholar]
- 58.Chu XY, Strauss JR, Mariano MA, Li J, Newton DJ, Cai X, Wang RW, Yabut J, Hartley DP, Evans DC, et al. Characterization of mice lacking the multidrug resistance protein MRP2 (ABCC2) J Pharmacol Exp Ther. 2006;317:579–589. doi: 10.1124/jpet.105.098665. [DOI] [PubMed] [Google Scholar]
- 59.Sugatani J, Yamakawa K, Yoshinari K, Machida T, Takagi H, Mori M, Kakizaki S, Sueyoshi T, Negishi M, Miwa M. Identification of a defect in the UGT1A1 gene promoter and its association with hyperbilirubinemia. Biochem Biophys Res Commun. 2002;292:492–497. doi: 10.1006/bbrc.2002.6683. [DOI] [PubMed] [Google Scholar]
- 60.Sugatani J, Kojima H, Ueda A, Kakizaki S, Yoshinari K, Gong QH, Owens IS, Negishi M, Sueyoshi T. The phenobarbital response enhancer module in the human bilirubin UDP-glucuronosyltransferase UGT1A1 gene and regulation by the nuclear receptor CA-R. Hepatology. 2001;33:1232–1238. doi: 10.1053/jhep.2001.24172. [DOI] [PubMed] [Google Scholar]
- 61.Crigler JF, Najjar VA. Congenital familial nonhemolytic jaundice with kernicterus. Pediatrics. 1952;10:169–180. [PubMed] [Google Scholar]
- 62.Ritter JK, Yeatman MT, Ferreira P, Owens IS. Identification of a genetic alteration in the code for bilirubin UDP-glucuronosyltransferase in the UGT1 gene complex of a Crigler-Najjar type I patient. J Clin Invest. 1992;90:150–155. doi: 10.1172/JCI115829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jansen PL. Diagnosis and management of Crigler-Najjar syndrome. Eur J Pediatr. 1999;158 Suppl 2:S89–S94. doi: 10.1007/pl00014330. [DOI] [PubMed] [Google Scholar]
- 64.Karon M, Imach D, Schwartz A. Effective phototherapy in congenital nonobstructive, nonhemolytic jaundice. N Engl J Med. 1970;282:377–380. doi: 10.1056/NEJM197002122820709. [DOI] [PubMed] [Google Scholar]
- 65.Shevell MI, Bernard B, Adelson JW, Doody DP, Laberge JM, Guttman FM. Crigler-Najjar syndrome type I: treatment by home phototherapy followed by orthotopic hepatic transplantation. J Pediatr. 1987;110:429–431. doi: 10.1016/s0022-3476(87)80510-3. [DOI] [PubMed] [Google Scholar]
- 66.Fox IJ, Chowdhury JR, Kaufman SS, Goertzen TC, Chowdhury NR, Warkentin PI, Dorko K, Sauter BV, Strom SC. Treatment of the Crigler-Najjar syndrome type I with hepatocyte transplantation. N Engl J Med. 1998;338:1422–1426. doi: 10.1056/NEJM199805143382004. [DOI] [PubMed] [Google Scholar]
- 67.Matas AJ, Sutherland DE, Steffes MW, Mauer SM, Sowe A, Simmons RL, Najarian JS. Hepatocellular transplantation for metabolic deficiencies: decrease of plasms bilirubin in Gunn rats. Science. 1976;192:892–894. doi: 10.1126/science.818706. [DOI] [PubMed] [Google Scholar]
- 68.Ilan Y, Attavar P, Takahashi M, Davidson A, Horwitz MS, Guida J, Chowdhury NR, Chowdhury JR. Induction of central tolerance by intrathymic inoculation of adenoviral antigens into the host thymus permits long-term gene therapy in Gunn rats. J Clin Invest. 1996;98:2640–2647. doi: 10.1172/JCI119085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takahashi M, Ilan Y, Chowdhury NR, Guida J, Horwitz M, Chowdhury JR. Long term correction of bilirubin-UDP-glucuronosyltransferase deficiency in Gunn rats by administration of a recombinant adenovirus during the neonatal period. J Biol Chem. 1996;271:26536–26542. doi: 10.1074/jbc.271.43.26536. [DOI] [PubMed] [Google Scholar]
- 70.Arias IM. Chronic unconjugated hyperbilirubinemia without overt signs of hemolysis in adolescents and adults. J Clin Invest. 1962;41:2233–2245. doi: 10.1172/JCI104682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gollan JL, Huang SN, Billing B, Sherlock S. Prolonged survival in three brothers with severe type 2 Crigler-Najjar syndrome. Ultrastructural and metabolic studies. Gastroenterology. 1975;68:1543–1555. [PubMed] [Google Scholar]
- 72.Moghrabi N, Clarke DJ, Boxer M, Burchell B. Identification of an A-to-G missense mutation in exon 2 of the UGT1 gene complex that causes Crigler-Najjar syndrome type 2. Genomics. 1993;18:171–173. doi: 10.1006/geno.1993.1451. [DOI] [PubMed] [Google Scholar]
- 73.Hunter JO, Thompson RP, Dunn PM, Williams R. Inheritance of type 2 Crigler-Najjar hyperbilirubinaemia. Gut. 1973;14:46–49. doi: 10.1136/gut.14.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Labrune P, Myara A, Hennion C, Gout JP, Trivin F, Odievre M. Crigler-Najjar type II disease inheritance: a family study. J Inherit Metab Dis. 1989;12:302–306. doi: 10.1007/BF01799221. [DOI] [PubMed] [Google Scholar]
- 75.Seppen J, Bosma PJ, Goldhoorn BG, Bakker CT, Chowdhury JR, Chowdhury NR, Jansen PL, Oude Elferink RP. Discrimination between Crigler-Najjar type I and II by expression of mutant bilirubin uridine diphosphate-glucuronosyltransferase. J Clin Invest. 1994;94:2385–2391. doi: 10.1172/JCI117604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gilbert A, Lereboullet P. La cholemie simple familiale. Semaine Medicale. 1901;21:241–243. [Google Scholar]
- 77.Nixon JC, Monahan GJ. Gilbert’s disease and the bilirubin tolerance test. Can Med Assoc J. 1967;96:370–373. [PMC free article] [PubMed] [Google Scholar]
- 78.Schmid R. Gilbert’s syndrome--a legitimate genetic anomaly? N Engl J Med. 1995;333:1217–1218. doi: 10.1056/NEJM199511023331812. [DOI] [PubMed] [Google Scholar]
- 79.Thomsen HF, Hardt F, Juhl E. Diagnosis of Gilbert’s syndrome. Reliability of the caloric restriction and phenobarbital stimulation tests. Scand J Gastroenterol. 1981;16:699–703. doi: 10.3109/00365528109182033. [DOI] [PubMed] [Google Scholar]
- 80.Barth RF, Grimley PM, Berk PD, Bloomer JR, Howe RB. Excess lipofuscin accumulation in constitutional hepatic dysfunction (Gilbert’s syndrome). Light and electron microscopic observations. Arch Pathol. 1971;91:41–47. [PubMed] [Google Scholar]
- 81.Dawson J, Seymour CA, Peters TJ. Gilbert’s syndrome: analytical subcellular fractionation of liver biopsy specimens. Enzyme activities, organelle pathology and evidence for subpopulations of the syndrome. Clin Sci (Lond) 1979;57:491–497. doi: 10.1042/cs0570491. [DOI] [PubMed] [Google Scholar]
- 82.Black M, Billing BH. Hepatic bilirubin udp-glucuronyl transferase activity in liver disease and gilbert’s syndrome. N Engl J Med. 1969;280:1266–1271. doi: 10.1056/NEJM196906052802303. [DOI] [PubMed] [Google Scholar]
- 83.Owens D, Evans J. Population studies on Gilbert’s syndrome. J Med Genet. 1975;12:152–156. doi: 10.1136/jmg.12.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bosma PJ, Chowdhury JR, Bakker C, Gantla S, de Boer A, Oostra BA, Lindhout D, Tytgat GN, Jansen PL, Oude Elferink RP. The genetic basis of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert’s syndrome. N Engl J Med. 1995;333:1171–1175. doi: 10.1056/NEJM199511023331802. [DOI] [PubMed] [Google Scholar]
- 85.Hsieh TY, Shiu TY, Huang SM, Lin HH, Lee TC, Chen PJ, Chu HC, Chang WK, Jeng KS, Lai MM, et al. Molecular pathogenesis of Gilbert’s syndrome: decreased TATA-binding protein binding affinity of UGT1A1 gene promoter. Pharmacogenet Genomics. 2007;17:229–236. doi: 10.1097/FPC.0b013e328012d0da. [DOI] [PubMed] [Google Scholar]
- 86.Burchell B, Hume R. Molecular genetic basis of Gilbert’s syndrome. J Gastroenterol Hepatol. 1999;14:960–966. doi: 10.1046/j.1440-1746.1999.01984.x. [DOI] [PubMed] [Google Scholar]
- 87.Beutler E, Gelbart T, Demina A. Racial variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter: a balanced polymorphism for regulation of bilirubin metabolism? Proc Natl Acad Sci USA. 1998;95:8170–8174. doi: 10.1073/pnas.95.14.8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ki CS, Lee KA, Lee SY, Kim HJ, Cho SS, Park JH, Cho S, Sohn KM, Kim JW. Haplotype structure of the UDP-glucuronosyltransferase 1A1 (UGT1A1) gene and its relationship to serum total bilirubin concentration in a male Korean population. Clin Chem. 2003;49:2078–2081. doi: 10.1373/clinchem.2003.024174. [DOI] [PubMed] [Google Scholar]
- 89.Ando Y, Chida M, Nakayama K, Saka H, Kamataki T. The UGT1A1*28 allele is relatively rare in a Japanese population. Pharmacogenetics. 1998;8:357–360. doi: 10.1097/00008571-199808000-00010. [DOI] [PubMed] [Google Scholar]
- 90.Takeuchi K, Kobayashi Y, Tamaki S, Ishihara T, Maruo Y, Araki J, Mifuji R, Itani T, Kuroda M, Sato H, et al. Genetic polymorphisms of bilirubin uridine diphosphate-glucuronosyltransferase gene in Japanese patients with Crigler-Najjar syndrome or Gilbert’s syndrome as well as in healthy Japanese subjects. J Gastroenterol Hepatol. 2004;19:1023–1028. doi: 10.1111/j.1440-1746.2004.03370.x. [DOI] [PubMed] [Google Scholar]
- 91.Udomuksorn W, Elliot DJ, Lewis BC, Mackenzie PI, Yoovathaworn K, Miners JO. Influence of mutations associated with Gilbert and Crigler-Najjar type II syndromes on the glucuronidation kinetics of bilirubin and other UDP-glucuronosyltransferase 1A substrates. Pharmacogenet Genomics. 2007;17:1017–1029. doi: 10.1097/FPC.0b013e328256b1b6. [DOI] [PubMed] [Google Scholar]
- 92.Kadakol A, Ghosh SS, Sappal BS, Sharma G, Chowdhury JR, Chowdhury NR. Genetic lesions of bilirubin uridine-diphosphoglucuronate glucuronosyltransferase (UGT1A1) causing Crigler-Najjar and Gilbert syndromes: correlation of genotype to phenotype. Hum Mutat. 2000;16:297–306. doi: 10.1002/1098-1004(200010)16:4<297::AID-HUMU2>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 93.Monaghan G, McLellan A, McGeehan A, Li Volti S, Mollica F, Salemi I, Din Z, Cassidy A, Hume R, Burchell B. Gilbert’s syndrome is a contributory factor in prolonged unconjugated hyperbilirubinemia of the newborn. J Pediatr. 1999;134:441–446. doi: 10.1016/s0022-3476(99)70201-5. [DOI] [PubMed] [Google Scholar]
- 94.Maruo Y, Nishizawa K, Sato H, Sawa H, Shimada M. Prolonged unconjugated hyperbilirubinemia associated with breast milk and mutations of the bilirubin uridine diphosphate- glucuronosyltransferase gene. Pediatrics. 2000;106:E59. doi: 10.1542/peds.106.5.e59. [DOI] [PubMed] [Google Scholar]
- 95.Burchell B, Soars M, Monaghan G, Cassidy A, Smith D, Ethell B. Drug-mediated toxicity caused by genetic deficiency of UDP-glucuronosyltransferases. Toxicol Lett. 2000;112-113:333–340. doi: 10.1016/s0378-4274(99)00209-x. [DOI] [PubMed] [Google Scholar]
- 96.Maruo Y, Iwai M, Mori A, Sato H, Takeuchi Y. Polymorphism of UDP-glucuronosyltransferase and drug metabolism. Curr Drug Metab. 2005;6:91–99. doi: 10.2174/1389200053586064. [DOI] [PubMed] [Google Scholar]
- 97.Rotger M, Taffe P, Bleiber G, Gunthard HF, Furrer H, Vernazza P, Drechsler H, Bernasconi E, Rickenbach M, Telenti A. Gilbert syndrome and the development of antiretroviral therapy-associated hyperbilirubinemia. J Infect Dis. 2005;192:1381–1386. doi: 10.1086/466531. [DOI] [PubMed] [Google Scholar]
- 98.Zucker SD, Qin X, Rouster SD, Yu F, Green RM, Keshavan P, Feinberg J, Sherman KE. Mechanism of indinavir-induced hyperbilirubinemia. Proc Natl Acad Sci USA. 2001;98:12671–12676. doi: 10.1073/pnas.231140698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang D, Chando TJ, Everett DW, Patten CJ, Dehal SS, Humphreys WG. In vitro inhibition of UDP glucuronosyltransferases by atazanavir and other HIV protease inhibitors and the relationship of this property to in vivo bilirubin glucuronidation. Drug Metab Dispos. 2005;33:1729–1739. doi: 10.1124/dmd.105.005447. [DOI] [PubMed] [Google Scholar]
- 100.Ando Y, Saka H, Asai G, Sugiura S, Shimokata K, Kamataki T. UGT1A1 genotypes and glucuronidation of SN-38, the active metabolite of irinotecan. Ann Oncol. 1998;9:845–847. doi: 10.1023/a:1008438109725. [DOI] [PubMed] [Google Scholar]
- 101.Iyer L, Das S, Janisch L, Wen M, Ramírez J, Karrison T, Fleming GF, Vokes EE, Schilsky RL, Ratain MJ. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J. 2002;2:43–47. doi: 10.1038/sj.tpj.6500072. [DOI] [PubMed] [Google Scholar]
- 102.Strassburg CP. Pharmacogenetics of Gilbert’s syndrome. Pharmacogenomics. 2008;9:703–715. doi: 10.2217/14622416.9.6.703. [DOI] [PubMed] [Google Scholar]
- 103.Dubin IN, Johnson FB. Chronic idiopathic jaundice with unidentified pigment in liver cells; a new clinicopathologic entity with a report of 12 cases. Medicine (Baltimore) 1954;33:155–197. doi: 10.1097/00005792-195409000-00001. [DOI] [PubMed] [Google Scholar]
- 104.Sprinz H, Nelson RS. Persistent non-hemolytic hyperbilirubinemia associated with lipochrome-like pigment in liver cells: report of four cases. Ann Intern Med. 1954;41:952–962. doi: 10.7326/0003-4819-41-5-952. [DOI] [PubMed] [Google Scholar]
- 105.Shani M, Seligsohn U, Gilon E, Sheba C, Adam A. Dubin-Johnson syndrome in Israel. I. Clinical, laboratory, and genetic aspects of 101 cases. Q J Med. 1970;39:549–567. [PubMed] [Google Scholar]
- 106.Erlinger S, Dhumeaux D, Desjeux JF, Benhamou JP. Hepatic handling of unconjugated dyes in the Dubin-Johnson syndrome. Gastroenterology. 1973;64:106–110. [PubMed] [Google Scholar]
- 107.Swartz HM, Chen K, Roth JA. Further evidence that the pigment in the Dubin-Johnson syndrome is not melanin. Pigment Cell Res. 1987;1:69–75. doi: 10.1111/j.1600-0749.1987.tb00392.x. [DOI] [PubMed] [Google Scholar]
- 108.Hunter FM, Sparks RD, Flinner RL. Hepatitis with resulting mobilization of hepatic pigment in a patient with dubin-johnson syndrome. Gastroenterology. 1964;47:631–635. [PubMed] [Google Scholar]
- 109.Watanabe S, Nishioka M, Kodama T, Ando K, Numa Y, Fukumoto Y, Okita K, Takemoto T, Mizuta M. Clinicopathological studies of the Dubin-Johnson syndrome complicated with chronic hepatitis. Gastroenterol Jpn. 1982;17:576–584. doi: 10.1007/BF02779135. [DOI] [PubMed] [Google Scholar]
- 110.Kartenbeck J, Leuschner U, Mayer R, Keppler D. Absence of the canalicular isoform of the MRP gene-encoded conjugate export pump from the hepatocytes in Dubin-Johnson syndrome. Hepatology. 1996;23:1061–1066. doi: 10.1053/jhep.1996.v23.pm0008621134. [DOI] [PubMed] [Google Scholar]
- 111.Paulusma CC, Bosma PJ, Zaman GJ, Bakker CT, Otter M, Scheffer GL, Scheper RJ, Borst P, Oude Elferink RP. Congenital jaundice in rats with a mutation in a multidrug resistance-associated protein gene. Science. 1996;271:1126–1128. doi: 10.1126/science.271.5252.1126. [DOI] [PubMed] [Google Scholar]
- 112.Paulusma CC, Kool M, Bosma PJ, Scheffer GL, ter Borg F, Scheper RJ, Tytgat GN, Borst P, Baas F, Oude Elferink RP. A mutation in the human canalicular multispecific organic anion transporter gene causes the Dubin-Johnson syndrome. Hepatology. 1997;25:1539–1542. doi: 10.1002/hep.510250635. [DOI] [PubMed] [Google Scholar]
- 113.Wada M, Toh S, Taniguchi K, Nakamura T, Uchiumi T, Kohno K, Yoshida I, Kimura A, Sakisaka S, Adachi Y, et al. Mutations in the canilicular multispecific organic anion transporter (cMOAT) gene, a novel ABC transporter, in patients with hyperbilirubinemia II/Dubin-Johnson syndrome. Hum Mol Genet. 1998;7:203–207. doi: 10.1093/hmg/7.2.203. [DOI] [PubMed] [Google Scholar]
- 114.Toh S, Wada M, Uchiumi T, Inokuchi A, Makino Y, Horie Y, Adachi Y, Sakisaka S, Kuwano M. Genomic structure of the canalicular multispecific organic anion-transporter gene (MRP2/cMOAT) and mutations in the ATP-binding-cassette region in Dubin-Johnson syndrome. Am J Hum Genet. 1999;64:739–746. doi: 10.1086/302292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Uchiumi T, Hinoshita E, Haga S, Nakamura T, Tanaka T, Toh S, Furukawa M, Kawabe T, Wada M, Kagotani K, et al. Isolation of a novel human canalicular multispecific organic anion transporter, cMOAT2/MRP3, and its expression in cisplatin-resistant cancer cells with decreased ATP-dependent drug transport. Biochem Biophys Res Commun. 1998;252:103–110. doi: 10.1006/bbrc.1998.9546. [DOI] [PubMed] [Google Scholar]
- 116.Hulot JS, Villard E, Maguy A, Morel V, Mir L, Tostivint I, William-Faltaos D, Fernandez C, Hatem S, Deray G, et al. A mutation in the drug transporter gene ABCC2 associated with impaired methotrexate elimination. Pharmacogenet Genomics. 2005;15:277–285. doi: 10.1097/01213011-200505000-00002. [DOI] [PubMed] [Google Scholar]
- 117.Ahmed S, Vo NT, Thalhammer T, Thalhammer F, Gattringer KB, Jäger W. Involvement of Mrp2 (Abcc2) in biliary excretion of moxifloxacin and its metabolites in the isolated perfused rat liver. J Pharm Pharmacol. 2008;60:55–62. doi: 10.1211/jpp.60.1.0007. [DOI] [PubMed] [Google Scholar]
- 118.Pedersen JM, Matsson P, Bergström CA, Norinder U, Hoogstraate J, Artursson P. Prediction and identification of drug interactions with the human ATP-binding cassette transporter multidrug-resistance associated protein 2 (MRP2; ABCC2) J Med Chem. 2008;51:3275–3287. doi: 10.1021/jm7015683. [DOI] [PubMed] [Google Scholar]
- 119.Jedlitschky G, Hoffmann U, Kroemer HK. Structure and function of the MRP2 (ABCC2) protein and its role in drug disposition. Expert Opin Drug Metab Toxicol. 2006;2:351–366. doi: 10.1517/17425255.2.3.351. [DOI] [PubMed] [Google Scholar]
- 120.Zhou SF, Wang LL, Di YM, Xue CC, Duan W, Li CG, Li Y. Substrates and inhibitors of human multidrug resistance associated proteins and the implications in drug development. Curr Med Chem. 2008;15:1981–2039. doi: 10.2174/092986708785132870. [DOI] [PubMed] [Google Scholar]
- 121.Cebecauerova D, Jirasek T, Budisova L, Mandys V, Volf V, Novotna Z, Subhanova I, Hrebicek M, Elleder M, Jirsa M. Dual hereditary jaundice: simultaneous occurrence of mutations causing Gilbert’s and Dubin-Johnson syndrome. Gastroenterology. 2005;129:315–320. doi: 10.1053/j.gastro.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 122.Rotor B, Manahan L, Florentin A. Familial non-hemolytic jaundice with direct van den Bergh reaction. Acta medica Philippina. 1948;5:37–49. [Google Scholar]
- 123.Wolkoff AW, Wolpert E, Pascasio FN, Arias IM. Rotor’s syndrome. A distinct inheritable pathophysiologic entity. Am J Med. 1976;60:173–179. doi: 10.1016/0002-9343(76)90426-5. [DOI] [PubMed] [Google Scholar]
- 124.Wolpert E, Pascasio FM, Wolkoff AW, Arias IM. Abnormal sulfobromophthalein metabolism in Rotor’s syndrome and obligate heterozygotes. N Engl J Med. 1977;296:1099–1101. doi: 10.1056/NEJM197705122961907. [DOI] [PubMed] [Google Scholar]
- 125.van de Steeg E, Stránecký V, Hartmannová H, Nosková L, Hřebíček M, Wagenaar E, van Esch A, de Waart DR, Oude Elferink RP, Kenworthy KE, et al. Complete OATP1B1 and OATP1B3 deficiency causes human Rotor syndrome by interrupting conjugated bilirubin reuptake into the liver. J Clin Invest. 2012;122:519–528. doi: 10.1172/JCI59526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kalliokoski A, Niemi M. Impact of OATP transporters on pharmacokinetics. Br J Pharmacol. 2009;158:693–705. doi: 10.1111/j.1476-5381.2009.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kalliokoski A, Neuvonen M, Neuvonen PJ, Niemi M. The effect of SLCO1B1 polymorphism on repaglinide pharmacokinetics persists over a wide dose range. Br J Clin Pharmacol. 2008;66:818–825. doi: 10.1111/j.1365-2125.2008.03287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Niemi M, Pasanen MK, Neuvonen PJ. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev. 2011;63:157–181. doi: 10.1124/pr.110.002857. [DOI] [PubMed] [Google Scholar]
- 129.Iusuf D, Sparidans RW, van Esch A, Hobbs M, Kenworthy KE, van de Steeg E, Wagenaar E, Beijnen JH, Schinkel AH. Organic anion-transporting polypeptides 1a/1b control the hepatic uptake of pravastatin in mice. Mol Pharm. 2012;9:2497–2504. doi: 10.1021/mp300108c. [DOI] [PubMed] [Google Scholar]
- 130.Trauner M, Fickert P, Stauber RE. [New molecular aspects of cholestatic liver diseases] Z Gastroenterol. 1999;37:639–647. [PubMed] [Google Scholar]
- 131.Wagner M, Trauner M. Transcriptional regulation of hepatobiliary transport systems in health and disease: implications for a rationale approach to the treatment of intrahepatic cholestasis. Ann Hepatol. 2005;4:77–99. [PubMed] [Google Scholar]
- 132.Kojima H, Nies AT, König J, Hagmann W, Spring H, Uemura M, Fukui H, Keppler D. Changes in the expression and localization of hepatocellular transporters and radixin in primary biliary cirrhosis. J Hepatol. 2003;39:693–702. doi: 10.1016/s0168-8278(03)00410-0. [DOI] [PubMed] [Google Scholar]
- 133.Zollner G, Fickert P, Silbert D, Fuchsbichler A, Marschall HU, Zatloukal K, Denk H, Trauner M. Adaptive changes in hepatobiliary transporter expression in primary biliary cirrhosis. J Hepatol. 2003;38:717–727. doi: 10.1016/s0168-8278(03)00096-5. [DOI] [PubMed] [Google Scholar]
- 134.Pauli-Magnus C, Kerb R, Fattinger K, Lang T, Anwald B, Kullak-Ublick GA, Beuers U, Meier PJ. BSEP and MDR3 haplotype structure in healthy Caucasians, primary biliary cirrhosis and primary sclerosing cholangitis. Hepatology. 2004;39:779–791. doi: 10.1002/hep.20159. [DOI] [PubMed] [Google Scholar]
- 135.Takeyama Y, Sakisaka S. Hepatobiliary membrane transporters in primary biliary cirrhosis. Hepatol Res. 2012;42:120–130. doi: 10.1111/j.1872-034X.2011.00912.x. [DOI] [PubMed] [Google Scholar]
- 136.Chen HL, Liu YJ, Chen HL, Wu SH, Ni YH, Ho MC, Lai HS, Hsu WM, Hsu HY, Tseng HC, et al. Expression of hepatocyte transporters and nuclear receptors in children with early and late-stage biliary atresia. Pediatr Res. 2008;63:667–673. doi: 10.1203/PDR.0b013e318170a6b5. [DOI] [PubMed] [Google Scholar]
- 137.Oswald M, Kullak-Ublick GA, Paumgartner G, Beuers U. Expression of hepatic transporters OATP-C and MRP2 in primary sclerosing cholangitis. Liver. 2001;21:247–253. doi: 10.1034/j.1600-0676.2001.021004247.x. [DOI] [PubMed] [Google Scholar]
- 138.Kiang TK, Ensom MH, Chang TK. UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharmacol Ther. 2005;106:97–132. doi: 10.1016/j.pharmthera.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 139.Roth M, Obaidat A, Hagenbuch B. OATPs, OATs and OCTs: the organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br J Pharmacol. 2012;165:1260–1287. doi: 10.1111/j.1476-5381.2011.01724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Karlgren M, Vildhede A, Norinder U, Wisniewski JR, Kimoto E, Lai Y, Haglund U, Artursson P. Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): influence of protein expression on drug-drug interactions. J Med Chem. 2012;55:4740–4763. doi: 10.1021/jm300212s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Shitara Y. Clinical importance of OATP1B1 and OATP1B3 in drug-drug interactions. Drug Metab Pharmacokinet. 2011;26:220–227. doi: 10.2133/dmpk.DMPK-10-RV-094. [DOI] [PubMed] [Google Scholar]
- 142.Sissung TM, Baum CE, Kirkland CT, Gao R, Gardner ER, Figg WD. Pharmacogenetics of membrane transporters: an update on current approaches. Mol Biotechnol. 2010;44:152–167. doi: 10.1007/s12033-009-9220-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sadée W, Dai Z. Pharmacogenetics/genomics and personalized medicine. Hum Mol Genet. 2005;14 Spec No. 2:R207–R214. doi: 10.1093/hmg/ddi261. [DOI] [PubMed] [Google Scholar]