

Abstract
Oxygen free radical and lipid peroxides (oxidative stress) are highly reactive and represent very damaging compounds. Oxidative stress could be a major contributing factor to the tissue injury and fibrosis that characterize Crohn’s disease. An imbalance between increased reactive oxygen species levels and decreased antioxidant defenses occurs in Crohn’s patients. Decreased blood levels of vitamins C and E and decreased intestinal mucosal levels of CuZn superoxide dismutase, glutathione, vitamin A, C, E, and β-carotene have been reported for Crohn’s patients. Increased levels of proinflammatory cytokines, such as interleukin-1 and -8 and tumor necrosis factor, have been detected in inflammatory bowel disease. Oxidative stress significantly increased the production of neutrophils, chemokines, and interleukin-8. These effects were inhibited by antioxidant vitamins and arachidonic acid metabolite inhibitors in human intestinal smooth muscle cells isolated from the bowels of Crohn’s disease patients. The main pathological feature of Crohn’s disease is an infiltration of polymorphonuclear neutrophils and mononuclear cells into the affected part of the intestine. Activated neutrophils produce noxious substances that cause inflammation and tissue injury. Due to the physiological and biochemical actions of reactive oxygen species and lipid peroxides, many of the clinical and pathophysiological features of Crohn’s disease might be explained by an imbalance of increased reactive oxygen species and a net decrease of antioxidant molecules. This review describes the general concepts of free radical, lipid peroxide and antioxidant activities and eventually illustrates their interferences in the development of Crohn’s strictures.
Keywords: Crohn’s disease, Reactive oxygen species, Antioxidant enzymes, Lipid peroxide
Core tip: Crohn’s disease is associated with an imbalance, comprising increased reactive oxygen species (ROS) and decreased net antioxidant activity. A deficiency in antioxidant molecules could lead to increased levels of lipid peroxides or ROS, which could act locally or be secreted into the circulation to produce different systemic effects in the patient. Future research should address the question of whether ROS are involved in increasing the production of the different extracellular proteins by enhancing the transcriptions of certain genes using specific transcription factors.
INTRODUCTION
Crohn’s disease (CD) is a debilitating illness of the bowel characterized by chronic inflammation of unknown etiology[1]. In the United States and Europe, the incidence ranges from 6-20 cases per 100000 population per year, and the prevalence, from 90-300 per 100000 population. This disease most often occurs between the ages of 15 and 20, with a secondary peak between the ages of 55 and 60[2]. It can affect any level of the alimentary tract, but the terminal ileum and proximal colon are most frequently involved. A classical feature of CD is the sharp demarcation of the affected bowel segment from adjacent uninvolved segments[1]. This disease is characterized pathologically as a recurrent granulomatous inflammation of all layers of the bowel wall[2-4]. Patients usually suffer from abdominal pain, cachexia, anorexia, chronic diarrhea, intestinal obstruction, malnutrition, weight loss, and growth failure.
The precise etiology of CD remains unclear. It is thought that interactions among various factors, including genetic factors, host immune system and environmental factors (including diets and microbiological agents), play crucial roles in disturbing the intestinal homeostasis, leading to the dysregulated inflammatory responses of the gut. As inflammation is intimately related to the formation of reactive intermediates, including reactive oxygen species (ROS), oxidative stress has been proposed as a mechanism underlying the pathophysiology of CD. The main pathological feature of CD is an infiltration of polymorphonuclear neutrophils and mononuclear cells into the affected part of the intestine[5-10]. As the disease becomes more established, neutrophils infiltrate isolated crypts, forming abscesses in the affected mucosa and underlying tissue layers[1]. Neutrophils, like other leukocytes, produce noxious substances, such as ROS, tumor necrosis factor alpha (TNFα), interleukin-1 (IL-1) and protease[11-14]. It has been shown that an imbalance in the levels of proinflammatory and anti-inflammatory cytokines occurs in CD[13,14]. Increased levels of proinflammatory cytokines, such as IL-1, IL-8 and TNFα, can be detected in CD.
Reactive oxygen species may play an important role in CD. As will be discussed in the following sections, the ROS generated from activated leukocytes or from other sources may affect various biological molecules, generating tissue damage that could have been prevented by dietary antioxidants. This review describes the general concepts of free radical, lipid peroxide and antioxidant activities and eventually illustrates their interference into the development of Crohn’s strictures.
CONCEPTS OF FREE RADICALS
A free radical can be defined as any chemical species that has one or more unpaired electrons in the outermost orbital electron shell[15]. Due to these chemical changes in the free radical atoms, they are chemically reactive. ROS include superoxide (•O2-), the hydroxyl radical (•OH), the hydroperoxyl radical (•O2H), nitric oxide (NO•), and singlet oxygen (1O2)[16]. In general, most free radicals are derivatives of oxygen, such as superoxide.
During the normal aerobic metabolism, oxygen is the final electron acceptor at the end of the mitochondrial respiratory chain, during which it is fully reduced to water[17-19]. A small percentage of the electrons do not travel to the end of the electron transport chain in the mitochondrial membrane (cytochrome oxidase); they escape and react directly with oxygen in the early part of the chain, forming •O2-[20]. It has been estimated based on studies of isolated brain mitochondria that 1%-2% of the electrons that travel down the respiratory chain leak out and reduce oxygen to superoxide and its dismutation product, hydrogen peroxide (H2O2)[21,22]. Hydrogen peroxide is not a radical, but it serves as a precursor to the generation of the hydroxyl radical (•OH) in the presence of iron ions. Superoxide is also produced endogenously through the action of oxidase enzymes (e.g., xanthine oxidase) in postischemic tissues[23,24]. Redox cycling substrates (e.g., catecholamines), the cytochrome P450 system and soluble oxidases, such as nicotinamide-adenine dinucleotide phosphate (NADPH) oxidase in phagocyte cells (e.g., neutrophils and monocytes), are endogenous sources of superoxide production[17-19,25-27]. Arachidonic acid metabolizing enzymes (cyclooxygenase and lipoxygenase) are other sources of intracellular superoxide production[28-30]. Although superoxide serves as a precursor to many ROS (e.g., •OH, •O2H), it is not the most reactive of the species[15]. Figure 1 illustrates the chemical interconversions that are driven by •O2-. Superoxide reacts with hydrogen to form hydrogen peroxide by nonenzymatic or by superoxide dismutase-catalyzed dismutation. One of the most important reactions for this is the •O2- driven Fenton reaction, in which ferric or cupric ions (Fe3+, Cu2+), usually Fe3+, are reduced by superoxide, and then, the reduced metal ion reacts with H2O2 to form •OH[17]. Another important free radical, NO•, is synthesized from L-arginine by the enzyme nitric oxide synthase. NO• can interact with •O2- to generate another damaging molecule, peroxynitrite (ONOO-) (Figure 1). Once ROS are formed by the mechanisms explained in Figure 1, they can generate oxidative damage to a variety of biological molecules, such as lipids, proteins, and nucleic acids. The most important ROS involved in cellular oxidative damage is •OH. Prime targets for ROS are the unsaturated fatty acids in membrane lipids, which will be discussed in the lipid peroxidation section. Proteins may also be damaged by ROS. This damage can be achieved through different stages, starting with site-specific lesions in the protein structure that leads to fragmentation products and cross-linked reaction products with other cellular components[25-27,31-33]. This oxidative damage to proteins by ROS can cause enzymatic dysfunction. For example, oxidation of the active site of the α-protease inhibitor inactivates the protein itself, causing tissue damage[31]. Nucleic acids can also be affected by ROS. Oxidative damage to the DNA can occur after the attack of hydroxyl radicals on the sugar residues, yielding strand break products[31]. This may occur indirectly, when oxidative stress activates endonuclease, or directly, through the reaction of H2O2 with DNA-bound metal ions and the formation of •OH, which fragments the DNA at site-specific spots[15,31,34].
Figure 1.
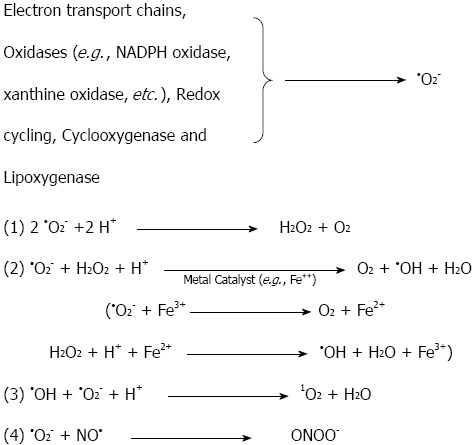
Reactions of superoxide (•O2-) that generate highly reactive oxygen species. •O2- can be dismutated into H2O2, which, in the presence of Fe2+, can generate the highly reactive hydroxyl radical, HO•. •O2- may react with NO• to form the strong pro-oxidant peroxynitrite (ONOO-); alternatively, it may react with HO• to form singlet oxygen (1O2); NADPH: Nicotinamide adenine dinucleotide phosphate.
ROS play an important role in increasing the intracellular levels of calcium. Ikebuchi et al[35] reported that the superoxide anion activates calcium channels in human amnion cells to increase the intracellular calcium concentrations. Hirosumi et al[36] and others have shown that the superoxide anion also increases the intracellular calcium concentration in endothelial cells[37,38]. ROS inhibit the Ca2+-ATPase, leading to elevated intracellular Ca2+ concentration[39]. Kimura et al[38] have found that •O2- attenuated smooth muscle contraction by impairing Ca2+ release-activated Ca2+ entry (CRAC), ATP-induced Ca2+ transient, and Ca2+ sensitivity in bovine aortic smooth muscle cells. Recent studies have shown that intracellular Ca2+ is involved in capsaicin-induced apoptosis. However, the molecular mechanisms of these intracellular Ca2+ signal pathways that lead to a sensitivity for the TNF-related induction of apoptosis remain unclear[40,41]. We will see in the next section that ROS are able to increase lipid peroxidation in the cell membrane to alter its permeability to ions and proteins.
CONCEPTS OF OXIDIZED LIPIDS
Oxidized lipids, also known as lipid hydroperoxides and lipid peroxides, can be formed in the presence of oxygen free radicals. The double bonds of polyunsaturated fatty acids (PUFA), such as linoleic acid, are most susceptible to oxidation by ROS[42]. The peroxidation chain reactions of membrane PUFA begin when oxygen free radicals extract hydrogen atoms from the methylene carbons adjacent to carbon-carbon double bonds. The presence of the double bond in the fatty acids weakens the carbon-hydrogen bonds in the carbon atoms adjacent to the double bonds, thus allowing hydrogen to be removed in the presence of ROS[15]. Because a hydrogen atom has only one electron, the loss of hydrogen from a methylene group leaves behind an unpaired electron on the carbon atom adjacent to the double bond, and the lipid becomes a lipid radical (L•). Next, the reaction of lipid radicals with oxygen (O2) molecules forms a peroxyl radical (LOO•). Peroxyl radicals are capable of abstracting a hydrogen atom from adjacent lipids to form lipid hydroperoxides (LOOH), sometimes called lipid peroxides. Once the chain of lipid peroxide formation is initiated, it becomes self-propagating. These events are illustrated in Figure 2. Reduced iron (Fe2+, Fe3+) complexes can react with lipid peroxides to form strong oxidizing agents, such as alkoxyl radicals (LO•) and LOO• (Figure 2D and E).
Figure 2.
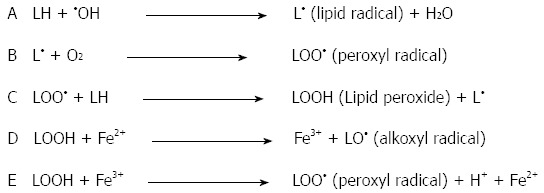
Initiation and propagation of the free-radical chain reaction of lipid peroxidation. Reactions D and E represent the metal-catalyzed Fenton reaction.
Lipid peroxides and oxygen radicals are responsible for many of the damaging reactions in the cell. They stimulate the peroxidation reactions that are toxic to cells and cell membranes. They can damage biological membranes, make the membrane leaky, and eventually cause complete membrane breakdown[43]. Furthermore, endothelial cell injury, the enhanced adhesion and activation of neutrophils, platelet aggregation, an increased uptake of low density lipoproteins by the endothelium, increased intracellular calcium and increased production of toxic aldehydes, as well as many other cellular dysfunctions, are due to the effects of LOOH and oxygen free radicals[42]. The breakdown products of lipid peroxidation, such as malondialdehyde, can affect membrane proteins by cross-linkage, rendering them useless as receptors or enzymes[44]. These different lipid peroxide effects can lead to cellular dysfunction and may result in the deaths of the affected cells.
CONCEPTS OF ANTIOXIDANTS
An antioxidant is any substance or compound that scavenges oxygen free radicals or inhibits the oxidation process in the cell[45]. The level of antioxidant defenses available within the cells and extracellularly should be sufficient to oppose the toxic effects of lipid peroxides and to maintain normal physiology. This balance can be lost due to the over-production of free radicals or the inadequate intake of nutrients containing antioxidant molecules. Antioxidants can be considered plasma antioxidants and intracellular antioxidants.
The major role of plasma antioxidant defense is to bind transition metal ions, such as iron and copper, thereby lowering their plasma concentration and capacity to stimulate free radical reactions[15]. This prevents the formation of hydroxyl radicals of the very potent variety that is able to generate lipid peroxides. This is achieved by plasma antioxidants, such as transferrin, lactoferrin, ceruloplasmin, albumin, uric acid, haptoglobins, and hemopexin[45]. Ascorbic acid is also a plasma antioxidant that can scavenge water-soluble peroxyl radicals and other ROS[17-19]. This is accomplished through the completion of two reactions. First, the ascorbic acid is converted into the ascorbyl radical and then to dehydroascorbate. Ascorbic acid can also reduce α-tocopheryl radicals to α-tocopherol at the cell membrane surface[46]. Vitamin E (α-tocopherol), a lipid peroxidation, chain-breaking antioxidant, is localized in membranes and lipoproteins[17]. It is the most important lipid-soluble antioxidant in the human plasma[47-50]. Vitamin E usually donates a hydrogen atom to the peroxyl radical and prevents the propagation of the chain reaction of lipid peroxidation. Each tocopherol can react with two LOO•, the first LOO• reacts with α-tocopherol to form the α-tocopherol radical that reacts with the second LOO• to form a stable adduct (LOO-α-tocopherol)[45]. Decreased blood and mucosal levels of vitamins A, C, E and β-carotene were reported in Crohn’s patients[51-60]. Reimund et al[61] showed that antioxidants inhibited the production of inflammatory cytokines, such as IL-1 and TNFα, in the colonic mucosa of inflammatory bowel disease patients. Vitamin C and E inhibited the levels of oxidative stress in human intestinal smooth muscle (HISM) cells isolated from Crohn’s bowels[62].
The main intracellular antioxidant enzymes are superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), and catalase. Human plasma has very little or no catalase, but it contains low activities of SOD and GSH-Px[15]. SOD is localized in both the cytosol (CuZn-SOD) and the mitochondria and acts to dismutate superoxides into hydrogen peroxide and molecular oxygen[17]. SOD is considered as the major intracellular enzyme because it is capable of reducing the most abundant free radical, •O2-. Catalase reacts rapidly with hydrogen peroxide, a precursor of •OH in the presence of iron, and converts it into water and molecular oxygen. Selenium glutathione peroxidase also converts hydrogen peroxide into water and lipid hydroperoxides into water and harmless fatty acid alcohols[45]. Decreased intestinal mucosal levels of CuZn superoxide dismutase and glutathione were reported in Crohn’s patients[63-69].
OXIDANT STRESS IN CROHN’S DISEASE
Oxidant stress is a major factor in Crohn’s disease. Reactive oxygen species can cause DNA modifications and damage, as briefly discussed in the previous section. The major sign of these modifications is the formation of 8-hydroxyguanine (8-OHDG). It was reported that 8-OHDG levels were significantly higher in the inflamed part of the bowel of Crohn’s patient[70]. These data show an important indication of the role and existence of ROS in the pathogenesis of Crohn’s disease.
There is direct and indirect evidence suggesting that the chronically inflamed intestines of Crohn’s patients may be subjected to considerable oxidative stress. The direct evidence includes the demonstration of the increased production of ROS in response to various stimuli by phagocytic cells (neutrophils, monocytes, and macrophages) isolated from the inflamed bowels of patients with IBD (both Crohn’s disease and ulcerative colitis)[7,71-73]. Kitahora et al[74] have demonstrated that monocytes from patients with Crohn’s disease release large amounts of ROS during stimulation. It is well known that active IBD is characterized by persistent neutrophil infiltration into the injured site. The influx of neutrophils and macrophages into the bowel wall during inflammation causes a large degree of tissue damage due to their release of myeloperoxidase, which constitutes 5% of their total protein load. Myeloperoxidase metabolizes H2O2 and chloride ions to form the potent oxidizing agent hypochlorous acid, commonly known as bleach, which is thought to be 100-1000 times more toxic than superoxide and hydrogen peroxide. Inflammatory phagocytes are activated by certain pro-inflammatory agents, such as leukotriene B4 (LTB4), platelet-activating factor (PAF), immune complexes, and bacterial products. As a result, the phagocytes release large amounts of ROS into the extracellular space[72-75]. Increased synthesis of LTB4 and PAF was detected in mucosal samples obtained from patients with active IBD[76-80]. Alzoghaibi et al[81] showed a significant increase in the production of the neutrophil-attracting chemokine, IL-8, LTB4 and other arachidonic acid metabolites in isolated HISM cells from Crohn’s strictures after stimulation by linoleic acid or oxidized linoleic acid (OxLA). The cyclooxygenase and lipoxygenase enzymes generate ROS when activated. Both cyclooxygenase and lipoxygenase have been involved in the production of IL-8 from HISM cells (Figure 3). OxLA increased the levels of lipid peroxidation, an indicator of oxidative stress, as quantified by thiobarbituric acid reactive substances in HISM cells. These pro-inflammatory mediators activate certain receptors on the phagocytes’ plasma membranes, resulting in the activation of plasma membrane-associated NADPH oxidase. NADPH oxidase reduces oxygen molecules to superoxide on the cell membrane and releases the superoxide extracellularly.
Figure 3.
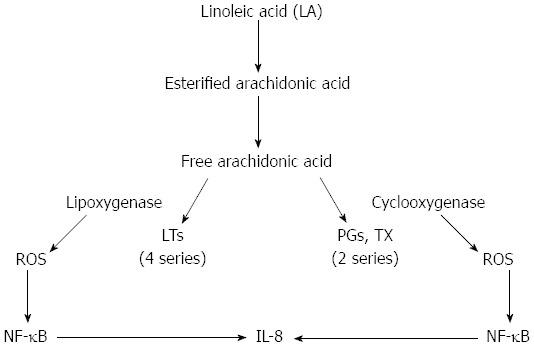
Two proposed mechanisms through which linoleic acid could increase the production of interleukin-8 via the activation of arachidonic acid pathways. By the generation of reactive oxygen species (ROS) from the cyclooxygenase and lipoxygenase enzymes to activate Nuclear factor-κB (NF-κB), increasing interleukin (IL)-8 production; or by the use of AA metabolites to increase IL-8 production. LTs: Leukotrienes; Tx: Thromboxane.
Most of the evidence for the role of the ROS in Crohn’s disease is indirect. Under normal physiological conditions, dietary and enzymatic antioxidants protect tissues from the damaging effects of ROS. However, a state of oxidative stress may exist when there is an imbalance in the levels of antioxidants and ROS. When the levels of antioxidants are low and ROS are high, increased cellular oxidative stress is present; such is the case in Crohn’s disease[63,82-89]. The intestinal mucosa is particularly favored, as it is the location of many different enzymes necessary for the production of large amounts of ROS, such as xanthine oxidase, cyclooxygenase, and 5-lipoxygenase[75]. In addition to these sources of ROS, the number of the main sources of ROS (phagocyte cells) is increased in IBD[72,82]. For example, neutrophils account for up to 20% of the cells found in the lamina propria in active IBD[86]. On the other hand, Crohn’s patients have low blood levels of vitamin C and vitamin E[63]. They also have low levels of CuZn superoxide dismutase, glutathione peroxidase, catalase, vitamin A and β-carotene in their intestinal mucosa, submucosa and muscularis-serosa[72,82-85,89]. This coincidence of increased levels of ROS and decreased levels of antioxidant defenses results in a state of oxidative stress in Crohn’s strictures. A study of mucosal biopsies obtained from patients with IBD by Simmonds et al[86] demonstrated a significant increase in chemiluminescence, a very sensitive measurement for ROS, in comparison to controls. The addition of various antioxidant enzymes and scavengers, such as SOD, catalase, dimethyl sulfoxide (hydroxyl radical scavenger) and taurine (hydrochloride scavenger), confirmed that this increase in chemiluminescence was due to ROS. A study of HISM cells demonstrated a significant inhibition in TBARS after the cells had been exposed to antioxidant vitamins, such as C and E. Vitamin C and E also inhibited the effect of oxidized linoleic acid on the production of the most potent neutrophil-attracting chemokine, IL-8, from HISM cells[62].
Clinical data suggest that ROS may play a role in the pathogenesis of Crohn’s disease. Emerit and coworkers reported that intramuscular injections of bovine CuZn-SOD had a beneficial effect in attenuating mucosal inflammation and injury in Crohn’s patients[90,91]. The beneficial effect of this antioxidant enzyme is a strong indication of the role of ROS in Crohn’s disease. Another piece of evidence of the involvement of ROS in this disease is the use of 5-aminosalicylic acid (5-ASA) in treating patients with IBD. It is well known that the oral administration of sulfasalazine (SAZ) is capable of reducing the mucosal injury and inflammation associated with Crohn’s disease. 5-ASA is the pharmacologically active moiety of SAZ[72,75]. It has been suggested that 5-ASA protects the gut mucosa by inhibiting cyclooxygenase and lipoxygenase activities. Several studies have reported that 5-ASA is a potent antioxidant and free radical scavenger. Research from several laboratories has demonstrated that 5-ASA is very effective in scavenging hydroxyl radicals, interacting with superoxides and causing the rapid decomposition of this free radical[92,93]. Nielsen et al[71] demonstrated a high rate of lipid peroxidation in the gut mucosa obtained from patients with IBD. They also reported a significant inhibition in lipid peroxidation among patients who had been treated with SAZ for 5 wk[94]. These data implicate the involvement of ROS in Crohn’s disease due to the antioxidant and free radical scavenging actions of 5-ASA or due to its role as an inhibitor of cyclooxygenase and lipoxygenase, which are enzymatic sources of ROS.
In summary, ROS could be an important factor in Crohn’s disease. Crohn’s disease is associated with an imbalance, comprising increased ROS and decreased net antioxidant activity. A deficiency in antioxidant molecules could lead to increased levels of lipid peroxides or ROS, which could act locally or be secreted into the circulation to produce different systemic effects in the patient. Obviously, more work is needed in this area to demonstrate the effects of ROS on the production of other neutrophil chemokines from different layers of Crohn’s strictures. Future research should also address the question of whether ROS are involved in increasing the production of the different extracellular proteins, such as collagen, that lead to increased thickness of the bowel wall and deceased bowel diameters by enhancing the transcriptions of certain genes using specific transcription factors.
Footnotes
P- Reviewers Bourke B, Grassi R, Maric I S- Editor Gou SX L- Editor A E- Editor Ma S
References
- 1.Cotran RS, Kumar V, Collins T. Robbins Pathologic Basis of Disease. 6th ed. Philadelphia: WB Saunders Company; 1999. pp. 816–821. [Google Scholar]
- 2.Porth C. Pathophysiology: Concepts of Altered Health States. 8th ed. New York: JB Lippincott Company; 1990. pp. 707–708. [Google Scholar]
- 3.Graham MF. Stricture formation: pathophysiologic and therapeutic concepts. Inflammatory Bowel Disease. RP MacDermott and WF Stenson. New York: Elsevier; 1992. pp. 323–335. [Google Scholar]
- 4.Graham MF. Stricture formation in Crohn’s disease: Wound healing in the intestine. In: Targan SR, Shanahan F, editors. Inflammatory Bowel Disease: From Bench to Bedside. Baltimore: Williams and Wilkins; 1994. pp. 171–179. [Google Scholar]
- 5.Yamada T, Grisham MB. Role of neutrophil-derived oxidants in the pathogenesis of intestinal inflammation. Klin Wochenschr. 1991;69:988–994. doi: 10.1007/BF01645144. [DOI] [PubMed] [Google Scholar]
- 6.Ina K, Kusugami K, Yamaguchi T, Imada A, Hosokawa T, Ohsuga M, Shinoda M, Ando T, Ito K, Yokoyama Y. Mucosal interleukin-8 is involved in neutrophil migration and binding to extracellular matrix in inflammatory bowel disease. Am J Gastroenterol. 1997;92:1342–1346. [PubMed] [Google Scholar]
- 7.Kruidenier L, Kuiper I, Lamers CB, Verspaget HW. Intestinal oxidative damage in inflammatory bowel disease: semi-quantification, localization, and association with mucosal antioxidants. J Pathol. 2003;201:28–36. doi: 10.1002/path.1409. [DOI] [PubMed] [Google Scholar]
- 8.Kristjánsson G, Venge P, Wanders A, Lööf L, Hällgren R. Clinical and subclinical intestinal inflammation assessed by the mucosal patch technique: studies of mucosal neutrophil and eosinophil activation in inflammatory bowel diseases and irritable bowel syndrome. Gut. 2004;53:1806–1812. doi: 10.1136/gut.2003.036418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kucharzik T, Walsh SV, Chen J, Parkos CA, Nusrat A. Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am J Pathol. 2001;159:2001–2009. doi: 10.1016/S0002-9440(10)63051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kraft M, Riedel S, Maaser C, Kucharzik T, Steinbuechel A, Domschke W, Luegering N. IFN-gamma synergizes with TNF-alpha but not with viable H. pylori in up-regulating CXC chemokine secretion in gastric epithelial cells. Clin Exp Immunol. 2001;126:474–481. doi: 10.1046/j.1365-2249.2001.01634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weiss SJ, LoBuglio AF. Phagocyte-generated oxygen metabolites and cellular injury. Lab Invest. 1982;47:5–18. [PubMed] [Google Scholar]
- 12.Ward PA, Mulligan MS. New insights into mechanisms of oxyradical and neutrophil mediated lung injury. Klin Wochenschr. 1991;69:1009–1011. doi: 10.1007/BF01645148. [DOI] [PubMed] [Google Scholar]
- 13.Rogler G, Andus T. Cytokines in inflammatory bowel disease. World J Surg. 1998;22:382–389. doi: 10.1007/s002689900401. [DOI] [PubMed] [Google Scholar]
- 14.José León A, Garrote JA, Arranz E. [Cytokines in the pathogenesis of inflammatory bowel diseases] Med Clin (Barc) 2006;127:145–152. doi: 10.1157/13090382. [DOI] [PubMed] [Google Scholar]
- 15.Halliwell B, Gutteridge JM. Role of free radicals and catalytic metal ions in human disease: an overview. Methods Enzymol. 1990;186:1–85. doi: 10.1016/0076-6879(90)86093-b. [DOI] [PubMed] [Google Scholar]
- 16.Rice-Evans C, Burdon R. Free radical-lipid interactions and their pathological consequences. Prog Lipid Res. 1993;32:71–110. doi: 10.1016/0163-7827(93)90006-i. [DOI] [PubMed] [Google Scholar]
- 17.Frei B. Reactive oxygen species and antioxidant vitamins: mechanisms of action. Am J Med. 1994;97:5S–13S; discussion 22S-28S. doi: 10.1016/0002-9343(94)90292-5. [DOI] [PubMed] [Google Scholar]
- 18.Marchioli R, Schweiger C, Levantesi G, Tavazzi L, Valagussa F. Antioxidant vitamins and prevention of cardiovascular disease: epidemiological and clinical trial data. Lipids. 2001;36 Suppl:S53–S63. doi: 10.1007/s11745-001-0683-y. [DOI] [PubMed] [Google Scholar]
- 19.Wendland BE, Aghdassi E, Tam C, Carrrier J, Steinhart AH, Wolman SL, Baron D, Allard JP. Lipid peroxidation and plasma antioxidant micronutrients in Crohn disease. Am J Clin Nutr. 2001;74:259–264. doi: 10.1093/ajcn/74.2.259. [DOI] [PubMed] [Google Scholar]
- 20.Halliwell B. Antioxidants and human disease: a general introduction. Nutr Rev. 1997;55:S44–S9; discussion S44-S9;. doi: 10.1111/j.1753-4887.1997.tb06100.x. [DOI] [PubMed] [Google Scholar]
- 21.Frei B, Stocker R, Ames BN. Antioxidant defenses and lipid peroxidation in human blood plasma. Proc Natl Acad Sci USA. 1988;85:9748–9752. doi: 10.1073/pnas.85.24.9748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas SR, Davies MJ, Stocker R. Oxidation and antioxidation of human low-density lipoprotein and plasma exposed to 3-morpholinosydnonimine and reagent peroxynitrite. Chem Res Toxicol. 1998;11:484–494. doi: 10.1021/tx970173a. [DOI] [PubMed] [Google Scholar]
- 23.McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med. 1985;312:159–163. doi: 10.1056/NEJM198501173120305. [DOI] [PubMed] [Google Scholar]
- 24.Cañas PE. The role of xanthine oxidase and the effects of antioxidants in ischemia reperfusion cell injury. Acta Physiol Pharmacol Ther Latinoam. 1999;49:13–20. [PubMed] [Google Scholar]
- 25.Machlin LJ, Bendich A. Free radical tissue damage: protective role of antioxidant nutrients. FASEB J. 1987;1:441–445. [PubMed] [Google Scholar]
- 26.Fang YZ, Yang S, Wu G. Free radicals, antioxidants, and nutrition. Nutrition. 2002;18:872–879. doi: 10.1016/s0899-9007(02)00916-4. [DOI] [PubMed] [Google Scholar]
- 27.Gaetke LM, Chow CK. Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology. 2003;189:147–163. doi: 10.1016/s0300-483x(03)00159-8. [DOI] [PubMed] [Google Scholar]
- 28.Kukreja RC, Kontos HA, Hess ML, Ellis EF. PGH synthase and lipoxygenase generate superoxide in the presence of NADH or NADPH. Circ Res. 1986;59:612–619. doi: 10.1161/01.res.59.6.612. [DOI] [PubMed] [Google Scholar]
- 29.Didion SP, Faraci FM. Effects of NADH and NADPH on superoxide levels and cerebral vascular tone. Am J Physiol Heart Circ Physiol. 2002;282:H688–H695. doi: 10.1152/ajpheart.00576.2001. [DOI] [PubMed] [Google Scholar]
- 30.Grunden AM, Jenney FE, Ma K, Ji M, Weinberg MV, Adams MW. In vitro reconstitution of an NADPH-dependent superoxide reduction pathway from Pyrococcus furiosus. Appl Environ Microbiol. 2005;71:1522–1530. doi: 10.1128/AEM.71.3.1522-1530.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Floyd RA. Role of oxygen free radicals in carcinogenesis and brain ischemia. FASEB J. 1990;4:2587–2597. [PubMed] [Google Scholar]
- 32.Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J. Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem. 2004;266:37–56. doi: 10.1023/b:mcbi.0000049134.69131.89. [DOI] [PubMed] [Google Scholar]
- 33.Lim P, Wuenschell GE, Holland V, Lee DH, Pfeifer GP, Rodriguez H, Termini J. Peroxyl radical mediated oxidative DNA base damage: implications for lipid peroxidation induced mutagenesis. Biochemistry. 2004;43:15339–15348. doi: 10.1021/bi048276x. [DOI] [PubMed] [Google Scholar]
- 34.Darley-Usmar V, Halliwell B. Blood radicals: reactive nitrogen species, reactive oxygen species, transition metal ions, and the vascular system. Pharm Res. 1996;13:649–662. doi: 10.1023/a:1016079012214. [DOI] [PubMed] [Google Scholar]
- 35.Ikebuchi Y, Masumoto N, Tasaka K, Koike K, Kasahara K, Miyake A, Tanizawa O. Superoxide anion increases intracellular pH, intracellular free calcium, and arachidonate release in human amnion cells. J Biol Chem. 1991;266:13233–13237. [PubMed] [Google Scholar]
- 36.Hirosumi J, Ouchi Y, Watanabe M, Kusunoki J, Nakamura T, Orimo H. Effect of superoxide and lipid peroxide on cytosolic free calcium concentration in cultured pig aortic endothelial cells. Biochem Biophys Res Commun. 1988;152:301–307. doi: 10.1016/s0006-291x(88)80714-9. [DOI] [PubMed] [Google Scholar]
- 37.Az-ma T, Saeki N, Yuge O. Cytosolic Ca2+ movements of endothelial cells exposed to reactive oxygen intermediates: role of hydroxyl radical-mediated redox alteration of cell-membrane Ca2+ channels. Br J Pharmacol. 1999;126:1462–1470. doi: 10.1038/sj.bjp.0702438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kimura C, Cheng W, Hisadome K, Wang YP, Koyama T, Karashima Y, Oike M, Ito Y. Superoxide anion impairs contractility in cultured aortic smooth muscle cells. Am J Physiol Heart Circ Physiol. 2002;283:H382–H390. doi: 10.1152/ajpheart.00574.2001. [DOI] [PubMed] [Google Scholar]
- 39.Brott T, Bogousslavsky J. Treatment of acute ischemic stroke. N Engl J Med. 2000;343:710–722. doi: 10.1056/NEJM200009073431007. [DOI] [PubMed] [Google Scholar]
- 40.Lee MJ, Kee KH, Suh CH, Lim SC, Oh SH. Capsaicin-induced apoptosis is regulated by endoplasmic reticulum stress- and calpain-mediated mitochondrial cell death pathways. Toxicology. 2009;264:205–214. doi: 10.1016/j.tox.2009.08.012. [DOI] [PubMed] [Google Scholar]
- 41.Moon DO, Kang CH, Kang SH, Choi YH, Hyun JW, Chang WY, Kang HK, Koh YS, Maeng YH, Kim YR, et al. Capsaicin sensitizes TRAIL-induced apoptosis through Sp1-mediated DR5 up-regulation: involvement of Ca(2+) influx. Toxicol Appl Pharmacol. 2012;259:87–95. doi: 10.1016/j.taap.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 42.Walsh SW. Lipid peroxidation in pregnancy. Hypertension in Pregnancy. 1994;13:1–32. [Google Scholar]
- 43.Halliwell B. Free radicals, antioxidants, and human disease: curiosity, cause, or consequence? Lancet. 1994;344:721–724. doi: 10.1016/s0140-6736(94)92211-x. [DOI] [PubMed] [Google Scholar]
- 44.Schmidley JW. Free radicals in central nervous system ischemia. Stroke. 1990;21:1086–1090. doi: 10.1161/01.str.21.7.1086. [DOI] [PubMed] [Google Scholar]
- 45.Krinsky NI. Mechanism of action of biological antioxidants. Proc Soc Exp Biol Med. 1992;200:248–254. doi: 10.3181/00379727-200-43429. [DOI] [PubMed] [Google Scholar]
- 46.May JM. Is ascorbic acid an antioxidant for the plasma membrane? FASEB J. 1999;13:995–1006. doi: 10.1096/fasebj.13.9.995. [DOI] [PubMed] [Google Scholar]
- 47.Ingold KU, Webb AC, Witter D, Burton GW, Metcalfe TA, Muller DP. Vitamin E remains the major lipid-soluble, chain-breaking antioxidant in human plasma even in individuals suffering severe vitamin E deficiency. Arch Biochem Biophys. 1987;259:224–225. doi: 10.1016/0003-9861(87)90489-9. [DOI] [PubMed] [Google Scholar]
- 48.Maes M, De Vos N, Pioli R, Demedts P, Wauters A, Neels H, Christophe A. Lower serum vitamin E concentrations in major depression. Another marker of lowered antioxidant defenses in that illness. J Affect Disord. 2000;58:241–246. doi: 10.1016/s0165-0327(99)00121-4. [DOI] [PubMed] [Google Scholar]
- 49.Owen AJ, Batterham MJ, Probst YC, Grenyer BF, Tapsell LC. Low plasma vitamin E levels in major depression: diet or disease? Eur J Clin Nutr. 2005;59:304–306. doi: 10.1038/sj.ejcn.1602072. [DOI] [PubMed] [Google Scholar]
- 50.Valk EE, Hornstra G. Relationship between vitamin E requirement and polyunsaturated fatty acid intake in man: a review. Int J Vitam Nutr Res. 2000;70:31–42. doi: 10.1024/0300-9831.70.2.31. [DOI] [PubMed] [Google Scholar]
- 51.Fernandez-Banares F, Abad-Lacruz A, Xiol X, Gine JJ, Dolz C, Cabre E, Esteve M, Gonzalez-Huix F, Gassull MA. Vitamin status in patients with inflammatory bowel disease. Am J Gastroenterol. 1989;84:744–748. [PubMed] [Google Scholar]
- 52.Waśko-Czopnik D, Paradowski L. The influence of deficiencies of essential trace elements and vitamins on the course of Crohn’s disease. Adv Clin Exp Med. 2012;21:5–11. [PubMed] [Google Scholar]
- 53.Abreu MT, Kantorovich V, Vasiliauskas EA, Gruntmanis U, Matuk R, Daigle K, Chen S, Zehnder D, Lin YC, Yang H, et al. Measurement of vitamin D levels in inflammatory bowel disease patients reveals a subset of Crohn’s disease patients with elevated 1,25-dihydroxyvitamin D and low bone mineral density. Gut. 2004;53:1129–1136. doi: 10.1136/gut.2003.036657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tajika M, Matsuura A, Nakamura T, Suzuki T, Sawaki A, Kato T, Hara K, Ookubo K, Yamao K, Kato M, et al. Risk factors for vitamin D deficiency in patients with Crohn’s disease. J Gastroenterol. 2004;39:527–533. doi: 10.1007/s00535-003-1338-x. [DOI] [PubMed] [Google Scholar]
- 55.Reimund JM, Arondel Y, Escalin G, Finck G, Baumann R, Duclos B. Immune activation and nutritional status in adult Crohn’s disease patients. Dig Liver Dis. 2005;37:424–431. doi: 10.1016/j.dld.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 56.Schoon EJ, Müller MC, Vermeer C, Schurgers LJ, Brummer RJ, Stockbrügger RW. Low serum and bone vitamin K status in patients with longstanding Crohn’s disease: another pathogenetic factor of osteoporosis in Crohn’s disease? Gut. 2001;48:473–477. doi: 10.1136/gut.48.4.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ghishan FK, Kiela PR. Advances in the understanding of mineral and bone metabolism in inflammatory bowel diseases. Am J Physiol Gastrointest Liver Physiol. 2011;300:G191–G201. doi: 10.1152/ajpgi.00496.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kuroki F, Iida M, Tominaga M, Matsumoto T, Hirakawa K, Sugiyama S, Fujishima M. Multiple vitamin status in Crohn’s disease. Correlation with disease activity. Dig Dis Sci. 1993;38:1614–1618. doi: 10.1007/BF01303168. [DOI] [PubMed] [Google Scholar]
- 59.Kuroki F, Iida M, Tominaga M, Matsumoto T, Kanamoto K, Fujishima M. Is vitamin E depleted in Crohn’s disease at initial diagnosis? Dig Dis. 1994;12:248–254. doi: 10.1159/000171459. [DOI] [PubMed] [Google Scholar]
- 60.Bousvaros A, Zurakowski D, Duggan C, Law T, Rifai N, Goldberg NE, Leichtner AM. Vitamins A and E serum levels in children and young adults with inflammatory bowel disease: effect of disease activity. J Pediatr Gastroenterol Nutr. 1998;26:129–135. doi: 10.1097/00005176-199802000-00002. [DOI] [PubMed] [Google Scholar]
- 61.Reimund JM, Allison AC, Muller CD, Dumont S, Kenney JS, Baumann R, Duclos B, Poindron P. Antioxidants inhibit the in vitro production of inflammatory cytokines in Crohn’s disease and ulcerative colitis. Eur J Clin Invest. 1998;28:145–150. doi: 10.1046/j.1365-2362.1998.00257.x. [DOI] [PubMed] [Google Scholar]
- 62.Alzoghaibi MA, Walsh SW, Willey A, Fowler AA, Graham MF. Linoleic acid, but not oleic acid, upregulates the production of interleukin-8 by human intestinal smooth muscle cells isolated from patients with Crohn’s disease. Clin Nutr. 2003;22:529–535. doi: 10.1016/s0261-5614(03)00083-9. [DOI] [PubMed] [Google Scholar]
- 63.Buffinton GD, Doe WF. Altered ascorbic acid status in the mucosa from inflammatory bowel disease patients. Free Radic Res. 1995;22:131–143. doi: 10.3109/10715769509147535. [DOI] [PubMed] [Google Scholar]
- 64.Hengstermann S, Valentini L, Schaper L, Buning C, Koernicke T, Maritschnegg M, Buhner S, Tillinger W, Regano N, Guglielmi F, et al. Altered status of antioxidant vitamins and fatty acids in patients with inactive inflammatory bowel disease. Clin Nutr. 2008;27:571–578. doi: 10.1016/j.clnu.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 65.Geerling BJ, Badart-Smook A, Stockbrügger RW, Brummer RJ. Comprehensive nutritional status in patients with long-standing Crohn disease currently in remission. Am J Clin Nutr. 1998;67:919–926. doi: 10.1093/ajcn/67.5.919. [DOI] [PubMed] [Google Scholar]
- 66.Filippi J, Al-Jaouni R, Wiroth JB, Hébuterne X, Schneider SM. Nutritional deficiencies in patients with Crohn’s disease in remission. Inflamm Bowel Dis. 2006;12:185–191. doi: 10.1097/01.MIB.0000206541.15963.c3. [DOI] [PubMed] [Google Scholar]
- 67.Rath HC, Caesar I, Roth M, Schölmerich J. [Nutritional deficiencies and complications in chronic inflammatory bowel diseases] Med Klin (Munich) 1998;93:6–10. doi: 10.1007/BF03045033. [DOI] [PubMed] [Google Scholar]
- 68.Valentini L, Schaper L, Buning C, Hengstermann S, Koernicke T, Tillinger W, Guglielmi FW, Norman K, Buhner S, Ockenga J, et al. Malnutrition and impaired muscle strength in patients with Crohn’s disease and ulcerative colitis in remission. Nutrition. 2008;24:694–702. doi: 10.1016/j.nut.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 69.Geerling BJ, v Houwelingen AC, Badart-Smook A, Stockbrügger RW, Brummer RJ. The relation between antioxidant status and alterations in fatty acid profile in patients with Crohn disease and controls. Scand J Gastroenterol. 1999;34:1108–1116. doi: 10.1080/003655299750024913. [DOI] [PubMed] [Google Scholar]
- 70.Lih-Brody L, Powell SR, Collier KP, Reddy GM, Cerchia R, Kahn E, Weissman GS, Katz S, Floyd RA, McKinley MJ, et al. Increased oxidative stress and decreased antioxidant defenses in mucosa of inflammatory bowel disease. Dig Dis Sci. 1996;41:2078–2086. doi: 10.1007/BF02093613. [DOI] [PubMed] [Google Scholar]
- 71.Nielsen OH, Ahnfelt-Rønne I. Involvement of oxygen-derived free radicals in the pathogenesis of chronic inflammatory bowel disease. Klin Wochenschr. 1991;69:995–1000. doi: 10.1007/BF01645145. [DOI] [PubMed] [Google Scholar]
- 72.Grisham MB. Role of reactive oxygen metabolites in inflammatory bowel disease. Curr Opin Gastroenterol. 1993;9:971–980. [Google Scholar]
- 73.Grisham MB. Oxidants and free radicals in inflammatory bowel disease. Lancet. 1994;344:859–861. doi: 10.1016/s0140-6736(94)92831-2. [DOI] [PubMed] [Google Scholar]
- 74.Kitahora T, Suzuki K, Asakura H, Yoshida T, Suematsu M, Watanabe M, Aiso S, Tsuchiya M. Active oxygen species generated by monocytes and polymorphonuclear cells in Crohn’s disease. Dig Dis Sci. 1988;33:951–955. doi: 10.1007/BF01535990. [DOI] [PubMed] [Google Scholar]
- 75.Yamada T, Grisham MB. Pathogenesis of tissue injury: Role of reactive metabolities of oxygen and nitrogen. In: Shanahan F, Targan S, editors. Inflammatory Bowel Disease: From Bench to Beside. Baltimore: Williams and Wilkins; 1994. pp. 133–150. [Google Scholar]
- 76.Sharon P, Stenson WF. Enhanced synthesis of leukotriene B4 by colonic mucosa in inflammatory bowel disease. Gastroenterology. 1984;86:453–460. [PubMed] [Google Scholar]
- 77.Shimizu T, Fujii T, Suzuki R, Igarashi J, Ohtsuka Y, Nagata S, Yamashiro Y. Effects of highly purified eicosapentaenoic acid on erythrocyte fatty acid composition and leukocyte and colonic mucosa leukotriene B4 production in children with ulcerative colitis. J Pediatr Gastroenterol Nutr. 2003;37:581–585. doi: 10.1097/00005176-200311000-00015. [DOI] [PubMed] [Google Scholar]
- 78.Ferraris L, Karmeli F, Eliakim R, Klein J, Fiocchi C, Rachmilewitz D. Intestinal epithelial cells contribute to the enhanced generation of platelet activating factor in ulcerative colitis. Gut. 1993;34:665–668. doi: 10.1136/gut.34.5.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Denizot Y, Chaussade S. Platelet activating factor and Crohn’s disease. Gut. 1994;35:141. doi: 10.1136/gut.35.1.141-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Saito S, Tsuno NH, Sunami E, Hori N, Kitayama J, Kazama S, Okaji Y, Kawai K, Kanazawa T, Watanabe T, et al. Expression of platelet-derived endothelial cell growth factor in inflammatory bowel disease. J Gastroenterol. 2003;38:229–237. doi: 10.1007/s005350300041. [DOI] [PubMed] [Google Scholar]
- 81.Alzoghaibi MA, Walsh SW, Willey A, Yager DR, Fowler AA, Graham MF. Linoleic acid induces interleukin-8 production by Crohn’s human intestinal smooth muscle cells via arachidonic acid metabolites. Am J Physiol Gastrointest Liver Physiol. 2004;286:G528–G537. doi: 10.1152/ajpgi.00189.2003. [DOI] [PubMed] [Google Scholar]
- 82.Grisham MB, MacDermott RP, Deitch EA. Oxidant defense mechanisms in the human colon. Inflammation. 1990;14:669–680. doi: 10.1007/BF00916370. [DOI] [PubMed] [Google Scholar]
- 83.Kruidenier L, Kuiper I, Van Duijn W, Mieremet-Ooms MA, van Hogezand RA, Lamers CB, Verspaget HW. Imbalanced secondary mucosal antioxidant response in inflammatory bowel disease. J Pathol. 2003;201:17–27. doi: 10.1002/path.1408. [DOI] [PubMed] [Google Scholar]
- 84.Wijeratne SS, Cuppett SL. Lipid hydroperoxide induced oxidative stress damage and antioxidant enzyme response in Caco-2 human colon cells. J Agric Food Chem. 2006;54:4476–4481. doi: 10.1021/jf060475v. [DOI] [PubMed] [Google Scholar]
- 85.Ohta Y, Nishida K. Protective effect of coadministered superoxide dismutase and catalase against stress-induced gastric mucosal lesions. Clin Exp Pharmacol Physiol. 2003;30:545–550. doi: 10.1046/j.1440-1681.2003.03871.x. [DOI] [PubMed] [Google Scholar]
- 86.Simmonds NJ, Allen RE, Stevens TR, Van Someren RN, Blake DR, Rampton DS. Chemiluminescence assay of mucosal reactive oxygen metabolites in inflammatory bowel disease. Gastroenterology. 1992;103:186–196. doi: 10.1016/0016-5085(92)91112-h. [DOI] [PubMed] [Google Scholar]
- 87.Zhu H, Li YR. Oxidative stress and redox signaling mechanisms of inflammatory bowel disease: updated experimental and clinical evidence. Exp Biol Med (Maywood) 2012;237:474–480. doi: 10.1258/ebm.2011.011358. [DOI] [PubMed] [Google Scholar]
- 88.Rezaie A, Parker RD, Abdollahi M. Oxidative stress and pathogenesis of inflammatory bowel disease: an epiphenomenon or the cause? Dig Dis Sci. 2007;52:2015–2021. doi: 10.1007/s10620-006-9622-2. [DOI] [PubMed] [Google Scholar]
- 89.Thomson A, Hemphill D, Jeejeebhoy KN. Oxidative stress and antioxidants in intestinal disease. Dig Dis. 1998;16:152–158. doi: 10.1159/000016859. [DOI] [PubMed] [Google Scholar]
- 90.Emerit J, Pelletier S, Likforman J, Pasquier C, Thuillier A. Phase II trial of copper zinc superoxide dismutase (CuZn SOD) in the treatment of Crohn’s disease. Free Radic Res Commun. 1991;12-13 Pt 2:563–569. doi: 10.3109/10715769109145831. [DOI] [PubMed] [Google Scholar]
- 91.Kruidenier L, Kuiper I, van Duijn W, Marklund SL, van Hogezand RA, Lamers CB, Verspaget HW. Differential mucosal expression of three superoxide dismutase isoforms in inflammatory bowel disease. J Pathol. 2003;201:7–16. doi: 10.1002/path.1407. [DOI] [PubMed] [Google Scholar]
- 92.Couto D, Ribeiro D, Freitas M, Gomes A, Lima JL, Fernandes E. Scavenging of reactive oxygen and nitrogen species by the prodrug sulfasalazine and its metabolites 5-aminosalicylic acid and sulfapyridine. Redox Rep. 2010;15:259–267. doi: 10.1179/135100010X12826446921707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.McKenzie SM, Doe WF, Buffinton GD. 5-aminosalicylic acid prevents oxidant mediated damage of glyceraldehyde-3-phosphate dehydrogenase in colon epithelial cells. Gut. 1999;44:180–185. doi: 10.1136/gut.44.2.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ahnfelt-Rønne I, Nielsen OH, Christensen A, Langholz E, Binder V, Riis P. Clinical evidence supporting the radical scavenger mechanism of 5-aminosalicylic acid. Gastroenterology. 1990;98:1162–1169. doi: 10.1016/0016-5085(90)90329-y. [DOI] [PubMed] [Google Scholar]