

Abstract
AIM: To investigate the association of macrophage migration inhibitory factor (MIF) promoter polymorphisms with inflammatory bowel disease (IBD) risk.
METHODS: One thousand and six New Zealand Caucasian cases and 540 Caucasian controls were genotyped for the MIF SNP -173G > C (rs755622) and the repeat polymorphism CATT5-8 (rs5844572) using a pre-designed TaqMan SNP assay and capillary electrophoresis, respectively. Data were analysed for single site and haplotype association with IBD risk and phenotype. Meta-analysis was employed, to assess cumulative evidence of association of MIF -173G > C with IBD. All published genotype data for MIF -173G > C in IBD were identified using PubMed and subsequently searching the references of all PubMed-identified studies. Imputed genotypes for MIF -173G > C were generated from the Wellcome Trust Case Control Consortium (and National Institute of Diabetes and Digestive and Kidney Diseases). Separate meta-analyses were performed on Caucasian Crohn’s disease (CD) (3863 patients, 6031 controls), Caucasian ulcerative colitis (UC) (1260 patients, 1987 controls), and East Asian UC (416 patients and 789 controls) datasets using the Mantel-Haenszel method. The New Zealand dataset had 93% power, and the meta-analyses had 100% power to detect an effect size of OR = 1.40 at α = 0.05, respectively.
RESULTS: In our New Zealand dataset, single-site analysis found no evidence of association of MIF polymorphisms with overall risk of CD, UC, and IBD or disease phenotype (all P values > 0.05). Haplotype analysis found the CATT5/-173C haplotype occurred at a higher frequency in New Zealand controls compared to IBD patients (0.6 vs 0.01; P = 0.03, OR = 0.22; 95%CI: 0.05-0.99), but this association did not survive bonferroni correction. Meta-analysis of our New Zealand MIF -173G > C data with data from seven additional Caucasian datasets using a random effects model found no association of MIF polymorphisms with CD, UC, or overall IBD. Similarly, meta-analysis of all published MIF -173G > C data from East Asian datasets (416 UC patients, 789 controls) found no association of this promoter polymorphism with UC.
CONCLUSION: We found no evidence of association of MIF promoter polymorphisms with IBD.
Keywords: Crohn’s disease, Ulcerative colitis, Migration inhibitory factor, rs755622, rs5844572, Genetic association study
Core tip: Migration inhibitory factor (MIF) is an important mediator of inflammatory bowel disease (IBD). However, whether promoter polymorphisms in MIF alter susceptibility to IBD is unclear. This study sought to clarify this, as definitively as possible, for Caucasians and East Asians. Analysis of a New Zealand Caucasian cohort found no association of the polymorphisms MIF -173G > C and CATT5-8 with IBD. Subsequent meta-analysis of the New Zealand data with published MIF -173G > C data from other Caucasian cohorts found no association. A separate meta-analysis of East Asian datasets also found no evidence of association of this promoter polymorphism with IBD.
INTRODUCTION
Macrophage migration inhibitory factor (MIF) is a pro-inflammatory cytokine implicated in the pathophysiology of numerous inflammatory conditions including inflammatory bowel disease (IBD)[1]. MIF is a widely expressed component of the immune system that is released in response to diverse stimuli including lipopolysaccharide (via toll-like receptor-4, TLR-4), pro-inflammatory cytokines such as tumor necrosis factor-α and interferon-gamma, and hypoxia[2,3]. MIF in turn up-regulates the expression of TLR-4, pro-inflammatory cytokines, and acts as a cofactor for the activation of T-cells[4]. MIF is elevated in the plasma of patients with active IBD and falls following successful treatment[5,6]. In murine models, transgenic over-expression of MIF increases susceptibility to IBD while MIF knockout mice are protected from the development of colitis[5-8]. Moreover, the ability of neutralising anti MIF antibodies to ameliorate murine colitis indicates the potential value of MIF as a therapeutic target[5,6,8].
A single nucleotide polymorphism (SNP) -173G > C (rs755622) and a tetra-nucleotide repeat CATT5-8 (rs5844572) have been identified in the MIF promoter[4,9]. These polymorphisms are associated with increased plasma concentrations of MIF, increased risk and severity of inflammatory disease, and reduced response to glucocorticoid medication[4,9,10]. The functional effect of MIF-173G > C is attributed to the creation of a binding site for the transcription factor AP4[11], whilst the mechanism by which CATT5-8 alters promoter activity is unknown. Despite several previous studies[12-19], the genetic contribution of MIF promoter polymorphisms to IBD susceptibility and phenotype is unclear. The overall aim of this study was to clarify, as definitively as possible, the contribution of MIF promoter polymorphisms to IBD risk and phenotype in Caucasians and East Asians. To achieve this aim, we conducted the largest single-dataset study of MIF promoter polymorphisms -173G > C and CATT5-8 in Caucasians to date, and meta-analysed these new data with previously published data to test for cumulative evidence of association with IBD risk and phenotype in Caucasians and East Asians.
MATERIALS AND METHODS
Ethics
All New Zealand study participants gave their informed written consent and approval for the study was obtained from the Upper and Lower South Regional Ethics Committees of New Zealand (Approval ID: CTY/03/01/011; Date: 05/06/2007). For the meta-analysis, genotype data from non-New Zealand subjects were obtained from review of published studies (Table 1).
Table 1.
Characteristics of previously published association studies of MIF -173G > C in inflammatory bowel disease
| Cohort | Sample size |
Demographics |
Ref. | |||
| Race | mean ± SD | % male | IBD diagnosis | |||
| 1 | Cases: 111 | Japanese | 39 ± 14.2 | 52.3 | UC | [18] |
| Controls: 209 | 44 ± 18.4 | 58.4 | NA | |||
| 2 | Cases: 221 | Japanese | 40.1 ± 14.0 | 46.2 | UC | [15] |
| Controls: 312 | 41.4 ± 18.1 | 50.3 | NA | |||
| 3 | Cases: 99 | Han Chinese | 38.4 ± 10.6 | 61.6 | CD (15); UC (84) | [13] |
| Controls: 142 | 42.3 ± 11.6 | 64.1 | NA | |||
| 4 | Cases: 623 | Caucasian | - | 48 (CD); 49.0 (UC) | CD (336); UC (287) | [16] |
| Controls: 361 | - | - | NA | |||
| 5 | Cases: 672 | Caucasian | - | 47.1 (CD); 43.0 (UC) | CD (325); UC (347) | [16] |
| Controls: 526 | - | - | NA | |||
| 6 | Cases: 99 | Caucasian | - | - | 41 (CD); 58 (UC) | [17] |
| Controls: 123 | - | - | NA | |||
| 7 | Cases: 198 | Caucasian | 39.5 ± 11.9 | 48.0 | CD | [12] |
| Controls: 159 | 44.1 ± 15.8 | 46.5 | NA | |||
| 8 | Cases: 259 | Caucasian | 42.3 ± 10.2 (CD); 44.1 ± 15.3 (UC) | - | 157 (CD), 102 (UC) | [14] |
| Controls: 489 | - | - | NA | |||
| 9 | Cases: 1234 | Caucasian | - | 47.2 | CD | [32] |
| Controls: 2112 | - | 51.0 | NA | |||
| 10 | Cases: 590 | Caucasian | 21 ± 10.8 | 47.2 | CD | [33,34] |
| Controls: 678 | - | 49.3 | NA | |||
| Controls: 488 | 44.9 ± 11.2 | 41.2 | NA | |||
| 12 | Cases: 139 | Caucasian (Indian) | 41.0 ± 14.11 | 59.3 | 139 (UC) | [19]1 |
| Controls: 176 | NA | |||||
The study of Sivaram et al[19] was not included in the meta-analyses as study participants were neither East Asian, nor Northern European or American Caucasians. IBD: Inflammatory bowel disease; UC: Ulcerative colitis; CD: Crohn’s disease; NA: Not available.
New Zealand controls and IBD patients
Study participants were selected from a population-based study of genetic and environmental determinants of the aetiology of IBD in the Canterbury region of New Zealand which has been described in detail elsewhere[20]. The population of Canterbury is largely of Northern European (United Kingdom and Irish) origin and participants were included in the current study if they were of self-reported European ancestry. Diagnosis of IBD was made by standard criteria[21]. Detailed phenotypic data according to the Montreal Classification system were available in addition to information regarding the presence of extra-intestinal manifestations of disease, history of immuno-modulator use and the need for surgery[21]. The control group comprised 540 healthy Caucasian New Zealanders over the age of 17 years with no history of inflammatory disorders[22].
MIF genotyping
DNA was collected from peripheral blood samples of the IBD patients and controls using guanidinium isothiocyanate-chloroform extraction[23]. Genotyping of MIF -173G > C (rs755622) was performed using a pre-designed Taqman® SNP genotyping assay (assay ID: C_2213785_10) as per the manufacturer’s instructions (Applied Biosystems, Foster City, CA, United States). The MIF CATT5-8 repeat, was amplified in a total volume of 10 μL containing 2 mM dNTPs (Fisher Biotec), 2 mM MgCl2, 10% betaine (Sigma, St Louis, Missouri, United States), 0.5 μmol/L of the primers MIFrepf (5’FAM-gcctgtgatccagttgctgccttgtc3’) and MIFrepr (5’ccactaatggtaaactcggggaccat3’), 1 U of Platinum Taq DNA Polymerase (Invitrogen, CA, United States), and 20 ng of genomic DNA. The forward primer, MIFrepf, was labelled to enable subsequent resolution on a DNA Analyzer (Applied Biosystems 3730xl DNA Analyzer). The polymerase chain reaction (PCR) conditions comprised an initial denaturation step of 2 min at 94 °C; followed by 30 cycles of 94 °C for 30 s, 65 °C for 30 s, 72 °C for 30 s; and a final extension step of 1 min at 72 °C. PCRs were diluted by the addition of 20 μL of water. Amplification of the MIF promoter was assessed by 3% agarose gel electrophoresis. To accurately size the PCR products and thus reliably assign CATT genotypes, 2 μL of each diluted PCR product was mixed with 1 μL of GeneScan™ 500 LIZ® Size Standard (Applied Biosystems, Foster City, CA, United States) and resolved on an ABI 3730xl DNA Analyzer. Samples containing 5, 6, 7, or 8 CATT repeats yielded PCR products of 152, 156, 160 and 164 bp, respectively. Genotyping results were then analysed using Peak Scanner™ Software version 1 (Applied Biosystems, Foster City, CA, United States). Accuracy of the TaqMan SNP and CATT assays was confirmed by repeat analysis of 10% of samples. The concordance between original and repeat genotypes was 100%.
Association testing
The software package PLINK[24] was used to test for deviations from Hardy-Weinberg Equilibrium (HWE), and to conduct single marker and haplotype association tests. The statistical significance of the observed allele and genotype associations were determined by Pearson’s χ2. Results were considered statistically significant if P was < 0.05. Bonferroni’s correction was applied to adjust for multiple testing (0.05/18 tests, adjusted P < 0.0028). As CATT5 is reported to have less promoter activity than longer repeat alleles[9,25], tests of association were performed on full genotype and following simplification to 55, 5X or XX, where X = CATT6-8, as previously described[9,26,27].
Imputation and meta-analysis
All published studies of MIF -173G > C and MIF CATT5-8 in IBD were identified using PubMed and subsequently searching the references of all PubMed-identified studies. Imputed genotypes for MIF -173G > C were generated from the Wellcome Trust Case Control Consortium (WTCCC)[28] and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Crohn’s disease (CD) datasets[29,30] using IMPUTE and the HapMap release [NCBI dbSNP Build 131 (Apr 2010, hg37.1)]. A quality threshold of 0.95 was set for imputation of both datasets.
Meta-analyses were conducted for the MIF SNP -173G > C in STATA (Stata Statistics/Data Analysis Software, version 8, Stata Corporation, Texas, United States) using the Mantel-Haenszel (MH) method with either a random or a fixed effects model depending on the presence or absence respectively of significant heterogeneity between studies (P < 0.05). Meta-analyses were considered significant if the pool MH P value was < 0.05. As published CATT5-8 frequency data was limited to a single Japanese ulcerative colitis (UC) dataset[18], meta-analysis was not conducted for this variant.
RESULTS
Single-site analysis of MIF polymorphisms
MIF-173G > C genotype was determined in 988 New Zealand patients (495 CD, 466 UC and 27 IBD-U) and 488 New Zealand controls, and CATT5-8 genotype was determined in 975 New Zealand patients (476 CD, 473 UC and 26 IBD-U) and 535 New Zealand controls. The baseline characteristics of the cases are shown in Table 2. Genotype and allele frequencies are shown in Table 3 (MIF-173G > C) and Table 4 (MIF CATT5-8). No significant deviation from HWE was observed in the patients or controls. Single-site analysis found no evidence of association of MIF-173G > C or CATT5-8, and CD, UC or overall risk of IBD (Tables 3 and 4). Furthermore, neither polymorphism was significantly associated with age at first diagnosis, disease location, or disease behaviour (penetrating, stricturing or non-penetrating/non-stricturing) in our New Zealand dataset.
Table 2.
Baseline characteristics of the New Zealand inflammatory bowel disease patients and controls n (%)
| Characteristic | Controls | CD | UC | IBD-U |
| Number of females | 340 (63.0) | 307 (63.0) | 242 (51.5) | 13 (50.0) |
| Age at first diagnosis (yr) | ||||
| < 17 | 57 (11.3) | 28 (5.9) | ||
| 17-40 | 277 (55.0) | 231 (48.6) | 15 (55) | |
| > 40 | 170 (33.7) | 216 (45.5) | 12 (45) | |
| CD location | ||||
| Colonic | 211 (41.8) | |||
| Ileocaecal | 125 (24.8) | |||
| Ileal | 64 (32.5) | |||
| Isolated upper GI disease | 3 (0.6) | |||
| Perianal disease modifier | 136 (27.0) | |||
| CD behaviour | ||||
| Non-stricturing/ non-penetrating | 286 (56.8) | |||
| Penetrating | 57 (11.3) | |||
| Stricturing | 161 (31.9) | |||
| UC location | ||||
| Proctitis | 164 (34.5) | 3 (11.1) | ||
| Left-sided | 125 (26.3) | 5 (18.5) | ||
| Extensive | 181 (38.1) | 19 (70.4) | ||
IBD: Inflammatory bowel disease; UC: Ulcerative colitis; CD: Crohn’s disease; GI: Gastrointestinal.
Table 3.
Allele and genotype frequencies of MIF-173G > C in New Zealand Caucasian patients with inflammatory bowel disease compared with controls
| Phenotype |
Genotype |
MAF | Allelic P (unadjusted) | OR (95%CI) | ||
| GG | GC | CC | ||||
| Control | 328 (0.672) | 143 (0.293) | 17 (0.035) | 177 (0.181) | ||
| IBD | 668 (0.676) | 287 (0.290) | 33 (0.033) | 353 (0.179) | 0.86 | 0.98 (0.80-1.20) |
| UC | 320 (0.687) | 129 (0.277) | 17 (0.036) | 163 (0.175) | 0.71 | 0.96 (0.76-1.21) |
| Age at first diagnosis (yr) | ||||||
| < 17 | 17 (0.610) | 11 (0.390) | 0 | 11 (0.196) | 0.78 | 1.10 (0.56-2.18) |
| 17-40 | 153 (0.680) | 60 (0.267) | 12 (0.053) | 84 (0.187) | 0.81 | 1.04 (0.78-1.38) |
| > 40 | 150 (0.704) | 58 (0.272) | 5 (0.023) | 68 (0.160) | 0.33 | 0.86 (0.63-1.17) |
| Disease location | ||||||
| Proctitis | 106 (0.658) | 49 (0.304) | 6 (0.037) | 61 (0.189) | 0.75 | 1.06 (0.76-1.46) |
| Left-sided | 92 (0.742) | 27 (0.218) | 5 (0.040) | 37 (0.149) | 0.23 | 0.79 (0.54-1.16) |
| Extensive | 118 (0.670) | 52 (0.295) | 6 (0.034) | 64 (0.182) | 0.99 | 1.00 (0.73-1.38) |
| CD | 331 (0.669) | 149 (0.301) | 15 (0.030) | 179 (0.181) | 0.98 | 0.00 (0.79-1.25) |
| Age at first diagnosis (yr) | ||||||
| < 17 | 36 (0.642) | 19 (0.339) | 1 (0.017) | 21 (0.188) | 0.87 | 1.04 (0.63-1.72) |
| 17-40 | 188 (0.683) | 81 (0.295) | 6 (0.220) | 93 (0.169) | 0.55 | 0.92 (0.70-1.21) |
| > 40 | 107 (0.652) | 49 (0.299) | 8 (0.049) | 65 (0.198) | 0.50 | 1.12 (0.81-1.53) |
| Disease location | ||||||
| Colonic | 131 (0.633) | 68 (0.328) | 8 (0.039) | 84 (0.203) | 0.35 | 1.15 (0.86-1.54) |
| Ileocaecal | 81 (0.653) | 37 (0.298) | 6 (0.048) | 49 (0.198) | 0.56 | 1.11 (0.78-1.58) |
| Ileal | 115 (0.719) | 44 (0.275) | 1 (0.006) | 46 (0.144) | 0.12 | 0.76 (0.53-1.08) |
| Disease behaviour | ||||||
| Non-stricturing/non-penetrating | 181 (0.644) | 92 (0.327) | 8 (0.028) | 108 (0.192) | 0.60 | 1.07 (0.82-1.40) |
| Penetrating | 37 (0.649) | 19 (0.333) | 1 (0.018) | 21 (0.184) | 0.94 | 1.02 (0.62-1.68) |
| Stricturing | 113 (0.720) | 38 (0.242) | 6 (0.038) | 50 (0.159) | 0.37 | 0.86 (0.61-1.21) |
IBD: Inflammatory bowel disease; UC: Ulcerative colitis; CD: Crohn’s disease.
Table 4.
Allele and genotype frequencies of MIF CATT5-8 haplotypes in New Zealand Caucasian controls and inflammatory bowel disease patients
| Phenotype |
Genotypea |
MAF | Allelic P (unadjusted) | OR (95%CI) | ||
| 5/5 | 5/X | X/X | ||||
| Control | 27 (0.050) | 187 (0.350) | 321 (0.600) | 241 (0.225) | ||
| IBD | 54 (0.055) | 366 (0.375) | 555 (0.569) | 474 (0.243) | 0.27 | 1.10 (0.93-1.32) |
| UC | 24 (0.051) | 172 (0.364) | 277 (0.586) | 220 (0.233) | 0.70 | 1.04 (0.85-1.28) |
| Age at first diagnosis (yr) | ||||||
| < 17 | 0 | 11 (0.393) | 17 (0.607) | 11 (0.196) | 0.61 | 1.04 (0.43-1.65) |
| 17-40 | 10 (0.043) | 91 (0.394) | 130 (0.563) | 111 (0.240) | 0.52 | 1.09 (0.84-1.41) |
| > 40 | 14 (0.065) | 70 (0.327) | 130 (0.607) | 98 (0.229) | 0.88 | 1.02 (0.78-1.33) |
| Disease location | ||||||
| Proctitis | 12 (0.073) | 56 (0.341) | 96 (0.585) | 80 (0.244) | 0.48 | 1.11 (0.83-1.48) |
| Left-sided | 5 (0.040) | 48 (0.387) | 71 (0.573) | 58 (0.234) | 0.77 | 1.05 (0.76-1.46) |
| Extensive | 7 (0.039) | 67 (0.372) | 106 (0.589) | 81 (0.225) | 0.99 | 1.00 (0.75-1.33) |
| CD | 30 (0.063) | 182 (0.555) | 264 (0.555) | 242 (0.254) | 0.12 | 1.18 (0.96-1.44) |
| Age at first diagnosis (yr) | ||||||
| < 17 | 4 (0.073) | 22 (0.400) | 29 (0.527) | 30 (0.273) | 0.26 | 1.29 (0.83-2.01) |
| 17-40 | 15 (0.057) | 104 (0.394) | 145 (0.549) | 134 (0.254) | 0.21 | 1.17 (0.92-1.49) |
| > 40 | 11 (0.070) | 56 (0.357) | 90 (0.573) | 78 (0.248) | 0.39 | 1.14 (0.85-1.52) |
| Disease location | ||||||
| Colonic | 14 (0.070) | 75 (0.375) | 111 (0.555) | 103 (0.258) | 0.19 | 1.19 (0.91-1.56) |
| Ileocaecal | 7 (0.058) | 48 (0.397) | 66 (0.545) | 62 (0.256) | 0.19 | 1.19 (0.86-1.64) |
| Ileal | 9 (0.060) | 56 (0.371) | 86 (0.570) | 74 (0.245) | 0.47 | 1.12 (0.83-1.51) |
| Disease behaviour | ||||||
| Non-stricturing/non-penetrating | 18 (0.066) | 104 (0.382) | 150 (0.551) | 140 (0.257) | 0.15 | 1.19 (0.94-1.51) |
| Penetrating | 1 (0.019) | 20 (0.377) | 32 (0.604) | 22 (0.208) | 0.68 | 0.90 (0.55-1.47) |
| Stricturing | 11 (0.073) | 58 (0.384) | 82 (0.543) | 80 (0.265) | 0.15 | 1.24 (0.93-1.66) |
X = 6, 7, or 8 CATT repeats. IBD: Inflammatory bowel disease; UC: Ulcerative colitis; CD: Crohn’s disease.
Haplotype analysis
Nine hundred and fifty-seven New Zealand patients (CD 467, UC 464, IBD-U 26) and 483 New Zealand controls were successfully genotyped for both MIF promoter polymorphisms. Haplotype analysis of these polymorphisms found the MIF CATT5/-173C haplotype occurred at a higher frequency in controls compared to IBD patients (0.6 vs 0.01; P = 0.03, OR = 0.22, 95%CI: 0.05-0.99). However, after bonferroni correction for multiple testing (0.05/18 tests) no association of haplotype with susceptibility to CD, UC or IBD, or with disease phenotype, behaviour or clinical course was found.
Meta-analyses of MIF-173G > C in Caucasian and East Asian IBD datasets
Meta-analysis was undertaken in order to determine the overall influence of MIF -173G > C on IBD susceptibility in Caucasians and East Asians. Published summary genotype data were available for five Caucasian[12,14,16,17], and three East Asian datasets[13,15,18]. In addition, MIF-173G > C genotypes were imputed from both the WTCCC and NIDDK Caucasian CD datasets. As the contribution that specific loci make to IBD susceptibility differs significantly with race, the East Asian and Caucasian datasets were meta-analysed separately. The Breslow-Day test revealed the existence of significant heterogeneity (Phet ≤ 0.05) among the Caucasian datasets but not among the East Asian datasets (Phet = 0.5). As a result a random effects model was applied to the Caucasian meta-analysis whilst a fixed effects model was applied to the East Asian meta-analysis. No cumulative evidence of association of MIF -173G > C with UC, CD or all IBD risk was detected in either Caucasians or East Asians (Figure 1).
Figure 1.
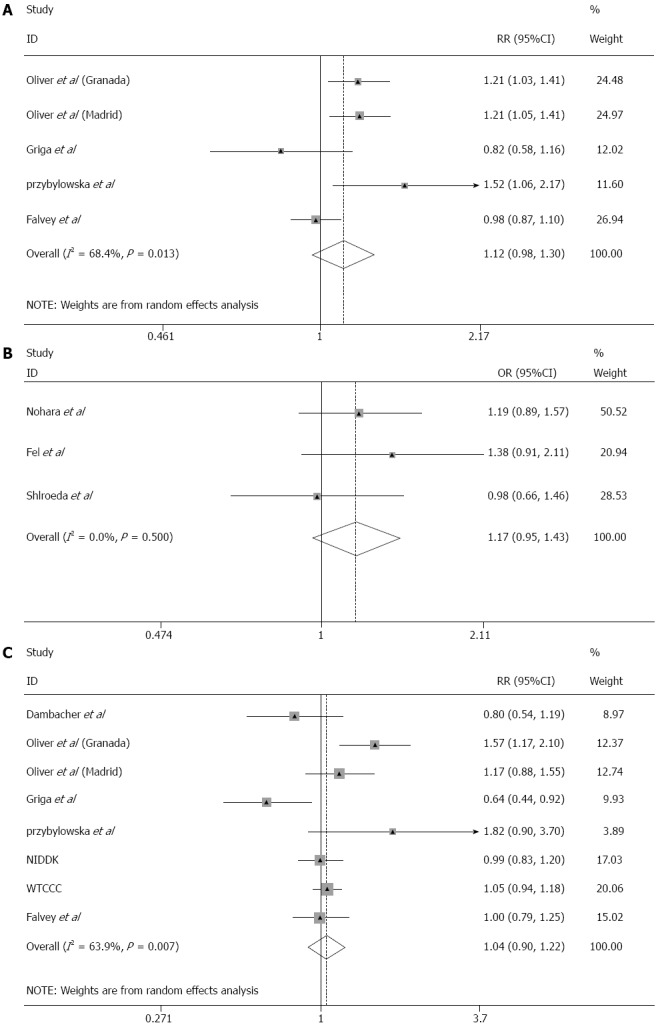
Meta-analyses of migration inhibitory factor-173G > C in Caucasian and East Asian inflammatory bowel disease datasets. Meta-analyses of migration inhibitory factor (MIF)-173G > C were performed by the Mantel-Haenszel method using a random effects model. On each Forest plot the 95%CI of the individual datasets are represented by horizontal lines and the total number of study participants in each dataset is proportional to the size of the square. The diamond represents the pooled OR with 95%CI delineated by the diamond’s width. A: The combined Caucasian ulcerative colitis (UC) dataset; B: The combined East Asian UC datasets; C: The combined Caucasian Crohn’s disease dataset had 100% power to detect an effect size of OR = 1.40 at α = 0.05.
DISCUSSION
Functional effects of MIF polymorphisms
MIF is a pleiotropic pro-inflammatory cytokine implicated in the pathophysiology of numerous diseases including IBD[5,9,31-33]. Promoter polymorphisms in MIF influence both basal and disease-associated MIF expression, and MIF expression has been shown to have profound effects on disease phenotype in experimental models[5,7,34]. In vitro MIF -173C allele exhibits greater expression than the MIF -173G allele in T lymphoblast cells, and expression of MIF increases with increasing length of the CATT repeat in COS-7 fibroblast like cells[9]. Despite these effects on expression, previous association studies investigating these polymorphisms in IBD have given discordant results[12-19].
Previously reported associations of MIF-173G > C and CATT5-8 polymorphisms with UC and CD
The MIF-173C allele has been significantly associated with increased UC susceptibility in Spanish and Polish datasets[16,17], but not in German[14] or Indian[19] datasets. The MIF-173 C/C genotype was significantly associated with pan-colitis compared with left-sided or distal disease (OR = 10.78, 95%CI: 1.34-86.62. P = 0.0074) in one Japanese UC dataset (n = 221)[15], but not a second Japanese dataset[18]. With respect to the length polymorphism, MIF CATT5/CATT5 genotype has been found to confer a protective effect against IBD in Japanese individuals aged over 20 years (OR = 0.33, 95%CI: 0.14-0.82. P = 0.013)[18], whilst the CATT7/CATT7 genotype was significantly associated with both a chronic continuous phenotype (OR = 5.49, 95%CI: 1.19-25.3, P = 0.015) and distal disease location (OR = 6.10, 95%CI: 1.32-28.3, P = 0.0091) in Japanese subjects. However, no association of CATT repeat length with UC risk was observed in two Spanish datasets[16].
The story is similar for CD. Of the previous studies to have investigated the association between MIF-173G > C and CD, four found no association[12,13,16,17], and one reported a protective effect of the minor allele, MIF-173C[14,16]. Furthermore, in one dataset, despite no association with overall CD susceptibility, further analysis revealed MIF-173C influenced CD phenotype; conferring a protective effect against the development of upper GI CD (OR = 0.31, 95%CI: 0.118-0.789, P = 0.01)[12].
MIF promoter polymorphisms in New Zealand Caucasians
Our study sought to resolve the discordance observed between previous investigations by conducting the single largest Caucasian dataset (1006 New Zealand cases, 540 New Zealand controls) study of MIF promoter polymorphisms, and employing meta-analysis. Single-site analysis found no evidence of association of MIF promoter polymorphisms with overall IBD susceptibility in New Zealand Caucasians. Haplotype analysis found preliminary evidence that a rare haplotype, CATT5/-173C, which is the least active haplotype in vitro[9], confers protection against IBD (OR = 0.22, 95%CI: 0.05-0.99, Punadjusted = 0.03). However, as the functional effects of MIF promoter polymorphisms are cell type specific and relevant experiments in monocytes, the primary cell type responsible for MIF dependent mucosal inflammation in IBD are lacking, no firm conclusions can be drawn from these observations[5].
Meta-analyses of MIF -173G > C in Caucasians and East Asians
Subsequent meta-analyses with additional Caucasian CD (3373 patients, 5543 controls), Caucasian UC (794 patients, 1499 controls), and Asian UC (416 patients, 789 controls) datasets also detected no association of MIF-173G > C with CD or UC in Caucasians, or with UC in East Asians (Figure 1). However, our Caucasian meta-analysis is limited by the presence of significant heterogeneity among the datasets, thus it is not possible to completely rule out the possibility that MIF promoter polymorphism may alter susceptibility to IBD in some Caucasian populations. In contrast, no significant heterogeneity was detected in the East Asian datasets (Phet = 0.50), therefore the discordance previously observed is likely to be due to sample size rather than to differences in baseline characteristics of the study participants.
In conclusion, based on the results of our study, neither MIF-173G > C nor MIF CATT5-8 are risk factors for IBD in New Zealand Caucasians, nor is MIF-173G > C a risk factor for UC in East Asians. The results of meta-analysis do not support MIF-173G > C as a susceptibility factor for IBD in Caucasian datasets however, because significant heterogeneity exists between the populations investigated, the possibility that a significant effect exists in a subgroup of these populations cannot be excluded. Additional well-powered studies in European Caucasian datasets would be required to clarify this point.
ACKNOWLEDGMENTS
We thank the people of Canterbury with IBD who generously gave of their time to take part in the study. We also thank Rhondda Brown and Judy Hoar for their assistance in coordinating the recruitment of patients to the Canterbury IBD dataset; Pip Shirley, Meagan Reilley, David Tan, Ramez Ailabouni and Charlotte Duncan for entering patient details into the clinical database. We are also very grateful to Dr. Michael Black for his expert review of our statistical analysis and manuscript. This study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk. Funding for the project was provided by the Wellcome Trust under award 076113 and 085475. The NIDDK IBD Genetics Consortium CD Genome-Wide Association Study was conducted and supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). This manuscript was not prepared in collaboration with Investigators of the NIDDK IBD Genetics Consortium CD Genome-Wide Association Study and does not necessarily reflect the opinions or views of the NIDDK IBD Genetics Consortium CD Genome-Wide Association Study or the NIDDK.
COMMENTS
Background
Macrophage migration inhibitory factor (MIF) is a pro-inflammatory cytokine implicated in the pathophysiology of inflammatory arthritis, systemic lupus erythematosus, atherosclerosis, and cancer. Evidence from animal models and human inflammatory bowel disease (IBD) indicates that MIF is also an important mediator of IBD; however few studies have considered its mechanism of action in these diseases. In recent years, evidence regarding the role of MIF in inflammatory disease has accumulated rapidly, and efforts have subsequently turned to the development of specific anti-MIF therapies for use in human disease.
Research frontiers
An international phase one trial of a neutralizing anti-MIF antibody is currently underway for lupus nephritis. On this background authors believe that critical appraisal of the role of MIF in IBD is needed in order for IBD patients to benefit from developments in this field. Two functional variants of the MIF promoter region have been identified. Despite clear association with other inflammatory diseases, the contribution of these polymorphisms to IBD risk and phenotype remains uncertain due to previous studies being under-powered.
Innovations and breakthroughs
This paper describes the largest single cohort study of MIF promoter polymorphisms to date, as well as meta-analysis of all available genotype data on Caucasians and East Asians. Their population based cohort study had 93% power, and the meta-analyses 100% power, to detect an effect size of OR = 1.40 (α = 0.05). They found no evidence of association of MIF promoter polymorphisms with IBD.
Applications
This study is important as it provides clear evidence that genetic variants of MIF do not influence IBD susceptibility. Their findings are in agreement with recent research that indicates the contribution of MIF to IBD pathophysiology is not determined at the level of nuclear transcription.
Peer review
The role of MIF in the pathogenesis of IBD has been intensively discussed during last couple of years, including the promoter polymorphism of this gene. It is also seen as one of the possible novel therapeutic targets for the management of IBD. The group has contributed to the research in this field by contributing data about the prevalence of the MIF promoter polymorphism in large population of patients and healthy controls. Furthermore, they have meta-analyzed this new data with previously published data, and concluded that there is no evidence of association of MIF promoter polymorphisms with IBD. Paper is well structured, comprehensive and clearly written.
Footnotes
Supported by The Health Research Council of New Zealand; Scholarships from the Canterbury Gastroenterology Research Trust, New Zealand Society of Gastroenterology Ferring Scholarship, the Bowel and Liver Trust Canterbury and from the University of Otago to Falvey JD
P- Reviewers Dambrauskas Z, Qin X, Wang F, Yan Y S- Editor Gou SX L- Editor A E- Editor Ma S
References
- 1.Renner P, Roger T, Calandra T. Macrophage migration inhibitory factor: gene polymorphisms and susceptibility to inflammatory diseases. Clin Infect Dis. 2005;41 Suppl 7:S513–S519. doi: 10.1086/432009. [DOI] [PubMed] [Google Scholar]
- 2.Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3:791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simons D, Grieb G, Hristov M, Pallua N, Weber C, Bernhagen J, Steffens G. Hypoxia-induced endothelial secretion of macrophage migration inhibitory factor and role in endothelial progenitor cell recruitment. J Cell Mol Med. 2011;15:668–678. doi: 10.1111/j.1582-4934.2010.01041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donn RP, Shelley E, Ollier WE, Thomson W. A novel 5’-flanking region polymorphism of macrophage migration inhibitory factor is associated with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2001;44:1782–1785. doi: 10.1002/1529-0131(200108)44:8<1782::AID-ART314>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 5.de Jong YP, Abadia-Molina AC, Satoskar AR, Clarke K, Rietdijk ST, Faubion WA, Mizoguchi E, Metz CN, Alsahli M, ten Hove T, et al. Development of chronic colitis is dependent on the cytokine MIF. Nat Immunol. 2001;2:1061–1066. doi: 10.1038/ni720. [DOI] [PubMed] [Google Scholar]
- 6.Murakami H, Akbar SM, Matsui H, Onji M. Macrophage migration inhibitory factor in the sera and at the colonic mucosa in patients with ulcerative colitis: clinical implications and pathogenic significance. Eur J Clin Invest. 2001;31:337–343. doi: 10.1046/j.1365-2362.2001.00796.x. [DOI] [PubMed] [Google Scholar]
- 7.Ohkawara T, Mitsuyama K, Takeda H, Asaka M, Fujiyama Y, Nishihira J. Lack of macrophage migration inhibitory factor suppresses innate immune response in murine dextran sulfate sodium-induced colitis. Scand J Gastroenterol. 2008;43:1497–1504. doi: 10.1080/00365520802273017. [DOI] [PubMed] [Google Scholar]
- 8.Ohkawara T, Nishihira J, Takeda H, Hige S, Kato M, Sugiyama T, Iwanaga T, Nakamura H, Mizue Y, Asaka M. Amelioration of dextran sulfate sodium-induced colitis by anti-macrophage migration inhibitory factor antibody in mice. Gastroenterology. 2002;123:256–270. doi: 10.1053/gast.2002.34236. [DOI] [PubMed] [Google Scholar]
- 9.Baugh JA, Chitnis S, Donnelly SC, Monteiro J, Lin X, Plant BJ, Wolfe F, Gregersen PK, Bucala R. A functional promoter polymorphism in the macrophage migration inhibitory factor (MIF) gene associated with disease severity in rheumatoid arthritis. Genes Immun. 2002;3:170–176. doi: 10.1038/sj.gene.6363867. [DOI] [PubMed] [Google Scholar]
- 10.Barton A, Lamb R, Symmons D, Silman A, Thomson W, Worthington J, Donn R. Macrophage migration inhibitory factor (MIF) gene polymorphism is associated with susceptibility to but not severity of inflammatory polyarthritis. Genes Immun. 2003;4:487–491. doi: 10.1038/sj.gene.6364014. [DOI] [PubMed] [Google Scholar]
- 11.De Benedetti F, Meazza C, Vivarelli M, Rossi F, Pistorio A, Lamb R, Lunt M, Thomson W, Ravelli A, Donn R, et al. Functional and prognostic relevance of the -173 polymorphism of the macrophage migration inhibitory factor gene in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2003;48:1398–1407. doi: 10.1002/art.10882. [DOI] [PubMed] [Google Scholar]
- 12.Dambacher J, Staudinger T, Seiderer J, Sisic Z, Schnitzler F, Pfennig S, Hofbauer K, Konrad A, Tillack C, Otte JM, et al. Macrophage migration inhibitory factor (MIF) -173G/C promoter polymorphism influences upper gastrointestinal tract involvement and disease activity in patients with Crohn’s disease. Inflamm Bowel Dis. 2007;13:71–82. doi: 10.1002/ibd.20008. [DOI] [PubMed] [Google Scholar]
- 13.Fei BY, Lv HX, Yang JM, Ye ZY. Association of MIF-173 gene polymorphism with inflammatory bowel disease in Chinese Han population. Cytokine. 2008;41:44–47. doi: 10.1016/j.cyto.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 14.Griga T, Wilkens C, Wirkus N, Epplen J, Schmiegel W, Klein W. A polymorphism in the macrophage migration inhibitory factor gene is involved in the genetic predisposition of Crohn’s disease and associated with cumulative steroid doses. Hepatogastroenterology. 2007;54:784–786. [PubMed] [Google Scholar]
- 15.Nohara H, Okayama N, Inoue N, Koike Y, Fujimura K, Suehiro Y, Hamanaka Y, Higaki S, Yanai H, Yoshida T, et al. Association of the -173 G/C polymorphism of the macrophage migration inhibitory factor gene with ulcerative colitis. J Gastroenterol. 2004;39:242–246. doi: 10.1007/s00535-003-1284-7. [DOI] [PubMed] [Google Scholar]
- 16.Latiano A, Palmieri O, Valvano MR, D’Incà R, Caprilli R, Cucchiara S, Sturniolo GC, Bossa F, Andriulli A, Annese V. The association of MYO9B gene in Italian patients with inflammatory bowel diseases. Aliment Pharmacol Ther. 2008;27:241–248. doi: 10.1111/j.1365-2036.2007.03551.x. [DOI] [PubMed] [Google Scholar]
- 17.Przybyłowska K, Mrowicki J, Sygut A, Narbutt P, Dziki Ł, Dziki A, Majsterek I. Contribution of the -173 G/C polymorphism of macrophage migration inhibitory factor gene to the risk of inflammatory bowel diseases. Pol Przegl Chir. 2011;83:76–80. doi: 10.2478/v10035-011-0012-x. [DOI] [PubMed] [Google Scholar]
- 18.Shiroeda H, Tahara T, Nakamura M, Shibata T, Nomura T, Yamada H, Hayashi R, Saito T, Yamada M, Fukuyama T, et al. Association between functional promoter polymorphisms of macrophage migration inhibitory factor (MIF) gene and ulcerative colitis in Japan. Cytokine. 2010;51:173–177. doi: 10.1016/j.cyto.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 19.Sivaram G, Tiwari SK, Bardia A, Anjum F, Vishnupriya S, Habeeb A, Khan AA. Macrophage migration inhibitory factor, Toll-like receptor 4, and CD14 polymorphisms with altered expression levels in patients with ulcerative colitis. Hum Immunol. 2012;73:201–205. doi: 10.1016/j.humimm.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 20.Gearry RB, Richardson A, Frampton CM, Collett JA, Burt MJ, Chapman BA, Barclay ML. High incidence of Crohn’s disease in Canterbury, New Zealand: results of an epidemiologic study. Inflamm Bowel Dis. 2006;12:936–943. doi: 10.1097/01.mib.0000231572.88806.b9. [DOI] [PubMed] [Google Scholar]
- 21.Silverberg MS, Satsangi J, Ahmad T, Arnott ID, Bernstein CN, Brant SR, Caprilli R, Colombel JF, Gasche C, Geboes K, et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can J Gastroenterol. 2005;19 Suppl A:5A–36A. doi: 10.1155/2005/269076. [DOI] [PubMed] [Google Scholar]
- 22.Roberts RL, Gearry RB, Hollis-Moffatt JE, Miller AL, Reid J, Abkevich V, Timms KM, Gutin A, Lanchbury JS, Merriman TR, et al. IL23R R381Q and ATG16L1 T300A are strongly associated with Crohn’s disease in a study of New Zealand Caucasians with inflammatory bowel disease. Am J Gastroenterol. 2007;102:2754–2761. doi: 10.1111/j.1572-0241.2007.01525.x. [DOI] [PubMed] [Google Scholar]
- 23.Ciulla TA, Sklar RM, Hauser SL. A simple method for DNA purification from peripheral blood. Anal Biochem. 1988;174:485–488. doi: 10.1016/0003-2697(88)90047-4. [DOI] [PubMed] [Google Scholar]
- 24.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donn R, Alourfi Z, Zeggini E, Lamb R, Jury F, Lunt M, Meazza C, De Benedetti F, Thomson W, Ray D. A functional promoter haplotype of macrophage migration inhibitory factor is linked and associated with juvenile idiopathic arthritis. Arthritis Rheum. 2004;50:1604–1610. doi: 10.1002/art.20178. [DOI] [PubMed] [Google Scholar]
- 26.Martin RJ, Savage DA, Carson DJ, McKnight AJ, Maxwell AP, Patterson CC. Polymorphisms of the macrophage migration inhibitory factor gene in a UK population with Type 1 diabetes mellitus. Diabet Med. 2010;27:143–149. doi: 10.1111/j.1464-5491.2009.02916.x. [DOI] [PubMed] [Google Scholar]
- 27.Sakaue S, Ishimaru S, Hizawa N, Ohtsuka Y, Tsujino I, Honda T, Suzuki J, Kawakami Y, Nishihira J, Nishimura M. Promoter polymorphism in the macrophage migration inhibitory factor gene is associated with obesity. Int J Obes (Lond) 2006;30:238–242. doi: 10.1038/sj.ijo.0803148. [DOI] [PubMed] [Google Scholar]
- 28.Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bucala R, Lolis E. Macrophage migration inhibitory factor: a critical component of autoimmune inflammatory diseases. Drug News Perspect. 2005;18:417–426. doi: 10.1358/dnp.2005.18.7.939345. [DOI] [PubMed] [Google Scholar]
- 32.Calandra T, Echtenacher B, Roy DL, Pugin J, Metz CN, Hültner L, Heumann D, Männel D, Bucala R, Glauser MP. Protection from septic shock by neutralization of macrophage migration inhibitory factor. Nat Med. 2000;6:164–170. doi: 10.1038/72262. [DOI] [PubMed] [Google Scholar]
- 33.Meyer-Siegler KL, Vera PL, Iczkowski KA, Bifulco C, Lee A, Gregersen PK, Leng L, Bucala R. Macrophage migration inhibitory factor (MIF) gene polymorphisms are associated with increased prostate cancer incidence. Genes Immun. 2007;8:646–652. doi: 10.1038/sj.gene.6364427. [DOI] [PubMed] [Google Scholar]
- 34.Radstake TR, Sweep FC, Welsing P, Franke B, Vermeulen SH, Geurts-Moespot A, Calandra T, Donn R, van Riel PL. Correlation of rheumatoid arthritis severity with the genetic functional variants and circulating levels of macrophage migration inhibitory factor. Arthritis Rheum. 2005;52:3020–3029. doi: 10.1002/art.21285. [DOI] [PubMed] [Google Scholar]