

Abstract
Sex chromosomes evolve from ordinary autosomes through the expansion and subsequent degeneration of a region of suppressed recombination that is inherited through one sex. Here we investigate the relative timing of these processes in the UV sex chromosomes of the moss Ceratodon purpureus using molecular population genetic analyses of eight newly discovered sex-linked loci. In this system recombination is suppressed on both the female-transmitted (U) sex chromosome and the male-transmitted (V) chromosome. Genes on both chromosomes therefore should show the deleterious effects of suppressed recombination and sex-limited transmission, while purifying selection should maintain homologs of genes essential for both sexes on both sex chromosomes. Based on analyses of eight sex-linked loci, we show that the non-recombining portions of the U and V-chromosomes expanded in at least two events (~0.6 – 1.3 MYA and ~2.8 – 3.5 MYA), after the divergence of C. purpureus from its dioecious sister species, Trichodon cylindricus and Cheilothela chloropus. Both U and V-linked copies showed reduced nucleotide diversity and limited population structure, compared to autosomal loci, suggesting that the sex chromosomes experienced more recent selective sweeps that the autosomes. Collectively these results highlight the dynamic nature of gene composition and molecular evolution on non-recombining portions of the U and V sex chromosomes.
Keywords: bryophyte, hitch-hiking, suppressed recombination, sex-linkage, degeneration
INTRODUCTION
Heteromorphic sex chromosomes are widely viewed as degenerated descendants of former autosomes, a view largely based on studies of vertebrate and insect model systems (Charlesworth and Charlesworth 2000). It is now clear, however, that the degree of sex chromosome degeneration may depend in complex ways on an organisms’ life history (Perrin 2009; Bachtrog et al. 2011; Otto et al. 2011). Degeneration appears to prevail in the male-specific non-recombining portions of the Y-chromosome (NRY) in mammals (Skaletsky et al. 2003) and the female-specific non-recombining portions of the W-chromosome (NRW) in birds and lepidopterans (Handley et al. 2004; Abe et al. 2005), but not in the NRY of the frogs Rana temporaria and Hyla spp (Matsuba et al. 2010; Stöck et al. 2011), and nor perhaps the flowering plant Silene latifolia (Bergero and Charlesworth 2011; Chibalina and Filatov 2011). A major challenge, therefore, is identifying the population genetic factors that govern the patterns of divergence and degeneration between formerly homologous sex chromosome pairs in different lineages. Here we study patterns of polymorphism and divergence on the UV sex chromosomes of the haploid dioecious moss Ceratodon purpureus, an under-explored class of sex chromosomes that may enable us to separate critical evolutionary factors that are confounded in other systems.
The major hypothesis to explain the origin and expansion of NRY (or NRW) involves loci with alleles that have opposing effects on male and female fitness, or sexually antagonistic (SA) alleles (Charlesworth and Charlesworth 1978; van Doorn and Kirkpatrick 2007). Theory shows that alleles that benefit one sex can increase in frequency more readily when they are linked to the locus that determines that sex, even if these alleles have deleterious effects on the opposite sex (Rice 1987). For example, a male beneficial allele that is linked to a male determining chromosome is more likely to fix in a population than an autosomal allele with similar effects. If linkage is incomplete, meiotic cross-overs will lead to such alleles migrating onto a female genetic background. If the allele is also detrimental in females, this in turn can generate selection to suppress recombination between the male beneficial allele and the fully male-linked region (the converse can occur for female-beneficial SA alleles). The repeated evolution of SA mutations may thus promote the expansion of suppressed recombination on sex chromosomes (Rice 1987), by favoring chromosomal inversions, translocations, or expansion of heterochromatin that ensure that male-benefit alleles are confined to a male chromosome (Charlesworth and Charlesworth 1980; Skaletsky et al. 2003; Koerich et al. 2008; Wang et al. 2012).
The sexual antagonism hypothesis, in spite of its intuitive appeal, is difficult to test empirically. The observation that genes near the boundary between the recombining and non-recombining portion of the sex chromosome have more recent X-Y coalescence times than genes more distant from the recombination boundary – that is, they form evolutionary strata (Lahn and Page 1999) – is consistent with this model, but does not exclude other mechanisms that do not require SA alleles (Ironside 2010). In particular, any selective force that favors the fixation of a chromosomal inversion (eg, an inversion that generates a beneficial mutation, or captures multiple locally adapted alleles, (Kirkpatrick and Barton 2006) can in principal lead to the incorporation of partially sex-linked genome regions into the sex-specific region of suppressed recombination.
In contrast, several well-understood processes contribute to gene loss from the non-recombining members of sex chromosome pairs, foremost among them suppressed recombination and transmission limited to one sex (Bachtrog 2006). These processes reduce the effective population size (Ne) of such chromosomes, making natural selection less effective at purging deleterious mutations or fixing adaptive ones. As a consequence, non-recombining portions of the genome exhibit decreased codon bias, increased amino acid substitution rates, and increased frequencies of transposable elements (Bachtrog 2005; Marais et al. 2008; Chibalina and Filatov 2011; Whittle et al. 2011). Moreover, the NRY (or NRW) chromosome is always heterozygous with its recombining partner, leading to weak purifying selection against deleterious mutations on NRY, since mutations are usually partially recessive (deleterious mutations in X- or Z-linked loci, in contrast, will be expressed and presumably purged in the homogametic sex). Therefore, deleterious Y-linked mutations can fix, and in practice many NRY chromosomes retain few homologs with their recombining partner.
The balance between the expansion and degeneration of the sex chromosomes of haploid dioecious species, such as bryophytes, should differ from that on the sex chromosomes of diploid dioecious animals and angiosperms (Bull 1978; Bachtrog et al. 2011). In dioecious mosses, sex determination happens at the haploid stage, which has important consequences for the evolution of sex chromosomes. Female and male gametophytes each carry a sex-specific chromosome, which, following Bachtrog et al. (2011), we refer to as U and V, respectively. The diploid part of the life cycle (the sporophyte) is always heterozygous (ie, UV) for the sex-determining chromosomes. At meiosis the U and V chromosomes segregate to haploid female and male spores, respectively, which develop into multicellular haploid female and male gametophytes, the dominant part of the moss life cycle. The non-recombining portions of the bryophyte U and V sex chromosomes (NRU and NRV) are genetically analogous to the mating type loci of some fungi (eg Microbotryum or Neurospora) and algae (eg Chlamydomonas), in that mating type is determined at meiosis (Ferris and Goodenough 1994; Hood 2002; Hood et al. 2004; Menkis et al. 2008). However, mosses additionally possess the gamete-size differences (and often extensive gametophyte sexual dimorphism) that generally are associated with male-female systems, while the best-studied fungi and algae lack such dimorphisms. Indeed, bryophytes experience biased sex ratios and sex differences in parental care, morphology and life history (Stark et al. 2001; Bisang and Hedenas 2005; McDaniel 2005; McDaniel et al. 2007), all traits that could involve sexually antagonistic alleles. Even in early developmental tissue like protonema, with only two cell types, growth is subtly sexually dimorphic (McDaniel et al. 2008), suggesting that males and females of C. purpureus are physiologically dimorphic almost right from spore germination, if not before.
In a haploid dioecious system, purifying selection is likely to be strong for many genes on both non-recombining sex chromosomes (Nishiyama et al. 2003; Szovenyi et al. 2011). Thus, size differences between the U and V chromosomes is unlikely to result from asymmetric gene loss, although genes expressed only in one sex can be lost from the sex chromosome of the other sex. Indeed, although cytological studies indicate that many bryophyte sex chromosomes are heteromorphic (suggesting differential gene loss or differential accumulation of repetitive sequences between the NRU and NRV), they are not generally smaller than the autosomes, and are sometimes larger (Allen 1935; Nakayama et al. 2001). In the moss Ceratodon purpureus, for example, both the male and female chromosomes appear approximately five times larger than the average autosome (Heitz 1932; McDaniel et al. 2007). Data on genetic differentiation between the NRU and NRV chromosomes of bryophyte species are currently very limited (Korpelainen et al. 2008). In the most thorough study, Yamato et al. (2007) reported 64 genes on the male chromosome of the liverwort Marchantia polymorpha, 50 with identifiable homologs on the female chromosome, although whether the differences between the NRU and NRV chromosomes resulted from gene addition or gene loss is unknown.
The bryophyte sex chromosome system also allows us to assess to role of suppressed recombination in sex chromosome evolution, independent of sex-limited inheritance and relaxed purifying selection. This is unlike XY or ZW systems where suppressed recombination is confounded with both sex-limited transmission (to males in XY systems or females in ZW systems) and relaxed purifying selection (because NRY- and NRW-linked mutations experience enforced heterozygosity with the recombining X or Z). In C. purpureus, meiotic chromosome staining and genetic mapping indicate that the UV chromosome pair has a large region over which crossing-over is suppressed (Heitz 1932; Ramsay and Berrie 1982; McDaniel et al. 2007). Levels of polymorphism in regions of suppressed recombination are strongly influenced by hitch-hiking effects of directional or purifying selection acting on the non-recombining chromosome (Bachtrog 2006). As adaptive mutations are fixed (hitch-hiking; Maynard-Smith and Haigh 1974), or as chromosomes containing deleterious mutations are purged (background selection; Charlesworth et al. 1993), diversity in the entire non-recombining region is reduced or lost. Assuming that beneficial and deleterious mutations occur similarly in male and female bryophytes, purifying and directional selection should have similar effects on nucleotide variation on non-recombining portions of the bryophyte U and V chromosomes.
Based on these inferences, we can construct three general predictions regarding the patterns of polymorphism and divergence on bryophyte sex chromosomes. First, because for every U or V chromosome in a sporophyte, there are two copies of each autosome, and we expect that the Ne of a sex-linked locus should be half that of an autosomal locus. Further reductions in the polymorphism on the NRU and NRV, relative to autosomes, may reflect hitchhiking effects as a consequence of selective sweeps or background selection. Second, because both sex chromosomes experience the same meiotic process (ie, suppressed recombination in a UV sporophyte), we expect that differences in patterns of polymorphism between the NRU and NRV will driven by sex differences in mutation rates, demography, or selection. Third, for genes that have homologs on both the U and V chromosomes the divergence times of the NRU-NRV pairs should reflect the times the loci stopped recombining (barring subsequent interchromosomal gene conversion, Perrin 2009). Thus, the ongoing expansion of the NRU-NRV may generate U-V divergence times of variable ages, potentially in the form of evolutionary strata.
To understand the processes shaping the evolution of the NRU and NRV we genetically mapped 71 ESTs from C. purpureus and identified eight sex-linked genes among them. We compared the patterns of polymorphism in these sex-linked loci to those for nine autosomal loci in a panel of wild isolates from eastern North America, including seven populations of C. purpureus and representatives of two closely related species. The divergence between the U and V allele pairs indicates that these loci ceased recombining in multiple events in the recent history of the species, but the polymorphism data suggest that subtle degenerative processes may be acting in spite of the strong purifying selection that results from haploid gene expression.
MATERIALS AND METHODS
Identifying candidate loci
To generate a pool of candidate genes to test for sex linkage in C. purpureus, we screened a set of 208 nuclear loci using primers described in McDaniel et al. (2013a). These primers were developed from 1,677 ESTs generated from 7-day old early developmental tissue (protonema) from a single male genotype (WT4) of C. purpureus. We selected ESTs with clear homology to the distantly related moss Physcomitrella patens to create a relatively conserved subset of the transcribed genes in this tissue. Here we targeted the 140 loci from C. purpureus that McDaniel et al. (2013a) reported to differ in sequence between the female lab strain GG1 (from Gross Gerunds, Austria) and the male lab strain R40 (from Petersburg Pass, New York, USA).
Generating a GG1 × R40 mapping population
To generate a mapping population for segregation analysis, we made an infraspecific cross in C. purpureus between the male R40 and female GG1 lab isolates. Tissue from each of the parental lines was generated on cellophane overlays using standard methods (Cove et al. 2009). Aliquots of blended tissue were inoculated onto wet Turface MVP soil amendment in greenhouse flats. The plants were bottom watered for two weeks to establish the cultures and were top watered subsequently. Plants were grown at 22C in 16 hr days in Conviron growth chambers. Following the initiation of sporophytes, the plants were stored in a dark coldroom at 4C for two - five weeks (the time mattered very little). These plants were then returned to the Conviron chamber where the sporophytes matured.
The diploid sporophyte produces recombinant spores by meiosis. To isolate the recombinants for mapping, hybrid sporophytes (each the product of a cross between two identical haploids) were individually surface sterilized in a 0.6% bleach (sodium hypochlorite) solution for 1 min., rinsed in sterile water, and placed in a microcentrifuge tube containing 1 ml of sterile water. The sporophyte was then ruptured, creating a suspension of spores. The spore suspension was plated on standard BCD media containing 0.5 g/L ammonium tartrate (Cove et al. 2009), and placed under 40 µmol/m2/s continuous light at approximately 20°C. Spores were germinated and allowed to produce protonemal filaments several cells long. Individual recombinants were isolated before the protonema overgrew one another and transferred to a new plate where they were grown under the same light conditions as the spore suspension plates. When the plants had reached approximately 1 cm in diameter, DNA was extracted from each recombinant.
Genotyping the mapping population
Tissue from each of the recombinants was disrupted using a Qiagen bead beater and genomic DNA was isolated using a Qiagen DNeasy plant 96 extraction kit following the manufactures instructions. Genotyping was carried out using the Sequenom platform at the Human Genetics Laboratory at the Washington University in St. Louis Genome Sequencing Center. Sequonom primers are listed in Supplementary Table 1. We also genotyped the mapping population at the sex linked des6 locus following McDaniel et al. (2007) to identify the sex chromosome.
Linkage map construction
The full mapping population consisted of 87 individuals genotyped for 71 polymorphic markers, with nearly all individuals genotyped at each marker. We constructed a linkage map using MAPMAKER 3.0 software (Lander et al. 1987; Lincoln and Lander 1992). We used the GROUP command (two-point linkage criteria set to a LOD > 6.0 and maximum distance between markers set to 37 cM) with the Kosambi mapping function to organize the markers into linkage groups. Where more than five markers formed a linkage group, the command ORDER, with error detection on, was used to find the most likely linear arrangement of markers. This function uses a starting subset of five markers, ordered with a threshold of LOD 3.0, and tries to place the remaining markers with a threshold of LOD 2.0. The error detection data and two-point distances were examined to identify potentially unreliable markers. These markers, as well as other markers that were linked but not placed, were evaluated using the TRY and MAP commands to determine whether their inclusion between other markers increased the map length by > 5 cM. Such markers, as well as those with more than eight errors, were discarded. For linkage groups with five or fewer markers, marker orders were constructed by hand, using the LOD TABLE and MAP commands. The same strategy to remove unreliable markers from the larger linkage groups was also used for the smaller linkage groups. This process resulted in three classes of markers: 1) markers ordered into linkage groups in the final map; 2) markers that are tightly linked to other markers, but could not be placed into single intervals with certainty; and 3) unlinked markers.
Population sampling in Eastern North America
Diploid sporophytes of C. purpureus were collected from seven populations in eastern North America (Table 1). Single spore isolates were generated as above, except that only one progeny gametophyte was chosen from each diploid sporophyte. Eight gametophytes from each population were chosen for genetic analysis. To root genealogies for non-recombining genes of Eastern North American C. purpureus, we chose isolates from a distantly related population of C. purpureus from Spain, and two isolates each of the two putative sister groups to C. purpureus, Cheilothela chloropus and Trichodon cylindricus (McDaniel and Shaw 2005; Cox et al. 2010). DNA was extracted from these individuals using a standard CTAB extraction, with modifications for a 96-well plate platform following McDaniel et al. (2007).
Table 1.
Sampling localities in Eastern North America
| Site | Latitude | Longitude | Elevation |
|---|---|---|---|
| Essex Co., NY | 44°15′02.63″ | 73°49′07.57″ | 1480′ |
| Rensselaer Co., NY | 42°43′23.57″ | 73°16′40.46″ | 1430′ |
| Westchester Co., NY | 41°16′28.85″ | 73°42′38.05″ | 80′ |
| Adams Co., PA | 39°54′22.83″ | 77°31′47.03″ | 250′ |
| St. Georges Co., MD | 38°49′45.76″ | 76°52′27.79″ | 20′ |
| Dinwiddie Co., VA | 37°06′06.27″ | 77°32′35.60″ | 40′ |
| Durham Co., NC | 36°01′42.68″ | 78°55′28.33″ | 300′ |
Sequencing sex linked and autosomal loci
Eight sex-linked loci were chosen for study from the pool of EST-derived loci (CDC2, CPU011109, ga03f07, PPR, rpS15A-1, rpS15A-2, rpS18, rpS18A). Nine autosomal loci were also chosen for comparison. Two of the autosomal loci were studied previously in C. purpureus (adk and phy2 McDaniel and Shaw 2005), one was studied in P. patens (ho1 McDaniel et al. 2010), and the remaining six (ga01f09, hp23.3, hp23.9, KIAA0187, rpS23, TBP) came from the pool of EST-derived loci and comprised a single linkage group. We attempted to amplify all loci from all individuals in the population sample in 16 µl PCR reactions using GoTaq Master Mix (Promega Co.). Because we extracted DNA from single haploid gametophytes, we could generate sequences from a single product without cloning. The cycling conditions were 94C for 2 min., then ten cycles of 94C for 15 sec., an annealing temperature of 65C that dropped one degree each cycle, and 72C for 1 min., followed by 20 cycles of 94C for 15 sec., 56C for 30 sec., and 72C for 1 min., and terminating with 72C for 7 min. Sequencing was accomplished, following exo-sap clean-up, on an ABI 3730 XL capillary sequencer at the University of Florida Interdisciplinary Center for Biotechnology Research core facility. Forward and reverse sequence fragments were edited and assembled using Sequencher 4.0 (Gene Codes, Ann Arbor, MI), and all polymorphisms were checked from the chromatograms. Edited sequences were aligned manually in Se-Al 2.0 (http://tree.bio.ed.ac.uk/software/seal/).
Analysis of divergence and polymorphism
For each locus we calculated population genetic summary statistics for the Eastern North American samples using the software Arlequin 3.5 (Excoffier and Lischer 2010). We also coded indels as a biallelic polymorphism in these calculations. For each autosomal locus, and for males and females separately in each sex-linked locus, we calculated the number of segregating sites and estimates of Neµ based on segregating sites (θw) and the average pairwise distance among sequences (θπ). We also calculated the mutation frequency spectrum statistic Tajima’s D, and evaluated whether the value was significantly different from neutral-equilibrium expectations using 1000 permutations. To test whether the males exhibited significantly lower nucleotide diversity than the females, we compared the θw values using a paired t-test. To test whether the autosomal and sex-linked loci differed in their population structure, we conducted an Analysis of Molecular Variance (AMOVA) using Arlequin (Excoffier et al. 1992). We estimated Φst for each autosomal locus separately, and all of the sex-linked loci combined, because several individual loci had no variation in males or females. We generated 95% confidence intervals for the Φst values by bootstrapping.
To identify suitable outgroup species for studying patterns of divergence, we generated a genealogy for each locus using PAUP* ver. 4.10b (Swofford 2003) using neighbor joining with an HKY model to correct for multiple hits. We first tested for evidence of historical recombination in each locus separately, and in the concatenated male and female sequences, using the software DNAsp ver. 5 (Librado and Rozas 2009), and found no recombination events in these analyses. We evaluated the support for the monophyly of the sexes using 1000 bootstrap replicates. To test whether the sex-linked loci had a different mutation rate from the autosomal loci, we compared the average pairwise divergence (DXY) between the C. purpureus sequences from the Eastern North American populations and the outgroup T. cylindricus, using Arlequin. To test whether males and females exhibited different rates of molecular evolution, we performed relative rates tests for each sex-linked locus, with either C. chloropus or T. cylindricus as an outgroup, using the software HyPhy (Nielsen et al. 2005). For these analyses we assumed a GTR model of molecular evolution with invariant sites and a discrete gamma distribution with four rate classes. Finally, to test whether the sex linked loci had experienced a selective sweep, we compared the patterns of polymorphism and divergence between the autosomes and U and V-linked loci using the software MLHKA (Wright and Charlesworth 2004). We conducted analyses with males and females separately, using a coalescent time of 25 and two chains of length 100,000 with different random starting seeds. In both cases we compared a model with no selected loci to a model with five selected loci (the five sex-linked loci for which we had outgroup data) and seven neutral loci (the seven autosomal loci for which we had outgroup data), using a likelihood ratio test with df = 5.
To examine when the sex-linked loci were captured by the sex chromosome (ie, when recombination ceased), we first compared the average pairwise divergence (DUV) between the male and female alignments. To estimate the divergence times we assumed that C. purpureus has an annual life cycle (one generation per year), and used a rate of 1 × 10−8 neutral mutations per site per generation; however, since we have no bryophyte-based calibration for this molecular clock, we provide the dates only as a rough estimate. We generated 1000 coalescent simulations, using the software SIMCOAL2 (Laval and Excoffier 2004), to generate 95% confidence intervals for the DUV divergence times for each locus to evaluate whether all the sex-linked loci were captured by the sex chromosome in a single event. For this analysis we assumed a transition bias of 0.90, a gamma parameter of 0.39, with four rates classes, and that evolving sex linkage resulted in a 10-fold decrease in effective population size, consistent with our nucleotide diversity estimates.
RESULTS
To identify sex-linked genes in C. purpureus, we genotyped 71 ESTs in the progeny of a cross between the GG1 and R40 laboratory isolates using the Sequonom platform, and genetically mapped these loci. We found eight sex linked loci. Transcriptomic data indicate that both the male and female-linked copies of these genes were expressed in protonemal tissue (http://www.ncbi.nlm.nih.gov/sra/?term=ceratodon). The full results of this mapping experiment and analyses of the transcriptomes will be reported elsewhere. Here we evaluate hypotheses regarding the patterns of polymorphism and divergence on the U and V chromosomes of this species using variation in the sex-linked loci and nine autosomal loci in 4 – 8 individuals from each of seven populations in Eastern North America (Table 1).
To understand the population genetic processes shaping variation on the sex chromosomes, we calculated several standard summary statistics (Table 2). For these calculations we included all site types (synonymous sites, non-synonymous sites, and introns) and we coded indels as a two-state character. We believe that the use of non-synonymous mutations is conservative in this case. Because the autosomes had more total variation than the sex-linked loci, the removal of all non-synonymous sites from the analysis disproportionally inflated the nucleotide diversity across the autosomes, potentially biasing our comparisons. Overall, non-synonymous mutations made a modest contribution to the total polymorphism (in four autosomal loci we found a single individual with a non-synonymous mutation, and in two autosomal loci we found two individuals each with a single non-synonymous mutation; in the sex linked loci we found a single locus with one individual with a non-synonymous mutation) and the mean divergence from the outgroup species (two autosomal loci had a single amino acid changing mutation, two loci had three, and one had four; two sex-linked loci each had two non-synonymous mutations). In our divergence analyses, the removal of the non-synonymous sites elevated the per-site distance from the outgroup, but did not qualitatively affect any of the tests we conducted.
Table 2.
Summary statistics for sex-linked and autosomal loci
| Locus | Length | Sample size | Segregating sites | θw | θx | Tajima’s D1 | D uv2 | D xy3 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex linked loci | |||||||||||||
| Female (U) | Male (V) | U | V | U | V | U | V | U | V | ||||
| ga03f07 | 592 | 21 | 26 | 3 | 0 | 0.0014 | 0.0000 | 0.0005 | 0.0000 | −1.77 | 0.00 | 0.012 (0) | 0.0854 (3) |
| rpS18A | 383 | 20 | 23 | 1 | 1 | 0.0007 | 0.0007 | 0.0003 | 0.0002 | −1.16 | −1.29 | 0.0162 (0) | 0.1047 (2) |
| PPR | 351 | 18 | 21 | 1 | 0 | 0.0008 | 0.0000 | 0.0009 | 0.0000 | −0.53 | 0.00 | 0.0264 (0) | |
| CPU011109 | 218 | 21 | 26 | 4 | 1 | 0.0051 | 0.0012 | 0.0018 | 0.0004 | −1.86 | −1.16 | 0.0267 (0) | 0.0577 (0) |
| rpS18 | 836 | 12 | 26 | 2 | 6 | 0.0008 | 0.0019 | 0.0004 | 0.0008 | −1.45 | −1.87 | 0.0562 (5) | |
| CDC2 | 437 | 20 | 20 | 0 | 2 | 0.0000 | 0.0013 | 0.0000 | 0.0011 | 0.00 | −0.29 | 0.057 (1) | |
| rpS15A-2 | 699 | 18 | 25 | 4 | 1 | 0.0017 | 0.0004 | 0.0011 | 0.0001 | −1.03 | −1.16 | 0.0675 (8) | 0.1701 (5) |
| rpS15A-1 | 121 | 15 | 26 | 1 | 0 | 0.0025 | 0.0000 | 0.0022 | 0.0000 | −1.16 | 0.00 | 0.0695 (1) | 0.0852 (0) |
| Autosomal loci | |||||||||||||
| Adk | 798 | 37 | 64 | 0.0192 | 0.0165 | −0.52 | 0.1454 (30) | ||||||
| ho1 | 868 | 32 | 19 | 0.0054 | 0.0021 | 2.10 | |||||||
| phy2 | 867 | 32 | 74 | 0.0212 | 0.0141 | −1.26 | 0.0742 (1) | ||||||
| rpS23 | 551 | 50 | 16 | 0.0065 | 0.0026 | 1.85 | 0.1182 (2) | ||||||
| hp23.9 | 215 | 44 | 13 | 0.0139 | 0.0090 | −1.09 | 0.1035 (2) | ||||||
| KIAA0187 | 523 | 51 | 20 | 0.0085 | 0.0032 | 1.97 | 0.0674 (2) | ||||||
| ga01f09 | 345 | 50 | 38 | 0.0246 | 0.0127 | 1.64 | |||||||
| hp23.3 | 298 | 54 | 16 | 0.0118 | 0.0133 | 0.40 | 0.0715 (1) | ||||||
| TBP | 402 | 53 | 7 | 0.0038 | 0.0008 | 2.05 | 0.0813 (0) | ||||||
Significant Tajima’s D values shown in bold
Average pairwise distance between U and V linked alleles; number of indels is shown in parentheses
Average pairswise distance between C. purpureus and T. cylindricus, with number of indels in parentheses; missing values indicate poor outgroup sequence quality or outgroup nested within ingroup
The mean autosomal nucleotide diversity, estimated as θw, was 0.013 (Table 2, Figure 1), and the values ranged from 0.004 (TPB) to 0.025 (ga01f09). The female values ranged from 0 – 0.0025 and the male values ranged from 0 – 0.0019. The levels of nucleotide diversity in males were lower than that in females, but not significantly so (p = 0.08). No polymorphic sites were shared between the sexes. To evaluate whether the decreased sex chromosome nucleotide diversity could represent a stronger response to a genome wide evolutionary process, such as a change in population size, we calculated Tajima’s D for all loci individually. The Tajima’s D values for the U and V-linked sequences of all the sex-linked loci were all negative (significantly so for rpS18 in males, and ga03f07 and CPU011109 in females), suggesting a recent population expansion or chromosome-wide selective sweep. In contrast, the autosomal loci had heterogeneous Tajima’s D values, with hp23.3 showing a positive value (0.40, potentially indicating some form of balancing selection at this locus, or a closely linked one), and five loci with significantly negative values (ga01f09, TPB, KIAA0187, ho1, rpS23).
Figure 1.
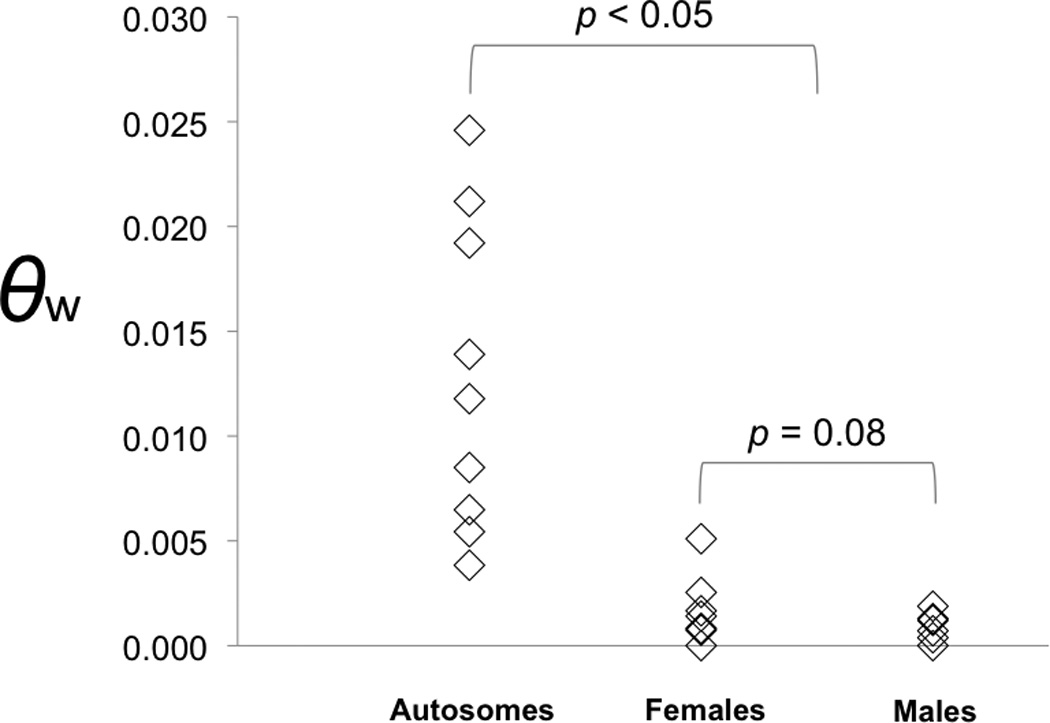
A plot of θw for each autosomal locus separately and the male and female alleles of each sex linked locus. The autosomal loci were significantly more diverse than the sex-linked loci (MLHKA test, p < 0.05); the male alleles were not significantly less diverse than the female alleles at the sex linked loci (paired t-test p = 0.08).
We also calculated the subdivision statistic Φst for each autosomal locus and the combined sex-linked loci. The mean autosomal value of Φst = 0.045, and the values of Φst ranged from 0 – 0.69. The value for the U and V-linked loci was Φst (UV) = 0.0042 (95% CI 0.0025 – 0.0060; Figure 2), significantly less than all but one of the autosomal values.
Figure 2.
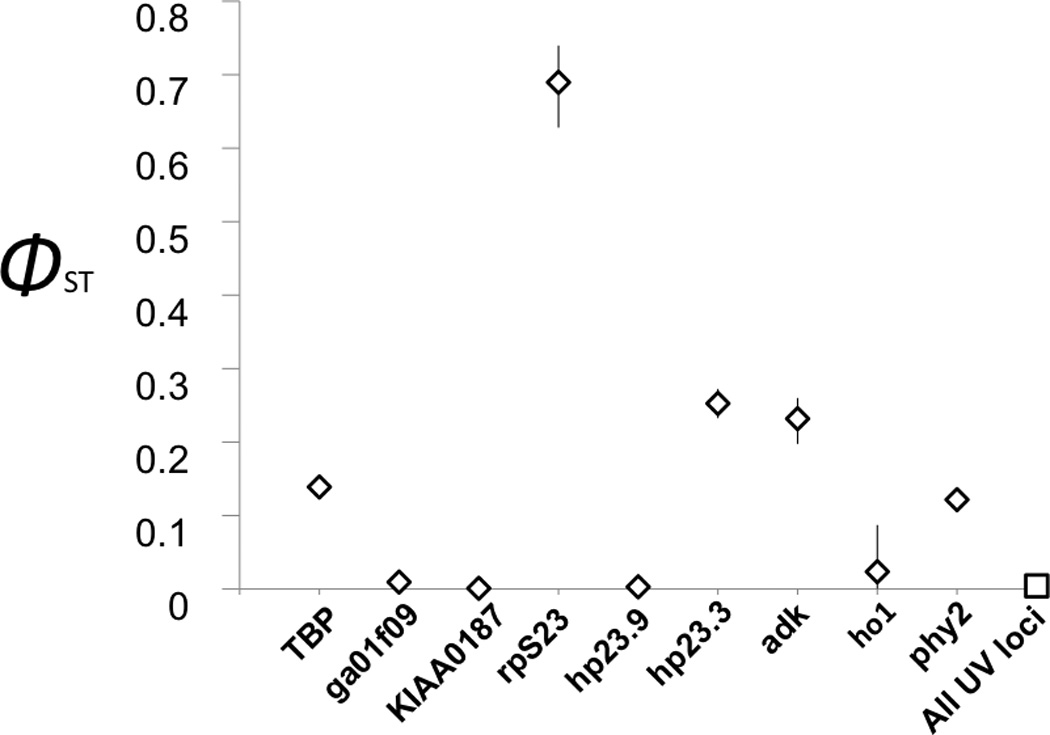
A plot of ΦST from an AMOVA of each autosomal locus separately (open diamonds) and the concatenated sex linked loci (open square), with 95% CI (generated by bootstrapping) indicated by vertical lines.
To test whether the difference in nucleotide diversity between the autosomes and non-recombining sex chromosomes resulted from differences in the mutation rates, we compared the divergence between C. purpureus and closely related species in the two genomic regions. This analysis was complicated by topological differences in placement of the outgroups relative to one another and to C. purpureus, potentially reflecting either low levels introgression between these three species or incomplete lineage sorting. Nevertheless, while the average corrected genetic divergences (DXY) between the C. purpureus sequences and the outgroups were heterogeneous among loci (Supplementary Figure 1a–q), the DXY values generated from loci with outgroup placements that were not obviously influenced by introgression or incomplete lineage sorting were similar for the autosomal (mean = 0.10, range = 0.056 – 0.170) and sex linked loci (mean = 0.095, range = 0.067 – 0.118), suggesting that the sex chromosomes and autosomes have similar ranges of mutation rates. In the five sex-linked loci with no evidence of introgression, the male and female alleles were reciprocally monophyletic with 100 percent bootstrap support (Supplementary Figure 1), and based on relative rates tests we found no evidence that males and females had different mutation rates (data not shown). The MLHKA test indicated that the sex-linked loci in both females and males had significantly less variation than autosomes, taking into account the differences in inheritance and mutation rate (likelihood ratio test for females and males, −2ΔL = 13.56, 18.64; df = 5; p < 0.05, 0.01, respectively), suggesting that the U and V chromosomes each recently experienced a selective sweep.
Our genealogical data showed that the male and female lineages coalesced with one another (Supplementary Figure 1a–h) in at least two discrete events: ga03f07 and rpS18A had U-V coalescence times of 0.6 – 0.8 MYA; PPR and CPU011109, 1.3 MYA (these four loci constitute Group 1 in Figure 3); rpS18 and CDC2 coalesced approximately 2.8 MYA, and rpS15A-1 and rpS15A-2, 3.5 MYA (these loci constitute Group 2). Figure 3 shows the divergence per nucleotide between the U and V allele pairs (DUV), as well as a 95% CI for DUV for each locus generated by simulations. We use the post hoc Groups 1 and 2 only to illustrate the minimum number of capture events necessary to explain the data. In fact, two loci with indistinguishable coalescence times could have been captured by separate events that occurred in rapid succession. The DUV for all loci in Group 1 was significantly more recent that that for all loci in Group 2 (except rpS15A-1 and CPU011109 which were short sequences, and therefore had higher variance). Loci in Group 2 also contained several fixed indel differences between the sexes, while length variation was absent from loci in Group 1. For six of the eight loci, the male and female alleles clearly coalesced after the C. purpureus lineage split from the sister species, while the remaining two loci appear to have experiences introgression events.
Figure 3.
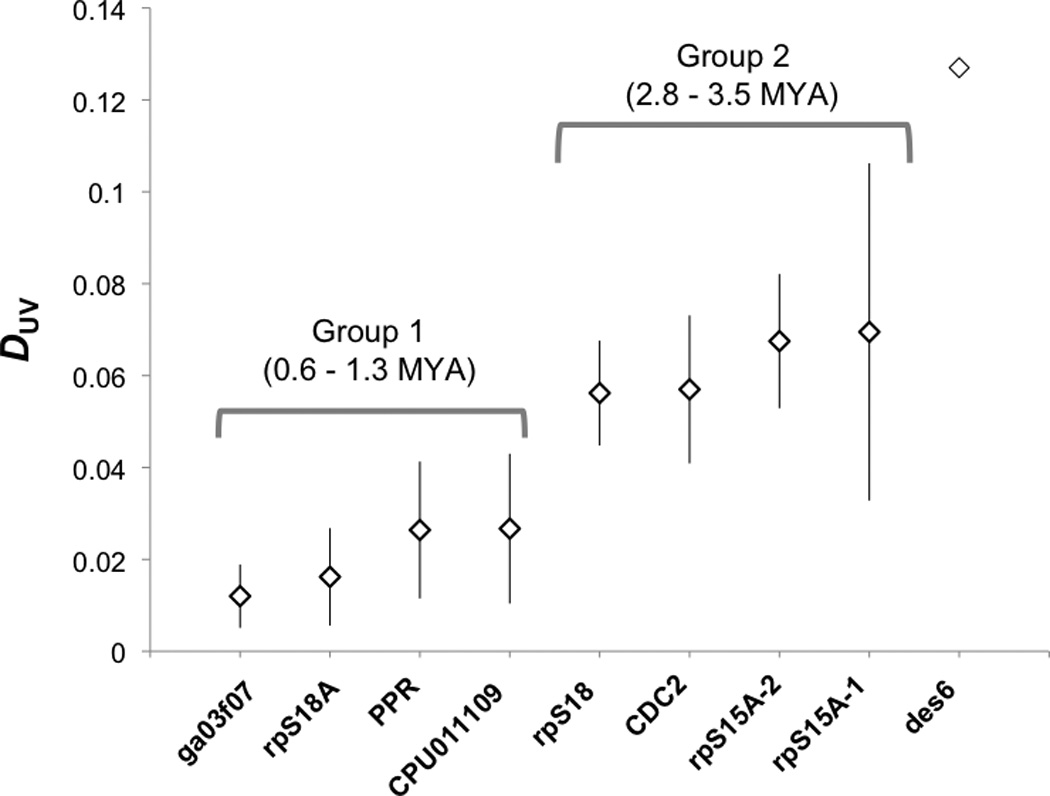
A plot of average pairwise distance (DUV) between the U and V alleles for each sex-linked locus, showing four proposed evolutionary strata on the sex chromosomes, with vertical lines showing the 95% confidence interval generated by simulations. The des6 locus (far right, from McDaniel et al. 2007) is shown for comparison.
DISCUSSION
Non-recombining sex chromosomes are widely expected to be gene-poor descendants of formerly recombining autosomes (Bull 1983; Charlesworth and Charlesworth 2000). Here we provide evidence that the non-recombining sex chromosomes in the moss C. purpureus recently have captured eight genes. The simplest explanation for this pattern is that the non-recombining region expanded stepwise into the adjacent and previously recombining pseudo-autosomal regions (PARs) in at least two events (~ 0.6 −1.3 MYA and 2.8 – 3.5 MYA (Figure 4); the previously studied des6 gene may represent another event at ~6 MYA (McDaniel et al. 2007). The fact that we found homologs on both the U and V chromosomes suggests that these were not translocation events to only the NRU or NRV. Nevertheless, we cannot exclude more complex scenarios involving a gradual expansion of heterochromatin, chromosomal inversions, or heterogeneous patterns of gene conversion between the sex chromosomes (or combinations of these processes) to explain the pattern of divergence times among the sex-linked loci.
Figure 4.

A hypothetical model for the expansion of the NRU-NRV boundary into the pseudoautosomal region (PAR) in two steps, generating loci with two groups of NRU-NRV coalescence times (~3 MYA and ~1 MYA). Because we do not know the physical location of the genes in Groups 1 and 2, our genealogical data are also consistent with more complex scenarios involving more numerous expansions of smaller size, chromosomal inversions, or localized UV gene conversion events.
Finding heterogeneous NRU-NRV divergence values is strikingly similar to the evolutionary strata on the mammalian XY pair (Lahn and Page 1999; Sandstedt and Tucker 2004), the avian ZW (Handley et al. 2004; Nam and Ellegren 2008), the XY chromosomes in the plants S. latifolia and C. papaya (Nicolas et al. 2005; Bergero et al. 2007; Gschwend et al. 2012; Wang et al. 2012) and the mating type loci of the fungus M. violaceum (Votintseva and Filatov 2009). In the other species where strata have been discovered, the genes with lowest divergence (i.e. most recent loss of recombination) cluster close to the PAR boundary, based on genetic map distances inferred from the recombining sex chromosome. In C. purpureus, it is not yet clear whether this is true, because genes in neither the U nor V regions can be genetically mapped due to recombination suppression on both homologous chromosomes. Our current physical map is insufficient to provide a linear order for the loci (McDaniel, SF, unpublished data); the limited physical map information available indicate that probes designed from rpS15A-1 and rpS15A-2 hybridize to the same BAC, perhaps representing a tandem duplication. These two loci do have very similar U-V divergence values, suggesting that the event that led to their ceasing to recombine may have affected both, consistent with their close proximity.
It is noteworthy that on animal sex chromosomes where the possession of separate sexes is ancient the evolutionary strata involve many genes and formed 10 to more than 100 MYA (Sandstedt and Tucker 2004; Nam and Ellegren 2008), while in the angiosperms C. papaya and S. latifolia the strata are quite young (Desfeux et al. 1996; Liu et al. 2004; Wang et al. 2012). The origin of the C. purpureus U-V sex chromosome pair almost certainly predates the gene-capture events that we report here, since the sister species C. chloropus and T. cylindricus are also dioecious (McDaniel et al. 2013b). In fact, our screen was probably biased towards indentifying young sex-linked genes. We sampled only loci that were expressed in a modestly sexually dimorphic tissue (protonema), that had clearly identifiable homologs between C. purpureus and the distantly related P. patens, and that were found in both a male and a female lab strain (McDaniel et al. 2013a). Older sex linked genes, in contrast, are likely to be expressed in more mature, sexually dimorphic tissues, and may be present in one sex and absent in the other. Nevertheless, in spite of these caveats, more than the 10% of genes tested here mapped to the approximately one quarter of the C. purpureus genome (~80 Mbps) that is occupied by the sex chromosome (McDaniel et al. 2007). This finding suggests that the C. purpureus sex chromosomes may be relatively rich in new sex-linked genes, much like Silene latifolia (Chibalina and Filatov 2011).
Whether the genes that have become linked to the U and V chromosomes are beginning to accumulate deleterious mutations is currently uncertain. Loci on these chromosomes may be prevented from degeneration as a result of purifying selection in the haploid gametophyte stage (Bergero and Charlesworth 2011; Chibalina and Filatov 2011). However, the low relative nucleotide variation on the C. purpureus sex chromosomes (Figure 1) suggests that degeneration processes may be occurring, given that the U and V diversity are both significantly lower than half the autosomal estimate (Qiu et al. 2010). Indeed, in the rpS15A-1 locus the male copy had a 14 bp insertion that generated a premature stop codon. Genetic degeneration of sex-linked loci that is unchecked by purifying selection clearly can operate on much shorter timescales than those relevant for the C. purpureus genes we evaluated. In Drosophila miranda, the neo-Y chromosome has lost genes rapidly following the cessation of recombination (Bachtrog 2005; Zhou and Bachtrog 2012). Similarly, genes on the Y chromosome in the flowering plant Silene latifolia show some signs of degeneration, as do non-recombining mating type loci in the fungus Neurospora tetrasperma, including transposable element accumulation, decreased codon bias, and decreased expression of Y-linked loci (Marais et al. 2008; Bergero and Charlesworth 2011; Chibalina and Filatov 2011; Whittle et al. 2011). However, other factors such as the number of genes captured at one time may be positively correlated with the strength of background selection or the probability of selective sweeps on the sex chromosome. For example, a recombination suppression event involving only a modest number of genes would present a smaller target for selected mutations, and therefore promote slower degeneration, than an event involving an entire Drosophila chromosome arm (Bachtrog 2008).
The low nucleotide diversity found in sex-linked genes is most often attributed to the combined effect of selection and low recombination (either purifying selection on variants within the sex-linked region, or fixation of adaptive changes in the NRU or NRV, both of which would result in loss of linked variants throughout these regions). Although distinguishing between positive and negative selection is challenging (Bachtrog 2004), three observations suggest that adaptive evolution is occurring on the sex chromosomes. First, the U and V-linked loci were significantly less diverse than autosomes, based on the MLHKA test, suggested that the sex chromosomes each experienced a recent selective sweep. Given that several autosomal loci also showed significantly negative Tajima’s D values, suggesting that they also may have experienced sweeps, we believe that this test is conservative.
Second, the statistic Φst indicated that the population structure among the Eastern North American populations was significantly lower for the sex-linked loci than for all but two of the autosomal loci (Table 2, Figure 2). This difference is difficult to evaluate in a rigorous framework without a genome-wide distribution of FST but the trend is unambiguous. The low values of Φst are inconsistent with background selection which is predicted to reduce within-population variation, thereby increasing FST (Charlesworth et al. 1997). Global adaptive sweeps, in contrast, increase the probability that migrant U or V chromosomes will replace local variants, homogenizing populations and reducing FST. In fact, we found similar or identical U and V-linked sequences in isolates from Australia and Antarctica (data not shown). Consistent with this interpretation, McDaniel et al. (2007, 2008) showed that two geographically isolated populations were partially reproductively isolated due to several autosomal factors, but that sex-linked loci were little involved; if sex chromosome variants frequently experience strong selection, incompatibilities would have little time to accumulate on these chromosomes.
Third, theory for the evolution of suppressed recombination, in inversions or as additions to sex chromosomes, generally involves either strong genetic drift or some form of adaptive evolution. The large population sizes of C. purpureus make drift-based explanations unlikely (McDaniel and Shaw 2005). Rather, the evidence recent gene capture suggests that there have been multiple additions, potentially followed by global selective sweeps, on the NRU and NRV in the recent past. While more data will almost certainly uncover more subtle evolutionary patterns, in particular regarding evolutionary strata, sex chromosome degeneration, and differences in nucleotide diversity between males and females, the analyses of the eight loci we report clearly illustrate the dynamism of the evolutionary processes shaping the sex chromosomes of C. purpureus.
Supplementary Material
ACKNOWLEDGEMENTS
DNA sequences generated for this work are deposited in GenBank under the numbers JY262665-JY263413
Deborah Charlesworth, Kelly Dyer, Mark Kirkpatrick, and three anonymous reviewers made many helpful comments on earlier drafts of this manuscript, and the Department of Energy – Joint Genome Institute provided advance access to transcript sequence from the C. purpureus genome project. This work was supported by a NIH National Research Service Award to SFM at Washington University in St. Louis, a Pilot Sequencing Grant from the Washington University Genome Sequencing Center to SFM and RSQ, and by research funds from the University of Florida.
LITERATURE CITED
- Abe H, Mita K, Yasukochi Y, Oshiki T, Shimada T. Retrotransposable elements on the W chromosome of the silkworm, Bombyx mori. Cytogenet. Genome Res. 2005;110:144–151. doi: 10.1159/000084946. [DOI] [PubMed] [Google Scholar]
- Allen C. Genetics of bryophytes. I. Bot. Rev. (Lancaster) 1935;1:269–291. [Google Scholar]
- Bachtrog D. Evidence that positive selection drives Y-chromosome degeneration in Drosophila miranda . Nature Genet. 2004;36:518–522. doi: 10.1038/ng1347. [DOI] [PubMed] [Google Scholar]
- Bachtrog D. Sex chromosome evolution: Molecular aspects of Y-chromosome degeneration in Drosophila . Genome Res. 2005;15:1393–1401. doi: 10.1101/gr.3543605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D. A dynamic view of sex chromosome evolution. Curr. Opin. Genet. Dev. 2006;16:578–585. doi: 10.1016/j.gde.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Bachtrog D. The temporal dynamics of processes underlying Y chromosome degeneration. Genetics. 2008;179:1513–1525. doi: 10.1534/genetics.107.084012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D, Kirkpatrick M, Mank JE, McDaniel SF, Pires JC, Rice WR, Valenzuela N. Are all sex chromosomes created equal? Trends in Genetics. 2011;27:350–357. doi: 10.1016/j.tig.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Bergero R, Charlesworth D. Preservation of the Y transcriptome in a 10-million-year-old plant sex chromosome system. Current Biology. 2011;21:1470–1474. doi: 10.1016/j.cub.2011.07.032. [DOI] [PubMed] [Google Scholar]
- Bergero R, Forrest A, Kamau E, Charlesworth D. Evolutionary strata on the X chromosomes of the dioecious plant Silene latifolia: Evidence from new sex-linked genes. Genetics. 2007;175:1945–1954. doi: 10.1534/genetics.106.070110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisang I, Hedenas L. Sex ratio patterns in dioicous bryophytes re-visited. J. Bryol. 2005;27:207–219. [Google Scholar]
- Bull JJ. Sex chromosomes in haploid dioecy: A unique contrast to Muller's Theory for diploid dioecy. Am. Nat. 1978;112:245–250. [Google Scholar]
- Bull JJ. Evolution of sex determining mechanisms. Menlo Park, CA: Benjamin/Cummings; 1983. [Google Scholar]
- Charlesworth B, Charlesworth D. A model for the evolution of dioecy and gynodioecy. Am. Nat. 1978;112:975–997. [Google Scholar]
- Charlesworth B, Charlesworth D. The degeneration of Y chromosomes. Phil. Trans. Roy. Soc. London Ser. B-Biol. Sci. 2000;355:1563–1572. doi: 10.1098/rstb.2000.0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Morgan MT, Charlesworth D. The effect of deleterious mutations on neutral molecular variation. Genetics. 1993;134:1289–1303. doi: 10.1093/genetics/134.4.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Nordborg M, Charlesworth D. The effects of local selection, balanced polymorphism and background selection on equilibrium patterns of genetic diversity in subdivided populations. Genetical Research. 1997;70:155–174. doi: 10.1017/s0016672397002954. [DOI] [PubMed] [Google Scholar]
- Charlesworth D, Charlesworth B. Sex differences in fitness and selection for centric fusions between sex-chromosomes and autosomes. Genet. Res. 1980;35:205–214. doi: 10.1017/s0016672300014051. [DOI] [PubMed] [Google Scholar]
- Chibalina MV, Filatov DA. Plant Y chromosome degeneration is retarded by haploid purifying selection. Curr. Biol. 2011;21:1475–1479. doi: 10.1016/j.cub.2011.07.045. [DOI] [PubMed] [Google Scholar]
- Cove D, Perroud P-F, Charron A, McDaniel S, Khandelwal A. Emerging Model Organisms. New York, NY: Cold Spring Harbor Laboratory Press; 2009. RQuatrano The moss Physcomitrella patens: a novel model system for plant development and genomic studies. [DOI] [PubMed] [Google Scholar]
- Cox CJ, Goffinet B, Wickett NJ, Boles SB, Shaw AJ. Moss diversity: a molecular phylogenetic analysis of genera. Phytotaxa. 2010;9:175–195. [Google Scholar]
- Desfeux C, Maurice S, Henry J-P, Lejeune B, Gouyon P-H. Evolution of reproductive systems in the genus Silene . Proc. Roy. Soc. B. 1996;263:409–414. doi: 10.1098/rspb.1996.0062. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Lischer HEL. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources. 2010;10:564–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Smouse PE, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris PJ, Goodenough UW. The mating-type locus of Chlamydomonas reinhardtii contains highly reaaranged DNA-sequences. Cell. 1994;76:1135–1145. doi: 10.1016/0092-8674(94)90389-1. [DOI] [PubMed] [Google Scholar]
- Gschwend AR, Yu QY, Tong EJ, Zeng FC, Han J, VanBuren R, Aryal R, Charlesworth D, Moore PH, Paterson AH, Ming R. Rapid divergence and expansion of the X chromosome in papaya. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:13716–13721. doi: 10.1073/pnas.1121096109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handley L-JL, Ceplitis H, Ellegren H. Evolutionary strata on the chicken Z chromosome: Implications for sex chromosome evolution. Genetics. 2004;167:367–376. doi: 10.1534/genetics.167.1.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitz E. Geschlectschromosomen bei einem Laubmoos. Berichte der Deutshen Botanischen Gesellshaft. 1932;30:204–206. [Google Scholar]
- Hood ME. Dimorphic mating-type chromosomes in the fungus Microbotryum violaceum . Genetics. 2002;160:457–461. doi: 10.1093/genetics/160.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood ME, Antonovics J, Koskella B. Shared forces of sex chromosome evolution in haploid-mating and diploid-mating organisms: Microbotryum violaceum and other model organisms. Genetics. 2004;168:141–146. doi: 10.1534/genetics.104.029900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ironside JE. No amicable divorce? Challenging the notion that sexual antagonism drives sex chromosome evolution. BioEssays. 2010;32:718–726. doi: 10.1002/bies.200900124. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick M, Barton N. Chromosome inversions, local adaptation and speciation. Genetics. 2006;173:419–434. doi: 10.1534/genetics.105.047985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerich LB, Wang X, Clark AG, Carvalho AB. Low conservation of gene content in the Drosophila Y chromosome. Nature. 2008;456:949–951. doi: 10.1038/nature07463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpelainen H, Bisang I, Hedenas L, Kolehmainen J. The first sex-specific molecular marker discovered in the moss Pseudocalliergon trifarium . Journal of Heredity. 2008;99:581–587. doi: 10.1093/jhered/esn036. [DOI] [PubMed] [Google Scholar]
- Lahn BT, Page DC. Four evolutionary strata on the human X chromosome. Science. 1999;286:964–967. doi: 10.1126/science.286.5441.964. [DOI] [PubMed] [Google Scholar]
- Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newburg L. MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics. 1987;1:174–181. doi: 10.1016/0888-7543(87)90010-3. [DOI] [PubMed] [Google Scholar]
- Laval G, Excoffier L. SIMCOAL 2.0: a program to simulate genomic diversity over large recombining regions in a subdivided population with a complex history. Bioinformatics. 2004;20:2485–2487. doi: 10.1093/bioinformatics/bth264. [DOI] [PubMed] [Google Scholar]
- Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- Lincoln SE, Lander ES. Systematic detection of errors in genetic linkage data. Genomics. 1992;14:604–610. doi: 10.1016/s0888-7543(05)80158-2. [DOI] [PubMed] [Google Scholar]
- Liu Z, Moore PH, Ma H, Ackerman CM, Ragiba M, Pearl HM. A primitive Y chromosome in papaya marks the beginning of sex chromosome evolution. Nature. 2004;427:348–352. doi: 10.1038/nature02228. [DOI] [PubMed] [Google Scholar]
- Marais AGB, Nicolas M, Bergero R, Chambrier P, Kejnovsky E, Moneger F, Hobza R, Widmer A, Charlesworth D. Evidence for degeneration of the Y chromosome in the dioecious plant Silene latifolia . Current Biology. 2008;18:545–549. doi: 10.1016/j.cub.2008.03.023. [DOI] [PubMed] [Google Scholar]
- Matsuba C, Alho JS, Merilä J. Recombination rate between sex chromosomes depends on phenotypic sex in the common frog. Evolution. 2010;64:3634–3637. doi: 10.1111/j.1558-5646.2010.01076.x. [DOI] [PubMed] [Google Scholar]
- Maynard-Smith J, Haigh J. The hitch-hiking effect of a favorable gene. Genetical Research. 1974;23:23–35. [PubMed] [Google Scholar]
- McDaniel S, van Baren M, Jones K, Payton A, Quatrano R. Estimating the nucleotide diversity in Ceratodon purpureus (Hedw.) Brid. from 208 conserved intron-spanning nuclear loci. Applications in Plant Sciences. 2013a doi: 10.3732/apps.1200387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel SF. Genetic correlations do not constrain the evolution of sexual dimorphism in the moss Ceratodon purpureus . Evolution. 2005;59:2353–2361. [PubMed] [Google Scholar]
- McDaniel SF, Atwood J, Burleigh JG. Recurrent evolution of dioecy in bryophytes. Evolution. 2013b;67:567–572. doi: 10.1111/j.1558-5646.2012.01808.x. [DOI] [PubMed] [Google Scholar]
- McDaniel SF, Shaw AJ. Selective sweeps and intercontinental migration in the cosmopolitan moss Ceratodon purpureus (Hedw.) Brid. Mol Ecol. 2005;14:1121–1132. doi: 10.1111/j.1365-294X.2005.02484.x. [DOI] [PubMed] [Google Scholar]
- McDaniel SF, von Stackelberg M, Richardt S, Quatrano RS, Reski R, Rensing SA. The speciation history of the Physcomitrium-Physcomitrella species complex. Evolution. 2010;64:217–231. doi: 10.1111/j.1558-5646.2009.00797.x. [DOI] [PubMed] [Google Scholar]
- McDaniel SF, Willis JH, Shaw AJ. A linkage map reveals a complex basis for segregation distortion in an interpopulation cross in the moss Ceratodon purpureus . Genetics. 2007;176:2489–2500. doi: 10.1534/genetics.107.075424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel SF, Willis JH, Shaw AJ. The genetic basis of developmental abnormalities in interpopulation hybrids of the moss Ceratodon purpureus . Genetics. 2008:1425–1435. doi: 10.1534/genetics.107.086314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menkis A, Jacobson DJ, Gustafsson T, Johannesson H. The mating-type chromosome in the filamentous ascomycete Neurospora tetrasperma represents a model for early evolution of sex chromosomes. Plos Genetics. 2008;4 doi: 10.1371/journal.pgen.1000030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama S, Fujishita M, Sone T, Ohyama K. Additional locus of rDNA sequence specific to the X chromosome of the liverwort, Marchantia polymorpha . Chromosome Research. 2001;9:469–473. doi: 10.1023/a:1011676328165. [DOI] [PubMed] [Google Scholar]
- Nam K, Ellegren H. The chicken (Gallus gallus) Z chromosome contains at least three nonlinear evolutionary strata. Genetics. 2008;180:1131–1136. doi: 10.1534/genetics.108.090324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas M, Marais G, Hykelova V, Janousek B, Laporte V, Vyskot B, Mouchiroud D, Negrutiu I, Charlesworth D, Moneger F. A gradual process of recombination restriction in the evolutionary history of the sex chromosomes in dioecious plants. PLoS Biology. 2005;3:e4. doi: 10.1371/journal.pbio.0030004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Pond SLK, Muse SV. Statistical Methods in Molecular Evolution. New York: Springer; 2005. HyPhy: Hypothesis testing using phylogenies; pp. 125–181. [Google Scholar]
- Nishiyama T, Fujita T, Shin-I T, Seki M, Nishide H, Uchiyama I, Kamiya A, Carninci P, Hayashizaki Y, Shinozaki K, Kohara Y, Hasebe M. Comparative genomics of Physcomitrella patens gametophytic transcriptome and Arabidopsis thaliana: Implication for land plant evolution. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8007–8012. doi: 10.1073/pnas.0932694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto SP, Pannell JR, Peichel CL, Ashman TL, Charlesworth D, Chippindale AK, Delph LF, Guerrero RF, Scarpino SV, McAllister BF. About PAR: The distinct evolutionary dynamics of the pseudoautosomal region. Trends in Genetics. 2011;27:358–367. doi: 10.1016/j.tig.2011.05.001. [DOI] [PubMed] [Google Scholar]
- Perrin N. Sex reversal: a fountain of youth for sex chromosomes? Evolution. 2009;63:3043–3049. doi: 10.1111/j.1558-5646.2009.00837.x. [DOI] [PubMed] [Google Scholar]
- Qiu S, Bergero R, Forrest A, Kaiser VB, Charlesworth D. Nucleotide diversity in Silene latifolia autosomal and sex-linked genes. Proceedings of the Royal Society B: Biological Sciences. 2010;277:3283–3290. doi: 10.1098/rspb.2010.0606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay H, Berrie G. Sex determination in bryophytes. Journal of the Hattori Botanical Laboratory. 1982;52:255–274. [Google Scholar]
- Rice WR. The accumulation of sexually antagonistic genes as a selective agent promoting the evolution of reduced recombination between primitive sex-chromosomes. Evolution. 1987;41:911–914. doi: 10.1111/j.1558-5646.1987.tb05864.x. [DOI] [PubMed] [Google Scholar]
- Sandstedt SA, Tucker PK. Evolutionary strata on the mouse X chromosome correspond to strata on the human X chromosome. Genome Research. 2004;14:267–272. doi: 10.1101/gr.1796204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature. 2003;423:825–837. doi: 10.1038/nature01722. [DOI] [PubMed] [Google Scholar]
- Stark L, McLetchie N, Mishler B. Sex expression and sex dimorphism in sporophytic populations of the desert moss Syntrichia caninervis . Plant Ecology. 2001;157:181–194. [Google Scholar]
- Stöck M, Horn A, Grossen C, Lindtke D, Sermier R, Betto-Colliard C, Dufresnes C, Bonjour E, Dumas Z, Luquet E, Maddalena T, Sousa H, Martinez-Solano I, Perrin N. Ever-young sex chromosomes in European tree frogs. PLoS Biology. 2011;9:e1001062. doi: 10.1371/journal.pbio.1001062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford D. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. Sunderland, MA: Sinauer Associates, Inc.; 2003. [Google Scholar]
- Szovenyi Pt, Rensing SA, Lang D, Wray GA, Shaw AJ. Generation-biased gene expression in a bryophyte model system. Molecular Biology and Evolution. 2011;28:803–812. doi: 10.1093/molbev/msq254. [DOI] [PubMed] [Google Scholar]
- van Doorn GS, Kirkpatrick M. Turnover of sex chromosomes induced by sexual conflict. Nature. 2007;449:909–912. doi: 10.1038/nature06178. [DOI] [PubMed] [Google Scholar]
- Votintseva AA, Filatov DA. Evolutionary strata in a small mating-type-specific region of the smut fungus Microbotryum violaceum . Genetics. 2009;182:1391–1396. doi: 10.1534/genetics.109.103192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JP, Na JK, Yu QY, Gschwend AR, Han J, Zeng FC, Aryal R, VanBuren R, Murray JE, Zhang WL, Navajas-Perez R, Feltus FA, Lemke C, Tong EJ, Chen CX, Wai CM, Singh R, Wang ML, Min XJ, Alam M, Charlesworth D, Moore PH, Jiang JM, Paterson AH, Ming R. Sequencing papaya X and Y-h chromosomes reveals molecular basis of incipient sex chromosome evolution. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:13710–13715. doi: 10.1073/pnas.1207833109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle CA, Sun Y, Johannesson H. Degeneration in codon usage within the region of suppressed recombination in the mating-type chromosomes of Neurospora tetrasperma . Eukaryotic Cell. 2011;10:594–603. doi: 10.1128/EC.00284-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright SI, Charlesworth B. The HKA test revisited: A maximum-likelihood-ratio test of the standard neutral model. Genetics. 2004;168:1071–1076. doi: 10.1534/genetics.104.026500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamato KT, Ishizaki K, Fujisawa M, Okada S, Nakayama S, Fujishita M, Bando H, Yodoya K, Hayashi K, Bando T, Hasumi A, Nishio T, Sakata R, Yamamoto M, Yamaki A, Kajikawa M, Yamano T, Nishide T, Choi SH, Shimizu-Ueda Y, Hanajiri T, Sakaida M, Kono K, Takenaka M, Yamaoka S, Kuriyama C, Kohzu Y, Nishida H, Brennicke A, Shin-i T, Kohara Y, Kohchi T, Fukuzawa H, Ohyama K. Gene organization of the liverwort Y chromosome reveals distinct sex chromosome evolution in a haploid system. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:6472–6477. doi: 10.1073/pnas.0609054104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Bachtrog D. Sex-specific adaptation drives early sex chromosome evolution in Drosophila . Science. 2012;337:341–345. doi: 10.1126/science.1225385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.