

Abstract
The various organs of the body harbor blood vessel networks that display unique structural and functional features; however, the mechanisms that control organ-specific vascular development and physiology remain mostly unknown. In the developing mouse brain, αvβ8 integrin-mediated TGF-β activation and signaling is essential for normal blood vessel growth and sprouting. Whether integrins activate TGF-β signaling pathways in vascular endothelial cells (ECs), neural cells, or both, has yet to be determined. Here, we have generated and characterized mice in which TGF-β receptors are specifically deleted in neuroepithelial cells via Nestin-Cre, or in ECs via a novel Cre transgenic strain (Alk1GFPCre) in which Cre is expressed under control of the endogenous activin receptor-like kinase 1 (Alk1) promoter. We report that deletion of Tgfbr2 in the neuroepithelium does not impact brain vascular development. In contrast, selective deletion of the Tgfbr2 or Alk5 genes in ECs result in embryonic lethality because of brain-specific vascular pathologies, including blood vessel morphogenesis and intracerebral hemorrhage. These data reveal for the first time that αvβ8 integrin-activated TGF-βs regulate angiogenesis in the developing brain via paracrine signaling to ECs.
Keywords: angiogenesis, animal model, cerebral vessel, integrin, TGF-β, vascular development
The elaborate vascular network within the brain develops mainly via angiogenesis, the formation of new blood vessels via endothelial cell (EC) growth and sprouting.1 Proper regulation of angiogenesis in the brain also involves recruitment of pericytes2,3 as well as adhesion and communication with neurons and glia in the local environment.4 These events are orchestrated by an assortment of growth factors, extracellular matrix (ECM) proteins as well as cell surface receptors and intracellular signaling effectors.5 Perturbation of various signaling pathways results in abnormal blood vessel morphogenesis and intracerebral hemorrhage. For example, mice genetically null for transcription factors such as inhibitors of differentiation (Id)1/3,6 Friend leukemia integration 1,7 or cAMP response element-binding protein-binding protein8 develop intracerebral hemorrhage phenotypes. Brain vascular phenotypes have been reported in mice lacking Notch signaling mediators such as presenilin-19 and Numb.10 In addition, canonical Wnt signaling pathways have shown to be essential for normal blood-brain barrier formation and vascular morphogenesis in the embryonic brain. 11–13 Genetic ablation of the EC-expressed orphan G protein-couple receptor, GPR124/Tem5, also causes brain-specific vascular pathologies likely due to impaired angiogenesis. 14
Integrins, which are heterodimeric receptors comprises α- and β-subunits, have important roles in development of most organs and tissues, including the brain.15 Many integrins are expressed in neural and vascular cells in the brain;16 however, αvβ8 integrin is absolutely essential for neural regulation of vascular development. Mouse embryos genetically null for integrin β8 (Itgb8−/−) develop disorganized and distended blood vessels within the ganglionic eminence and display severe intracerebral hemorrhage beginning at embryonic day (E)11.5.17 Adult Itgb8−/− mice also display progressive neurological deficits likely due to prior vascular insults.18 Mice deficient in integrin αv (Itgav−/−) also develop abnormal angiogenesis and intracerebral hemorrhage.19 Furthermore, selective deletion of αv or β8 integrin genes in neuroepithelial cells, but not ECs, causes cerebral hemorrhage phenotypes similar to those that develop in complete knockouts. These data reveal that neuroepithelial cells are the primary cell types in which integrin αvβ8 functions during brain vascular development.20
In vitro experiments have shown that αvβ8 integrin can activate ECM-bound latent TGF-β1 and TGF-β3 by recognizing specific arginine-glycine-aspartic acid (RGD) sequences found within the latent-associated proteins.21,22 Recently, it was shown that mice harboring homozygous mutations of the RGD sequence in TGF-β1 combined with homozygous null alleles of TGF-β3 (Tgfb1RGE/RGETgfb3−/−) developed embryonic intracerebral hemorrhage phenotypes.23 Although this TGF-β1/3 double knockout study supports the hypothesis that neural-expressed integrin αvβ8 mediates TGF-β activation, which in turn controls the properties of neighboring ECs, additional experiments have been necessary for determining whether TGF-β signaling in ECs, neural cells, or both cell types in combination is required for normal angiogenesis in the brain.
TGF-β subfamily signals are transduced by the TGF-β type II receptor (TGFBR2) and the TGF-β type I receptor (TGFBR1; ALK5). TGFBR2 and ALK5 are expressed in numerous cell types and are involved in a variety of cellular processes. 24–27 There have been previous reports of conditional knockout (cKO) mouse strains in which each receptor has been ablated in ECs using several Cre transgenes; for example, when Tgfbr2 or Alk5 was deleted by Tie1-Cre in which Cre activity begins around E8,28Tie1-Cre;Alk5f/f and Tie1-Cre;Tgfbr2f/f mice were lethal before E10.5, recapitulating the null phenotypes. In contrast, EC-specific knockouts of Alk5 or Tgfbr2 by the L1Cre transgenic mice, in which Cre is active beginning at E13.5 within ECs of certain organs including the lungs and brain, were viable and did not exhibit any apparent vascular phenotypes.30 Studies of these TGF-β receptors with Tie2-Cre and Cdh5-CreERT2 showed some brain vascular phenotypes, but these studies were mostly focused on the role of TGF-β receptor signaling in heart development, and did not closely analyze the roles of these receptors in neural cells versus ECs in brain vascular development.31–33
In this study, we report the generation of a novel EC-specific Cre (Alk1GFPCre), in which Cre is active at appropriate developmental stages to allow analysis of cerebral vascular development. Utilizing the Alk1GFPCre and Nestin-Cre transgenes, we demonstrate for the first time that the TGF-β receptors have an essential role in ECs, but not in neural cells, for proper regulation of cerebral vascular development.
Materials and Methods
Mouse Strains and Mating Scheme
Mice were maintained under standard specific-pathogen-free conditions, and all animal procedures performed were reviewed and approved by the University of Florida and University of Texas MD Anderson Cancer Center Institutional Animal Care and Use Committee. Generation of mouse strains (Tgfbr2-floxed, Alk5-floxed, L1Cre, Nestin-Cre, and ROSA26 reporter mice) used in this study was previously described.30,34–37 Mice were on a mixed genetic background of C57BL/6J (B6) and 129SvJ (129). For monitoring the Cre activity, male Alk1GFPCre mice were crossed with female ROSA26 reporter (R26R) line, and embryos at various developmental stages, neonatal pups, and adult mice were collected for X-gal staining. When female mice had a vaginal plug, the stage of the embryo was judged as at E0.5. For obtaining conditional homozygous mutant embryos, Alk1GFPCre/+;Alk5f/+ and Alk1GFPCre/+;Tgfbr2f/+ (or Nestin-Cre;Tgfbr2f/+) males were mated with Alk5f/f and Tgfbr2f/f females, respectively.
Isolation of Primary Neonatal Astrocytes
Astrocytes were cultured as previously described.38 Briefly, neonatal mouse brains (PN1–PN3) were removed from the skull and placed in sterile, ice-cold PBS. Whole neocortices were dissected and the hippocampus was removed to leave only the cortical sheets. The meninges were stripped away, and the cortical sheets were minced with a razorblade and digested for 30 min at 37°C in DMEM containing 150 units/ml collagenase (Worthington) and 40 μg/ml deoxyribonuclease (Sigma). The cortical tissue was then triturated in DMEM containing 10% calf serum (Sigma) and filtered through a 50 μm sterile mesh. The resulting single-cell suspension was plated onto T-75 tissue culture flasks that had been pre-coated with 3 mg/ml mouse laminin (Sigma). After 7–10 days, the astroglial cells formed a confluent monolayer with neurons, oligodendrocytes, and fibroblasts growing on top. These contaminating cells were removed by rotary shaking the flasks overnight at 250 rpm.
Analysis of Tgfbr2-Dependent SMAD2/3 Phosphorylation in Astrocytes
To interrogate TGF-β-induced SMAD phosphorylation, astrocytes were grown on laminin-coated glass coverslips in DMEM containing 10% calf serum (Sigma). Cells were serum starved for 16 h and then stimulated with 1 ng/ml TGF-β1 (R&D Systems). Cells were then fixed in 4% PFA/PBS and immunofluorescently labeled with a rabbit polyclonal antibody recognizing total SMAD2/3. Subcellular localization of SMAD2/3 was then visualized with goat anti-rabbit secondary antibodies conjugated to Alexa488 (Molecular Probes). Alternatively, cells were stimulated with TGF-β1 and then lysed in RIPA buffer (10mM Tris, pH 7.4, 1% NP40, 0.5% deoxycholate, 0.1% SDS, 150mM NaCl, and 1mM EDTA). Protein concentrations in detergent-soluble fractions were determined using a BCA assay kit (Thermo Scientific). Ly-sates (5–10 μg protein per lane) were mixed with 2 × SDS sample buffer, resolved on 10% polyacrylamide gels and then immunoblotted with an anti-phosphorylated-SMAD2 rabbit polyclonal antibody (Cell Signaling Technologies).
X-Gal Staining
Embryos were collected from pregnant R26R female crossed with Alk1GFPCre male mice at various embryonic stages such as E9.5, 10.5, 11.5, 12.5, 15.5, and 17.5 embryonic stages and postnatal stage. For the embryos older than E11.5, internal organs such as lung, liver, heart, intestine, eyes, brain, kidney, and sternum were dissected out for whole mount X-gal staining as described previously.39 Whole mount stained embryos and organs were subjected to paraffin embedding for histological analyses. Histological sections for X-gal stained tissues were counterstained with nuclear fast red.
Histology and Immunohistochemistry
Paraffin-embedded tissues were sectioned in 5–7 μm slices for hematoxylin and eosin or immunohistochemical staining. For EC staining, 1:100 α-PECAM (BioCare, Santa Cruz) using BioCare's Rat on Mouse AP staining kit were used according to the manufacturer's protocol with some modifications. Briefly, after dehydration the sections were treated with 3% H2O2 for 10min, washed with 1 × Tris-buffered saline then treated with trypsin (BioCare, Santa Cruz) mix for 7min at 37 °C The sections were incubated in the primary antibody overnight at 4 °C The alkaline phospho-tase reaction was carried out using BioCare's Vulcan Fast Red Staining kit. For staining of smooth muscle layers, 1:800 α-SMA (Sigma) was carried out using VectaStains's Mouse-On-Mouse kit according to the manufacturer's protocol. Vector DAB staining kit (Vector Laboratory) was used to visualize HRP reaction. All immunostained sections were counterstained with hematoxylin.
Generation and Analysis ofAlk1GFPCre Mice
An Alk1-eGFPCre-knock-in (Alk1GFPCre-KI) vector was constructed as following. SV40 splice donor and acceptor signals (SD/SA), internal ribosomal entry sequence (IRES) and poly(A) signal (pA) were sequentially subcloned into pBluescriptIISK(+), of which XhoI site was deleted, and the eGFPCre fusion gene was subcloned between IRES and pA. This SD/SA-IRES-eGFPCre-pA fragment was replaced with SD/SA-IRES-LacZ fragment in the Alk1-SIBN2 knock-in vector previously generated.39 After electroporation of the linearlized knock-in vector, 365 G418 and FIAU-resistant clones were screened by Southern hybridization analysis by digesting genomic DNA with EcoRI followed by hybridization with a [32P]dCTP labeled 5′ probe (Supplementary Figure 1) for the homologous recombination. Three positive ES clones were obtained. Positive ES cells were injected into B6 blastocysts to generate chimeric mice. The chimeric mice were mated with B6 females and the Alk1GFPCre line was maintained on a mixed 129/B6 hybrid background.
Results
Selective Ablation of TGF-β Receptor in the Embryonic Neuroepithelium (NE) does not Impact Cerebral Vascular Development
Mouse embryos lacking integrin-activated TGF-β1 and TGF-β3 (Tgfb1RGE/RGETgfb3−/−) develop cerebral hemorrhage phenotypes, similar to those phenotypes reported in αv and β8 integrin-deficient mice.20,40 In order to address whether vascular pathologies of the central nervous system (CNS) in Tgfb1RGE/RGETgfb3−/− mice were due to the defects in TGF-β receptor signaling in neural cells we selectively ablated TGF-β receptors in the NE using Nestin-Cre transgenic mice in which the Cre recombinase is expressed in CNS neural progenitor cells beginning at approximately E10. 37,41,42 We analyzed control (Nestin-Cre(+);Tgfbr2f/+ or Nestin-Cre(—);Tgfbr2f/f) and mutant (Nestin-Cre(+); Tgfbr2f/f) embryos, and found that the mutant mice did not display CNS vascular defects at E12.5 (Figures 1a and b), a stage when intracerebral hemorrhage was evident in Nestin-Cre(+);Itgavf/− cKOs (Figure 1c).
Figure 1.

Selective ablation of Tgfbr2 gene expression in embryonic neuroepithelial cells does not lead to brain vascular pathologies. (a, b) Images of E12.5 control (a) and Nestin-Cre(+);Tgfbr2f/f mutant (b) embryos reveal a lack of brain vascular pathologies in the absence of TGF-β receptor signaling in the neuroepithelium. In contrast, E12.5 Nestin-Cre(+);Itgavf/f embryo (c) display intracerebral hemorrhages in the forebrain area (arrow). (d) Genomic DNA isolated from primary neonatal astrocytes was amplified with primers to monitor the presence of the Cre transgene (lower panel) or Cre-mediated deletion of the Tgfbr2-flox allele (top panel). The genotypes of the cells are listed above the top panel. (e) Control and mutant primary astrocytes were stimulated with TGF-β1 for 0, 30, and 60 min. Detergent-soluble cell lysates were immunoblotted with anti-phospho-SMAD2 or anti-actin antibodies. Note the TGF-β-dependent phosphorylation of SMAD2 in control cells, whereas there is no pSMAD2 detected in mutant cell lysates. (f, g) Primary astrocytes isolated from control (f) and mutant (g) P0 pups were stimulated with TGF-β1 for 0, 30, and 60 min. Cells were then immunofluorescently labeled with an anti-SMAD2/3 antibody. Note the TGF-β-mediated accumulation of SMAD2/3 protein in the nuclei of control cells, whereas no SMAD2/3 nuclear accumulation is detected in mutant cells.
Nestin-Cre(+);Tgfbr2f/f cKOs were born in the expected Mendelian ratios. Of 231 neonatal animals analyzed, 55 mice (23%) were Nestin-Cre(+);Tfgbr2f/f. Analysis of postnatal life span in control (n=15) and mutant (n=13) littermates revealed that conditional Tgfbr2 knockouts survived for up to 2 years without developing obvious phenotypes. cKO mice did not display neurological deficits resulting from long-term or acute neurovascular abnormalities or blood-brain barrier disruption. Analysis of neurovascular unit and blood-brain barrier integrity by immuno staining fixed brain sections with anti-mouse IgG and anti-transferrin did not reveal pathological extravasation of these endogenous circulating proteins into the brain parenchyma of Tgfbr2-control of cKO mice (data not shown). These data reveal that ablation of TGF-β receptor signaling in neural cells of the brain does not significantly impact neurovascular development or physiology.
Cre-mediated recombination of the Tgfbr2-flox allele was confirmed by PCR-based analysis of genomic DNA isolated from astrocytes cultured from Nestin-Cre(+);Tgfbr2f/+ newborn mice (Figure 1d). Loss of TGFBR2 signaling was verified in primary astrocytes. We stimulated control and mutant primary astrocytes with TGF-β1 (1 ng/ml), prepared detergent-soluble cell lysates, and immunoblotted resolved proteins with anti-pSMAD2 antibodies. As shown in Figure 1e, we detected a TGF-β1-dependent increase in SMAD2 phosphorylation in control cells, whereas there was no increase in SMAD2 phosphorylation levels in Tgfbr2 mutant astrocytes. These data demonstrate that signaling mediated by TGFBR2 in the embryonic CNS NE is not essential for cerebral vascular development. In addition, TGF-β1 stimulation of control cells resulted in SMAD2/3 translocation from the cytoplasm to the nucleus (Figure 1f); in contrast, mutant cells did not show nuclear translocation of SMAD2/3 (Figure 1g).
Generation and Characterization of Alk1GFPCre-Knockin Mouse Strain as a Novel EC-Specific Cre Transgene
In order to investigate whether TGF-β signaling in ECs has an essential role in cerebral vascular development, we utilized a novel EC-specific Cre deleter line. The Alk1-eGFPCre-knock-in (Alk1GFPCre) mouse line was generated by inserting the eGFPCre fusion gene into the endogenous Alk1 (activin receptor-like kinase 1) locus via homologous recombination (Supplementary Figure 1). This strategy allows for expression of the eGFPCre fusion protein in cells where endogenous Alk1 gene products are normally expressed. The detailed information for vector construction and targeting scheme is described in Materials and methods section (Supplementary Figure 1). Cre activity during development was monitored by analyzing the X-gal staining pattern of Alk1GFPCre/+ R26R mice. Littermates of Alk1GFPCre/+ R26R and Alk1+/+;R26R were used as a control for endogenous lacZ activity at all stages. Consistent to our previous data from Alk1lacZ and Tg(Alk1-lacZ) mice,39,43 the Cre activity was primarily detected in endothelium of developing blood vessels of Alk1GFPCreR26R embryos throughout development.
X-gal-positive cells were detected in the vascular tree of E9.5 embryos with patchy and spotty staining patterns (Figure 2a), and they were detected in more uniform patterns within the vasculature as development progressed (Figures 2b–d). Histological sections of whole mount stained E11.5 embryos demonstrated that the X-gal-positive cells were vascular ECs within large and small vessels surrounding and invading the NE of the embryonic brain (Figure 2e). In the heart, X-gal was found in endocardial cells and mesenchymal cells of the atrioventicular (AV) cushion (Figure 2f). As endogenous Alk1 is not expressed in the AV cushion cells,39 this data indicate that Cre-medicated recombination has occurred in endocardial cells before the endocardium–mesenchymal transition in the artrioventricular region. X-gal-positive cells were also present in the blood vessels in the brain (Figures 2g and h). Strong lacZ expression/activity was detected in the vasculature of most organs during developmental and neonatal stages (Figures 2i–l). In the lungs, vascular ECs were X-gal-positive while the airway epithelial cells and smooth muscle layers of the airways and blood vessels were negative (Figure 2j). The vessels in the glomeruli of developing and neonatal kidney showed X-gal-positive (Figures 2k and l).
Figure 2.

Cre-mediated recombination occurs primarily in vascular ECs of Alk1GFPCre mice. (a–d) Whole mount X-gal staining of Alk1GFPCre/+ ;R26R heads (a–c) and back (d) at E9.5 (a), E10.5 (b), and E11.5 (c, d). X-gal-positive cells were found in the developing vasculature. X-gal staining patterns were heterogenous in E9.5 embryos, but becomes more uniform by E11.5 (c, d). (e, f) Histological sections of whole mount X-gal stained E11.5 embryos. ECs surrounding and invading into the neural tube (NT) are X-gal-positive (e). Mesenchymal cells in the AV cushion as well as endocardial cells of ventricles and atria show X-gal-positive (f). (g, h) Whole mount staining of postnatal (PN)1 (g) and PN30 (h) brains show Cre activity in the brain vasculature. (i, j) Whole mount staining (i) and sections (j) of PN1 lung. (k, l) X-gal staining was detected in the vasculature of nephrons of embryos (k) and PN14 mice (l). Abbreviations: A, atrium; AV, atrioventicular cushion; V, ventricle.
Endothelial-Specific Deletion of Tgfbr2 or Alk5 Results in Intracerebral Hemorrhage
When Tgfbr2 or Alk5 was selectively deleted by Alk1GFPCre mice, no Alk1GFPCre/+Tgfbr2f/f and Alk1GFPCre/+Alk5f/f pups were recovered at birth, indicating these mutant mice were embryonic lethal. Timed mating studies revealed that these mutant mice begin to show obvious phenotypes at E11.5 and died at around E13.5–E15.5 (Table 1).
Table 1. Summary of mutant phenotypes at various developmental stages.
| Stages | No. of mutantsa | Hemorrhages | Cardiac defects | Dead or resorbed* | ||
|---|---|---|---|---|---|---|
|
| ||||||
| Forebrain | Spinal | |||||
| Tgfbr2 | E10.5 | 6 (10) | 0 | 0 | 0 | 0 |
| E11.5 | 17 (23) | 17 | 0 | 0 | 0 | |
| E12.5 | 5 (11) | 5 | 2 | 0 | 0 | |
| E13.5 | 7 (9) | 7 | 5 | 0 | 0 | |
| E14.5 | 4 (4) | 4 | 4 | 0 | 3 | |
| E15.5 | 5 (15) | ND | ND | ND | 5* | |
| PN1 | 0 (110) | 0 | 0 | 0 | 0 | |
| Alk5 | E10.5 | 8 (10) | 0 | 0 | 0 | 0 |
| E11.5 | 12 (14) | 12 | 0 | 0 | 0 | |
| E12.5 | 8 (14) | 8 | 5 | 6 | 0 | |
| E13.5 | 3 (3) | 3 | 2 | 3 | 2 | |
| E14.5 | 3 (5) | ND | ND | ND | 3* | |
| E15.5 | 5 (5) | ND | ND | ND | 5* | |
| PN1 | 0 (106) | 0 | 0 | 0 | 0 | |
Mutants are embryos with Alk1GFPCre/+;Tgfbr2f/f or Alk1GFPCre/+;Alk5f/f genotypes, and the number in parenthesis indicates the number of embryos with control genotypes.
ND, not determined: all of the resorbed mutants exhibited some degrees of cerebral and spinal hemorrhages.
Indicates number of embryos found resorbed.
At E10.5, neither Alk1GFPCre/+Tgfbr2f/f nor Alk1GFPCre/+Alk5f/f embryos exhibited any noticeable phenotypes and were indistinguishable from control littermates (data not shown). At E11.5, however, 100% of mutant embryos exhibited bilateral hemorrhage in forebrain regions (Figures 3a, b and f; Table 1). Intracerebral hemorrhages were progressively more severe in mutant embryos at advanced developmental stages (Figures 3c–e, g and h). The hemorrhage was largely confined to areas of the forebrain (Figures 3c and d), but in some E13.5 and E14.5 embryos hemorrhage was detected in the developing midbrain, hindbrain, and spinal cord (Supplementary Figure 2; Table 1). It is important to note that these mutants did not exhibit obvious hemorrhage in organs other than the brain and spinal cord. Although the phenotypes of Tgfbr2 and Alk5 mutants are remarkably similar to each other, Alk5 mutants died at approximately E13.5, 1 day earlier than Tgfbr2 mutants that died at around E14.5 (Table 1). E13.5 Alk5 mutants showed abnormalities in heart development: pericardial cavity was enlarged and the heart appeared to be underdeveloped (Figure 3d).
Figure 3.

Endothelial cell-specific deletion of Tgfbr2 or Alk5 gene during embryogenesis results in intracerebral hemorrhage. (a–e) Morphologies of Tgfbr2-(a, c, e) and Alk5- (b, d) conditional mutant embryos along with their controls at E11.5 (a, b), E13.5 (c, d), and E14.5 (e). Bilateral hemorrhages in the developing brain were initiated in the ganglionic eminences of the forebrain (arrows). Also note the pericardial edema in Alk5-conditional knockouts at E13.5 (asterisks in d). (f–h) Bilateral hemorrhage develops in brains of mutant embryos at E11.5 (f) and E13.5 (g, h).
Blood Vessels Fail to Invade into the Neuroepithelial Layers in Tgfbr2 and Alk5 Mutants
During development blood vessels in the perineural vascular plexus invade the NE by migrating along neuroepithelial and radial glial cell processes. Histological sections of the Tgfbr2- and Alk5-conditional knockout (cKO) mutant embryos revealed that TGF-β signaling in ECs is necessary for these processes. At E11.5, regularly spaced blood vessels are apparent in the NE layer of control embryos (Figures 4a and b). In both Alk5- and Tgfbr2-cKO mutants, patterns of blood vessel sprouting were abnormal. At E11.5, glomeruloid-like aggregates of CD31-positive microvessels were found within the NE (Figures 4d and f) and these pathologies were even more apparent E12.5 (Supplementary Figures 3g and h). In addition, at E11.5 cavitations had formed in the ventricular zone of mutant animals (Figure 4f). Interestingly, in the mutants no apparent abnormalities in blood vessel architecture outside of the NE, for example, in the perineural vascular plexus, were observed, indicating that TGF-β signaling in ECs has an essential role for the invasion into the NE. Along these lines, angiogenesis in the other organs such as lung and peripheral tissues was also unaffected in the Tgfbr2-and Alk5-cKO mutants (Supplementary Figure 4; Figure 5). As shown in Figure 3d, Alk5-cKO mutants showed defective heart development. Previously, it was shown that atrioventricular (AV) cushions of Tie2-Cre;Alk5f/f mice were hypoplastic.32 Histological analysis revealed that Alk5-cKO heart may have a defect in completion of turning, which results in delayed development of the right ventricle. Abnormal positioning of AV cushion and impaired closure of ventricular septum were found most consistently in the Alk5-cKO mutants (Figures 5d and f), but no apparent hypoplasia of AV cushions was observed. These differences are likely due to differences in the temporal expression of Cre during heart development between Tie2-Cre and Alk1GFPCre lines.
Figure 4.
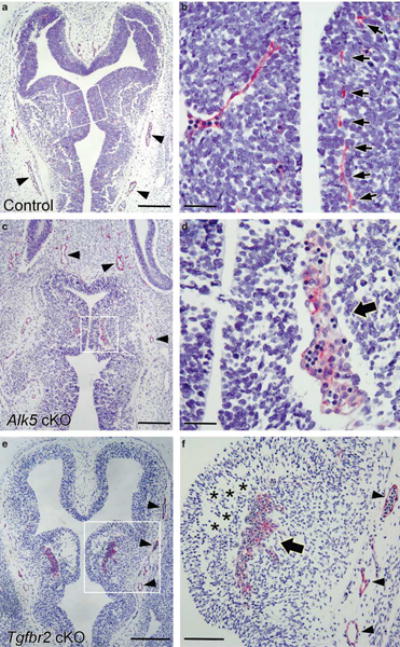
Abnormal blood vessel morphogenesis in the neuroepithelium of Alk5 and Tgfbr2 mutants. Transverse sections through heads of E11.5 control (a, b), Alk1GFPCre/+;Alk5f/f(c, d), and Alk1GFPCre/+;Tgfbr2f/f(e, f) mutants stained with anti-CD31/PECAM antibodies (red). (a, b) In control embryos blood vessels invade into the brain parenchyma and form regularly spaced vascular network (arrows in b). (c–f) In Alk5 and Tgfbr2 mutants an abnormal vascular network is detected in the NE. Note the development of cavitations within the periventricular regions of the ganglionic eminences (asterisks in f). CD31-positive cells at the subventricular zone display abnormal clusters in both mutants (shown by thick arrows in d, f). In contrast, note the apparently normal mutant blood vessels within the perineural vascular plexus (shown by arrowheads in a, c, e, f). Scale bars indicate 500μm (a, c, e), 100 μm (b, d), and 200 μm (f).
Figure 5.
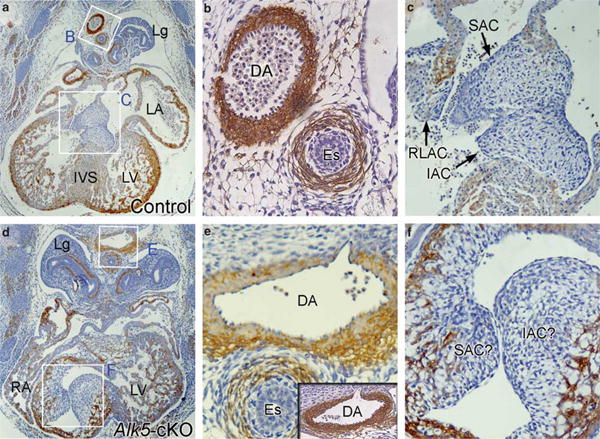
Defective heart development in Alk5-cKO mutants. Transverse sections of an E12.5 control (a–c) and Alk1GFPCre/+;Alk5f/f mutant (d–f) stained with anti-αSMA antibodies (brown color). (a–c) Section of a control embryo show normal development of heart with atrioventricular (AV) cushion, interventricular septum, and trabeculation. Magnified views of dorsal aorta (DA) and AV cushion areas are shown in (b) and (c), respectively. (d–f) Abnormal heart development in Alk5-cKO embryos. Magnified views of DA and AV cushion areas are shown in (e) and (f), respectively. Endocardial cushions are found in an area where the right ventricle should be located, and the ventricular septum did not develop properly. Development of DA is unaffected. Inset in (e) show αSMA-Ab stained DA in the subsequent section of the same Alk5-cKO embryo. Abbreviations: Es, esophagus; IAC, inferior AV cushion; IVS, interventricular septum; Lg, lung; LA, left atrium; LV, left ventricle; RA, right atrium; RLAC, right lateral AV cushion; SAC, superior AV cushion.
Discussion
Here, we have used gene knockout strategies in mice to determine cell type-specific roles for TGF-β receptors in brain vascular development. We show that genetic ablation of Tgfbr2 in CNS neuroepithelial cells does not result in obvious vascular pathologies (Figure 1). In contrast, ablation of Tgfbr2 or Alk5 in ECs leads to abnormal cerebral blood vessel morphogenesis and intracerebral hemorrhage (Figures 3 and 4). These data are the first in vivo evidence demonstrating that TGF-β signaling in ECs, but not in neural cells, is required for normal angiogenesis and blood–brain barrier development during embryogenesis.
Prior reports have shown that both TGF-β1 and TGF-β3 are required for normal regulation of brain vascular development.23 Furthermore, the neuroepithelial cell-expressed integrin αvβ8 has been shown to be a major activator of TGF-β signaling in the developing brain.20,40 However, it remained uncertain whether autocrine or paracrine TGF-β signaling pathways were involved in these processes. The data we report herein provide direct in vivo evidence to support a model in which a paracrine signaling axis—comprises αvβ8 integrin in neuroepithelial cells,17,20 latent TGF-β1 and β3 in vascular basement membranes and canonical TGF-β receptors in vascular ECs—is essential for normal angiogenesis and vascular barrier formation in the embryonic brain.
Numerous knockout mouse models exhibit intracerebral hemorrhage phenotype without apparent defects in vascular beds within other organs, suggesting that brain vascular development is regulated via specific signaling pathways. Our study demonstrates that TGF-β signaling via TGFBR2 and ALK5 in ECs is one such important pathway. Our study also shows that TGF-β signaling in ECs is crucial during specific developmental stages. Genetic ablation of Tgfbr2 or Alk5 using Tie1-Cre leads to yolk sac vascular pathologies at E9.5 and lethality by E11.44 In the Alk1GFPCre strain used in this study, Cre expression occurs approximately 1–2 days later than the other commonly used Tie1-Cre and Tie2-Cre transgenic strains. Owing to the temporal difference in Cre activation, the Alk1GFPCre mice allows for Alk5- and Tgfbr2-cKO embryos survival to E14.5 and beyond, thus revealing the importance of TGF-β signaling in ECs of the brain vasculature. In contrast, cerebral vascular development was unaffected when Tgfbr2 or Alk5 was deleted in ECs with the L1Cre line, which begins to express Cre in brain ECs after E13.5.30 These results indicate that TGF-β signaling in ECs, likely initiated via αvβ8 integrin in perivascular neuroepithelial cells, is crucial for active stages of angiogenesis, but may be dispensable for the maintenance of a more established brain vasculature.
Angiogenesis involves both EC proliferation and directional sprouting, with specialized endothelial tip cells having important roles in directional navigation by interpreting extracellular cues within the tissue microenvironment. We propose that TGF-β receptors in brain endothelial tip cells may serve vital roles in sensing integrin-activated TGF-β1 and TGF-β3 within the neural environment. Various other transmembrane proteins, such as the VEGF receptors Neuropilin 1 and VEGFR2, as well as Notch/Delta family members are expressed in tip cells where they have essential roles in blood vessel sprouting.45 It will be interesting to determine whether TGF-β signaling is integrated with these other pathways and how these various ligand-receptor pairs coordinately control angiogenesis and barrier functions in the brain.
TGF-βs modulate intracellular signaling via SMAD-dependent (canonical) and SMAD-independent (non-canonical) pathways.46 Recent data reveal that cell polarity and directional migration are regulated by TGF-β receptor signaling through components of the Par complex, comprised of Cdc42, Par6, and aPKCζ.47 Hence, it is enticing to speculate that TGF-β-mediated activation of the Par complex, possible in combination with SMAD activation, is necessary for directional navigation of blood vessels as they invade through the brain microenvironment. It remains uncertain whether there is a differential requirement for canonical versus non-canonical TGF-β signaling in angiogenesis in the developing brain; however, the Alk1GFPCre transgenic strain we describe herein will enable us to decipher the exact TGF-β signaling pathways within ECs that are necessary for angiogenesis within the developing brain. The orphan G protein-coupled receptor, GPR124/Tem5, regulates EC directional migration via a Cdc42-dependent pathway, and genetic ablation of GPR124 in ECs leads to brain-specific vascular pathologies.14,48 Hence, TGF-β receptors may function in coordination with GPR124 to regulate the Par complex during EC growth and sprouting in the developing brain.
Supplementary Material
Acknowledgments
We thank Harold L Moses and Stefan Karlsson for Tgfbr2- and Alk5 conditional knockout mouse lines, respectively. We also thank Se-Woon Choi for technical assistance in enhanced imaging process. This work was supported by grants from Novartis Pharmaceuticals Corporation and NHLBI (HL64024) to SPO, NIDDK predoctoral fellowship to H-LN, The Ellison Medical Foundation (AG-NS-0324-06), the NINDS (R01NS059876) and the NCI (P50CA127001) to JHM, and in part by World Class University (WCU) program by Korean Ministry of Education, Science and Technology to SPO.
Footnotes
Supplementary Information accompanies the paper on the Laboratory Investigation website (http://www.laboratoryinvestigation.org)
DISCLOSURE/CONFLICT OF INTEREST: The authors declare no conflict of interest.
References
- 1.Mancuso MR, Kuhnert F, Kuo CJ. Developmental angiogenesis of the central nervous system. Lymphat Res Biol. 2008;6:173–180. doi: 10.1089/lrb.2008.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armulik A, Genove G, Mae M, et al. Pericytes regulate the blood-brain barrier. Nature. 2010;468:557–561. doi: 10.1038/nature09522. [DOI] [PubMed] [Google Scholar]
- 3.Daneman R, Zhou L, Kebede AA, et al. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McCarty JH. Integrin-mediated regulation of neurovascular development, physiology and disease. Cell Adh Migr. 2009;3:211–215. doi: 10.4161/cam.3.2.7767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCarty JH. Cell adhesion and signaling networks in brain neurovascular units. Curr Opin Hematol. 2009;16:209–214. doi: 10.1097/MOH.0b013e32832a07eb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lyden D, Young AZ, Zagzag D, et al. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature. 1999;401:670–677. doi: 10.1038/44334. [DOI] [PubMed] [Google Scholar]
- 7.Spyropoulos DD, Pharr PN, Lavenburg KR, et al. Hemorrhage, impaired hematopoiesis, and lethality in mouse embryos carrying a targeted disruption of the Fli1 transcription factor. Mol Cell Biol. 2000;20:5643–5652. doi: 10.1128/mcb.20.15.5643-5652.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanaka Y, Naruse I, Hongo T, et al. Extensive brain hemorrhage and embryonic lethality in a mouse null mutant of CREB-binding protein. Mech Dev. 2000;95:133–145. doi: 10.1016/s0925-4773(00)00360-9. [DOI] [PubMed] [Google Scholar]
- 9.Shen J, Bronson RT, Chen DF, et al. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- 10.Zhong W, Jiang MM, Schonemann MD, et al. Mouse numb is an essential gene involved in cortical neurogenesis. Proc Natl Acad Sci USA. 2000;97:6844–6849. doi: 10.1073/pnas.97.12.6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liebner S, Corada M, Bangsow T, et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol. 2008;183:409–417. doi: 10.1083/jcb.200806024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stenman JM, Rajagopal J, Carroll TJ, et al. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science. 2008;322:1247–1250. doi: 10.1126/science.1164594. [DOI] [PubMed] [Google Scholar]
- 13.Daneman R, Agalliu D, Zhou L, et al. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci USA. 2009;106:641–646. doi: 10.1073/pnas.0805165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuhnert F, Mancuso MR, Shamloo A, et al. Essential regulation of CNS angiogenesis by the orphan G protein-coupled receptor GPR124. Science. 2010;330:985–989. doi: 10.1126/science.1196554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 16.Milner R, Campbell IL. The integrin family of cell adhesion molecules has multiple functions within the CNS. J Neurosci Res. 2002;69:286–291. doi: 10.1002/jnr.10321. [DOI] [PubMed] [Google Scholar]
- 17.Zhu J, Motejlek K, Wang D, et al. beta8 integrins are required for vascular morphogenesis in mouse embryos. Development. 2002;129:2891–2903. doi: 10.1242/dev.129.12.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mobley AK, Tchaicha JH, Shin J, et al. Beta8 integrin regulates neurogenesis and neurovascular homeostasis in the adult brain. J Cell Sci. 2009;122:1842–1851. doi: 10.1242/jcs.043257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bader BL, Rayburn H, Crowley D, et al. Extensive vasculogenesis, angiogenesis, and organogenesis precede lethality in mice lacking all alpha v integrins. Cell. 1998;95:507–519. doi: 10.1016/s0092-8674(00)81618-9. [DOI] [PubMed] [Google Scholar]
- 20.McCarty JH, Lacy-Hulbert A, Charest A, et al. Selective ablation of alphav integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development. 2005;132:165–176. doi: 10.1242/dev.01551. [DOI] [PubMed] [Google Scholar]
- 21.Mu D, Cambier S, Fjellbirkeland L, et al. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cambier S, Gline S, Mu D, et al. Integrin alpha(v)beta8-mediated activation of transforming growth factor-beta by perivascular astrocytes: an angiogenic control switch. Am J Pathol. 2005;166:1883–1894. doi: 10.1016/s0002-9440(10)62497-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mu Z, Yang Z, Yu D, et al. TGFbeta1 and TGFbeta3 are partially redundant effectors in brain vascular morphogenesis. Mech Dev. 2008;125:508–516. doi: 10.1016/j.mod.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 24.Wang YQ, Sizeland A, Wang XF, et al. Restricted expression of type-II TGF beta receptor in murine embryonic development suggests a central role in tissue modeling and CNS patterning. Mech Dev. 1995;52:275–289. doi: 10.1016/0925-4773(95)00408-s. [DOI] [PubMed] [Google Scholar]
- 25.Iseki S, Osumi-Yamashita N, Miyazono K, et al. Localization of transforming growth factor-beta type I and type II receptors in mouse development. Exp Cell Res. 1995;219:339–347. doi: 10.1006/excr.1995.1237. [DOI] [PubMed] [Google Scholar]
- 26.Tomoda T, Shirasawa T, Yahagi YI, et al. Transforming growth factor-beta is a survival factor for neonate cortical neurons: coincident expression of type I receptors in developing cerebral cortices. Dev Biol. 1996;179:79–90. doi: 10.1006/dbio.1996.0242. [DOI] [PubMed] [Google Scholar]
- 27.Seki T, Hong KH, Oh SP. Nonoverlapping expression patterns of ALK1 and ALK5 reveal distinct roles of each receptor in vascular development. Lab Invest. 2006;86:116–129. doi: 10.1038/labinvest.3700376. [DOI] [PubMed] [Google Scholar]
- 28.Gustafsson E, Brakebusch C, Hietanen K, et al. Tie-1-directed expression of Cre recombinase in endothelial cells of embryoid bodies and transgenic mice. J Cell Sci. 2001;114:671–676. doi: 10.1242/jcs.114.4.671. [DOI] [PubMed] [Google Scholar]
- 29.Carvalho RL, Jonker L, Goumans MJ, et al. Defective paracrine signalling by TGFbeta in yolk sac vasculature of endoglin mutant mice: a paradigm for hereditary haemorrhagic telangiectasia. Development. 2004;131:6237–6247. doi: 10.1242/dev.01529. [DOI] [PubMed] [Google Scholar]
- 30.Park SO, Lee YJ, Seki T, et al. ALK5- and TGFBR2-independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2 (HHT2) Blood. 2008;111:633–642. doi: 10.1182/blood-2007-08-107359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiao K, Langworthy M, Batts L, et al. Tgfbeta signaling is required for atrioventricular cushion mesenchyme remodeling during in vivo cardiac development. Development. 2006;133:4585–4593. doi: 10.1242/dev.02597. [DOI] [PubMed] [Google Scholar]
- 32.Sridurongrit S, Larsson J, Schwartz R, et al. Signaling via the Tgf-beta type I receptor Alk5 in heart development. Dev Biol. 2008;322:208–218. doi: 10.1016/j.ydbio.2008.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robson A, Allinson KR, Anderson RH, et al. The TGFbeta type II receptor plays a critical role in the endothelial cells during cardiac development. Dev Dyn. 2010;239:2435–2442. doi: 10.1002/dvdy.22376. [DOI] [PubMed] [Google Scholar]
- 34.Chytil A, Magnuson MA, Wright CV, et al. Conditional inactivation of the TGF-beta type II receptor using Cre:Lox. Genesis. 2002;32:73–75. doi: 10.1002/gene.10046. [DOI] [PubMed] [Google Scholar]
- 35.Larsson J, Blank U, Helgadottir H, et al. TGF-beta signaling-deficient hematopoietic stem cells have normal self-renewal and regenerative ability in vivo despite increased proliferative capacity in vitro. Blood. 2003;102:3129–3135. doi: 10.1182/blood-2003-04-1300. [DOI] [PubMed] [Google Scholar]
- 36.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 37.Tronche F, Kellendonk C, Kretz O, et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- 38.Tchaicha JH, Mobley AK, Hossain MG, et al. A mosaic mouse model of astrocytoma identifies alphavbeta8 integrin as a negative regulator of tumor angiogenesis. Oncogene. 2010;29:4460–4472. doi: 10.1038/onc.2010.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seki T, Yun J, Oh SP. Arterial endothelium-specific activin receptor-like kinase 1 expression suggests its role in arterialization and vascular remodeling. Circ Res. 2003;93:682–689. doi: 10.1161/01.RES.0000095246.40391.3B. [DOI] [PubMed] [Google Scholar]
- 40.Proctor JM, Zang K, Wang D, et al. Vascular development of the brain requires beta8 integrin expression in the neuroepithelium. J Neurosci. 2005;25:9940–9948. doi: 10.1523/JNEUROSCI.3467-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graus-Porta D, Blaess S, Senften M, et al. Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron. 2001;31:367–379. doi: 10.1016/s0896-6273(01)00374-9. [DOI] [PubMed] [Google Scholar]
- 42.Haigh JJ, Morelli PI, Gerhardt H, et al. Cortical and retinal defects caused by dosage-dependent reductions in VEGF-A paracrine signaling. Dev Biol. 2003;262:225–241. doi: 10.1016/s0012-1606(03)00356-7. [DOI] [PubMed] [Google Scholar]
- 43.Seki T, Hong KH, Yun J, et al. Isolation of a regulatory region of activin receptor-like kinase 1 gene sufficient for arterial endothelium-specific expression. Circ Res. 2004;94:e72–e77. doi: 10.1161/01.RES.0000127048.81744.31. [DOI] [PubMed] [Google Scholar]
- 44.Carvalho RL, Itoh F, Goumans MJ, et al. Compensatory signalling induced in the yolk sac vasculature by deletion of TGFbeta receptors in mice. J Cell Sci. 2007;120:4269–4277. doi: 10.1242/jcs.013169. [DOI] [PubMed] [Google Scholar]
- 45.Phng LK, Gerhardt H. Angiogenesis: a team effort coordinated by notch. Dev Cell. 2009;16:196–208. doi: 10.1016/j.devcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 46.Massague J. TGFbeta in cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bose R, Wrana JL. Regulation of Par6 by extracellular signals. Curr Opin Cell Biol. 2006;18:206–212. doi: 10.1016/j.ceb.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 48.Anderson KD, Pan L, Yang XM, et al. Angiogenic sprouting into neural tissue requires Gpr124, an orphan G protein-coupled receptor. Proc Natl Acad Sci U SA. 2011;108:2807–2812. doi: 10.1073/pnas.1019761108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.