

Abstract
Purpose.
We examined the signaling mechanisms involved in δ-opioid–receptor agonist, SNC-121–mediated attenuation of TNF-α–induced matrix metalloproteinase-2 (MMP-2) secretion from human optic nerve head (ONH) astrocytes.
Methods.
Human ONH astrocytes were treated with SNC-121 (1 μmol/L) for 15 minutes followed by TNF-α (25 ng/mL) treatment for 6 or 24 hours. Cells were pretreated with inhibitors of p38 mitogen-activated protein (MAP) kinase (SB-203580) or NF-κB (Helenalin) prior to TNF-α treatment. Changes in phosphorylation and expression of p38 MAP kinase, IκBα, NF-κB, and MMP-2 were measured by Western blotting. Translocation of NF-κB was determined by immunocytochemistry.
Results.
TNF-α treatment increased MMP-2 secretion from ONH astrocytes to 236% ± 17% and 142% ± 8% at 6 and 24 hours, respectively; while SNC-121 treatment reduced MMP-2 secretion to 149% ± 11% and 108% ± 7% at 6 and 24 hours, respectively. The SNC-121-mediated inhibitory response was blocked by the δ-opioid–receptor antagonist naltrindole. TNF-α treatment resulted in a sustained phosphorylation of p38 MAP kinase up to 24 hours (226% ± 15% over control levels), which was reduced to 150% ± 20% by SNC-121 treatment. TNF-α treatment increased the expression of NF-κB to 179% ± 21% and 139% ± 6% at 6 and 24 hours, respectively, which was significantly blocked by SNC-121 treatment. Furthermore, TNF-α–induced MMP-2 secretion was blocked by 100% and 78% in the presence of SB-203580 and Helenalin, respectively.
Conclusions.
Evidence is provided that SNC-121 attenuated TNF-α–induced MMP-2 secretion from ONH astrocytes. Data also supported the idea that p38 MAP kinase and NF-κB played central roles in TNF-α–induced MMP-2 secretion, and both were negatively regulated by SNC-121.
Keywords: optic nerve head, astrocytes, glaucoma, matrix metalloproteinases, opioids
δ-opioid agonist can directly mitigate the destabilization/remodeling of the optic nerve by blocking the TNF-α–induced production of MMP-2.
Introduction
Glaucoma is an optic neuropathy and a leading cause of blindness worldwide. It is characterized by loss of the retinal ganglion cells leading to progressive loss of vision. It is believed that the site of damage to the neurons is at the level of the axons as they pass through the lamina cribrosa in the optic nerve head (ONH). Studies in glaucomatous humans and primates have established a relationship between chronic elevation of intraocular pressure (IOP) and extensive remodeling of the ONH tissues, which is clinically known as cupping of the optic disc. The ONH astrocytes are the major glial cell type in the nonmyelinated region of the optic nerve head in most mammalian species. As a consequence of injury, such as that seen in glaucoma, astrocytes become activated, produce proinflammatory cytokines, chemokines, reactive oxygen species, and matrix metalloproteinases (MMPs).1–3 Astrocyte-activation initiates both protective and neurotoxic pathways, and it is increasingly associated with worsened outcome in the injured CNS.4 However, the signaling mechanisms that are initiated in response to astrocyte activation remain largely unknown.
Matrix metalloproteinases are a family of neutral zinc endopeptidases that regulate cellular activity in various ways. These include extracellular proteolytic release of extracellular matrix (ECM)-sequestered molecules and shedding of cell surface proteins that transduce signals from the extracellular environment. Currently, more than 25 MMPs have been described.5 Soluble MMPs are secreted as proenzymes (latent forms) and activated in the extracellular environment. Latent MMPs can be activated by plasmin, urokinase-type plasminogen activator (uPA), and tissue-type plasminogen activator (tPA).6 Matrix metalloproteinase-2 (MMP-2) or gelatinase-A degrades gelatin, type IV and V collagen, fibronectin, and elastin.7 Activation of MMP-2 occurs primarily at the cell surface through a unique mechanism that involves MT-MMPs and TIMP-2.8 In this process, TIMP-2 tethers pro-MMP-2 to the membrane region by binding and inhibiting one MT-MMP protein through the TIMP-2 N-terminus, followed by binding of the TIMP-2 C-terminal domain to the hemopexin domain of pro-MMP-2. The pro-MMP-2 is then cleaved and activated by an adjacent MT-MMP enzyme. The increased expression of MMP-2, along with other MMPs and cytokines in the glaucomatous optic nerve head, suggests a possible role for MMP-2 in the tissue remodeling of the ONH.9,10 Although a direct pathological role for MMPs in glaucoma is unclear, they have been suggested to play key roles in destabilization of the optic nerve head by excessive remodeling, thereby predisposing retinal ganglion cells (RGCs) to damage.9,11,12 The cellular source and signaling pathways that regulate MMP-2 production during glaucomatous injury have remained poorly defined.
TNF-α upregulation has been reported in numerous neurodegenerative diseases.13,14 We and others have shown that TNF-α is upregulated in the ischemic retina,15 and the glaucomatous optic nerve head and retina.9,16–19 In addition, the expression of TNF-α receptors on astrocytes and axons of the glaucomatous optic nerve head suggest that TNF-α is stimulating cytodestructive processes in both the astrocytes and axons in the optic nerve head. It is believed that TNF-α plays a crucial role in the RGC death process during glaucomatous neurodegeneration.19,20
Recently, we have demonstrated that δ-opioid–receptor activation provides retina neuroprotection against glaucomatous injuries16 via inhibition of TNF-α production and p38 MAP kinase activation in the optic nerve. However, the cellular events that are involved in δ-opioid receptor–mediated retina neuroprotection against glaucomatous injury have remained poorly described. To throw more light on the signaling aspects, we determined the active participation of p38 MAP kinase and NF-κB in TNF-α–induced MMP-2 secretion from ONH astrocytes. The data presented herein provided evidence that δ-opioid-receptor activation by the exogenous ligand, SNC-121, attenuated the TNF-α–induced MMP-2 production from ONH astrocytes. The mechanistic data provided clues that SNC-121 blocked the sustained activation of p38 MAP kinase and NF-κB, which subsequently reduced the TNF-α–induced MMP-2 secretion from ONH astrocytes.
Methods
Optic Nerve Head Astrocyte Cultures
Ten human eyes from five donors were obtained from the tissue bank (National Disease Research Interchange, Philadelphia, PA) for isolation of ONH astrocytes as previously described.15 Briefly, optic nerve head tissue free of scleral tissue and central retinal vessels was cut into four pieces and placed onto a collagen-I–coated cell culture plate and allowed to grow 2 to 3 weeks in DMEM (Cellgro, Manassas, VA) with 10% fetal bovine serum (Hyclone, Logan, UT) and antibiotics (Cellgro, Manassas, VA). ONH astrocytes from the mixed cell culture were purified by immunopanning as described earlier.15 Briefly, culture plates were coated with the C5-neuroepithelial monoclonal antibody, which was obtained from the Developmental Studies Hybridoma Bank at the University of Iowa (Iowa City, IA). The mixed population of cells derived from tissue explants was trypsinized and resuspended in DMEM and placed on the C5-antibody–coated plates for 40 minutes. Nonadherent cells were removed and adherent cells were cultured in astrocyte growth medium (Lonza, Walkersville, MD) containing 3% fetal bovine serum. The purity of the astrocyte culture was determined by positive immunostaining for the astrocyte markers GFAP (glial fibrillary-acidic protein) and NCAM (a cell surface adhesion-molecule) in each batch. ONH astrocytes were pretreated with the δ-opioid agonist, SNC-121 (1 μmol/L), for 15 minutes, followed by treatment with 25 ng/mL TNF-α for 6 or 24 hours. Cells from 3 to 6 passages were used in these studies. Each experimental condition was evaluated using astrocyte cultures obtained from at least three different donors.
Immunocytochemistry
Primary cultures of human ONH astrocytes grown in two-chamber slides were treated with SNC-121 (1 μM/L) for 15 minutes followed by TNF-α treatment for 6 hours. After incubation, the cells were washed with Tris-buffered saline (TBS), fixed with 4% paraformaldehyde for 15 minutes, and subsequently washed three times with TBS followed by treatment with 0.2% Triton X-100 for 3 minutes. After washing, cells were blocked by 5% BSA for 1 hour at room temperature. Cells were then incubated with primary antibodies (e.g., anti-NF-κB; Cell Signaling, Danvers, MA) at a 1:100 dilution in 0.2% BSA for 3 hours at room temperature. Cells were then washed with TBS and incubated with AlexaFlour 488 conjugated secondary antibody (anti-rabbit; Jackson Immuno Research Laboratories, Inc., West Grove, PA) at a 1:400 dilution at room temperature for 1 hour. Negative control slides were incubated with 0.2% BSA in place of the primary antibody. The cells were observed under a bright-field microscope equipped with epifluorescence, and digitized images were captured by a digital camera. All experiments were performed at least in triplicate in astrocyte cultures from three different donors.
Western Blot Analysis
After drug treatment for 6 or 24 hours, medium was collected and concentrated by using an ultrafiltration centrifugal concentrator (10-kDa cutoff, Centricon; Amicon, Beverly, MA) and adjusted to a final concentration ratio of 10:1. Equivalent amounts of cell lysates of human ONH astrocytes (15 μg protein/lane), or equal volumes of conditioned media (40 μL) were subjected to 10% SDS-PAGE, where proteins were separated and transferred to nitrocellulose membranes as described earlier.15 After blocking, membranes were incubated for 12 hours at 4°C with appropriate primary antibodies (e.g., anti-MMP-2 [Calbiochem EMD, Billerica, MA]; anti-phospho-p38 MAP kinase; anti-NF-κB [Cell Signaling]; anti-IκBα [Cell Signaling]; or anti-β-actin [Sigma-Aldrich, St. Louis, MO]) at 1:1000 dilution. After washing, membranes were incubated for 1 hour at 20°C with appropriate secondary antibodies (horseradish peroxidase [HRP]-conjugated; dilution 1:3000, Sigma-Aldrich, St. Louis, MO). For chemiluminescent detection, the membranes were treated with enhanced chemiluminescent reagent, and the signal was monitored using a commercial imaging system (Bio-Rad VersaDoc; Bio-Rad, Hercules, CA).
Zymography
For zymography, concentrated medium was subjected to 10% SDS-PAGE containing 1 mg/mL gelatin under nonreducing conditions. After electrophoresis, gels were washed twice in 50 mM Tris-HCl buffer (pH 7.5) containing 2.5% Triton X-100 for 30 minutes followed by incubation in activation buffer (50 mM Tris-HCl, pH 7.5 containing 10 mM CaCl2) for 18 hours at 37°C to allow enzymatic degradation of the substrate. Gels were stained with Coomassie blue R-250 and destained. Digestion of the substrate (gelatin) at the position of the enzyme was observed as a clear area in the otherwise uniformly dark-staining gel. The density of digested areas was measured by densitometry and normalized with total cellular protein.
Statistical Analysis
Statistical comparisons were made using the Student's t-test for paired data or ANOVA using the Bonferroni posttest for multiple comparisons (GraphPad Software, Inc., San Diego, CA), and P ≤ 0.05 was considered significant.
Results
TNF-α–Induced-Secretion of MMP-2 From ONH Astrocytes
ONH astrocytes were treated with TNF-α and MMP-2 secretion was measured at 6 and 24 hours. Preliminary studies in our laboratory demonstrated that 25 ng/mL of TNF-α provided optimal secretion of MMP-2 (data not shown), thus this concentration was chosen for all subsequent studies. As shown in Figure 1, TNF-α (25 ng/mL) increased the secretion of MMP-2 to 236% ± 17% (Fig. 1A) and 142% ± 8% (Fig. 1C) over basal levels at 6 and 24 hours, respectively. To determine if δ-opioid–receptor activation by a selective agonist, SNC-121, counterbalanced the TNF-α–induced secretion of MMP-2, ONH astrocytes were pretreated with 1 μM SNC-121 prior to the TNF-α treatment. Treatment of the cells with SNC-121 significantly reduced the secretion of MMP-2 to 149% ± 11% (P < 0.001) and 108% ± 7% (P < 0.016) at 6 and 24 hours, respectively (Figs. 1A, 1B). The activity of MMP-2 (molecular weight, 66 kDa; active-MMP-2) was measured by zymogram. As shown in Figure 2, TNF-α increased the MMP-2 activity to 222% ± 23% (Fig. 2A) and 139% ± 7% (Fig. 2B) over basal levels at 6 and 24 hours, respectively. The TNF-α–induced increase in MMP-2 activity was reduced to 112% ± 3% and 94% ± 12% at 6 and 24 hours, respectively, in the presence of SNC-121. To confirm the participation of δ-opioid receptors in this response, ONH astrocytes were pretreated with a highly selective δ-opioid–receptor antagonist, naltrindole, prior to SNC-121 treatment. As shown in the Table, naltrindole itself had no significant effect on MMP-2 secretion between 6 and 24 hours; however, SNC-121-mediated inhibition of TNF-α–induced-MMP-2 secretion was completely reversed by naltrindole treatment.
Figure 1.
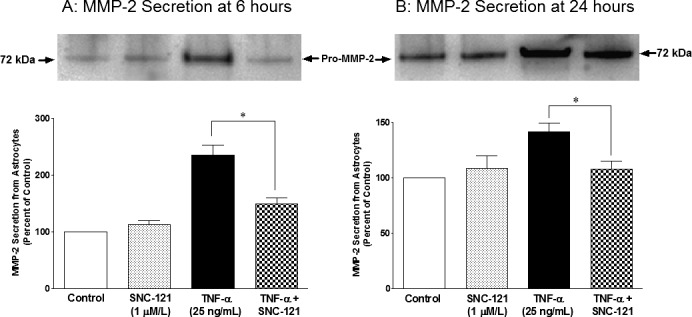
TNF-α–induced secretion of MMP-2 from ONH astrocytes at 6 (A) and 24 (B) hours. ONH astrocytes were starved overnight in serum-free medium. Cells were then pretreated with SNC-121 (1 μmol/L) for 15 minutes followed by TNF-α (25 ng/mL) treatment for 6 or 24 hours, respectively. Media was collected, concentrated 10-fold, and analyzed by Western blotting using selective anti-MMP-2 antibodies followed by incubation with appropriate secondary antibodies (HRP-conjugated; dilution 1:3000). The signal was captured using enhanced chemiluminescent reagent and a commercial imaging system (Bio-Rad). Data are expressed as mean ± SE. *P < 0.05. n = 6 to 7.
Figure 2.
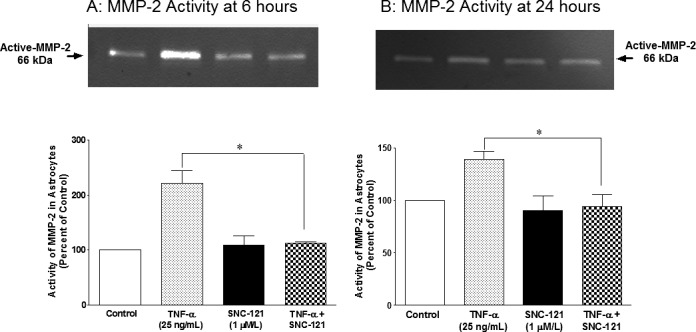
TNF-α–induced changes in MMP-2 activity in ONH astrocytes at 6 (A) and 24 (B) hours. ONH astrocytes were starved overnight in serum-free medium. Cells were then pretreated with SNC-121 (1 μmol/L) for 15 minutes followed by TNF-α (25 ng/mL) treatment for 6 or 24 hours, respectively. Media was collected, concentrated 10-fold, and analyzed by zymography as described in the Methods section. Data are expressed as mean ± SE. *P < 0.05. n = 4.
Table.
Effects of Naltrindole on TNF-α–Induced MMP-2 Secretion in the Absence or Presence of SNC-121
|
Treatment |
MMP-2 Secretion From ONH Astrocytes at 6 h |
MMP-2 Secretion From ONH Astrocytes at 24 h |
| Control | 100 ± 00 | 100 ± 00 |
| TNF-α, 25 ng/mL | 212 ± 27* | 142 ± 8* |
| SNC-121, 1 μm/L | 113 ± 7 | 109 ± 11 |
| SNC-121 + TNF-α | 120 ± 19 | 108 ± 7 |
| Naltrindole, 1 μm/L | 105 ± 12 | 106 ± 9 |
| Naltrindole + SNC-121 + TNF-α | 210 ± 47* | 152 ± 13* |
Data are mean ± SEM (where SEM are shown). ONH astrocytes were pretreated with naltrindole (1 μm/L) for 15 minutes followed by SNC-121 (1 μm/L) and TNF- α (25 ng/mL) treatment as described in the Methods section.
* Statistically significant when compared with control (P < 0.05).
Inhibitory Effects of SNC-121 on TNF-α–Induced p38 MAP Kinase Activation and NF-κB Expression in ONH Astrocytes
To identify downstream signaling targets of δ-opioid receptor–mediated inhibition in MMP-2 secretion, we chose to determine the roles of p38 MAP kinase and NF-κB in this process. The rationale for choosing p38 MAP kinase was based upon our recently published studies in which we have shown a sustained activation of p38 MAP kinase in response to glaucomatous injury.16 Moreover, TNF-α–induced p38 MAP kinase activation is inhibited by SNC-121 treatment in isolated ONH astrocytes at 6 hours.16 The activity of p38 MAP kinase was measured by its phosphorylation state. We used selective phospho-p38 MAP kinase antibodies, which recognizes the dual phosphorylation sites at threonine 180 and tyrosine 182 (Thr180/Tyr182). This antibody does not cross-react with the phosphorylated forms of either p42/44 MAP kinase or SAPK/JNK. As shown in Figure 3, TNF-α treatment resulted in a sustained phosphorylation of p38 MAP kinase up to 24 hours (226% ± 15% over control levels; P = 0.014). This increase in p38 MAP kinase activation was reduced to 150% ± 20% (P = 0.037) in the presence of SNC-121. In contrast, the protein expression level of p38 MAP kinase was not changed significantly by TNF-α treatment (Fig. 3, upper panel).
Figure 3.
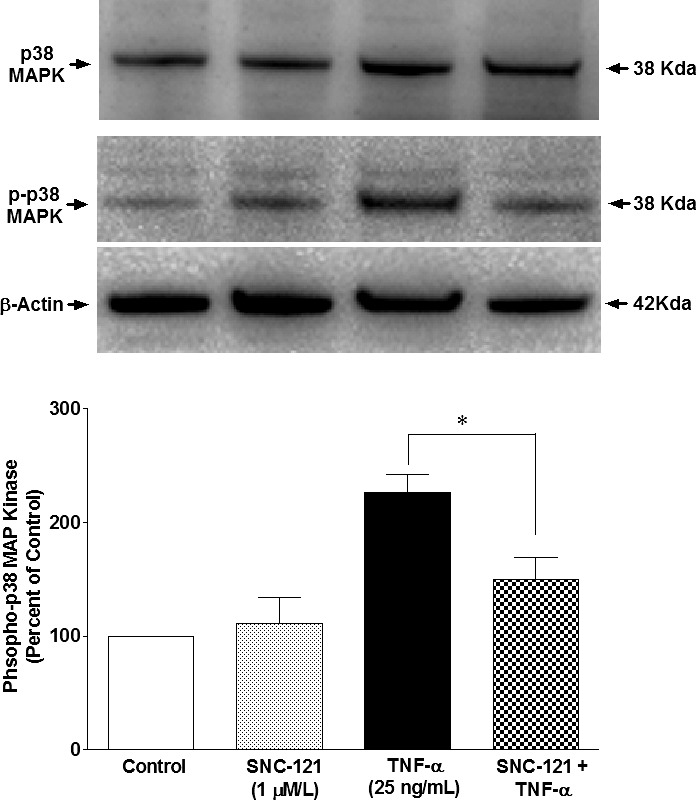
TNF-α–induced activation of p38 MAPK in ONH astrocytes. ONH astrocytes were starved overnight in serum-free medium. Cells were then pretreated with SNC-121 (1 μmol/L) for 15 minutes followed by TNF-α (25 ng/mL) treatment for 24 hours. Cell lysate (15 μg protein) was analyzed by Western blotting using selective anti-p38 MAPK or anti-phospho–p38 MAPK antibodies followed by incubation with appropriate secondary antibodies (HRP-conjugated; dilution 1:3000). The signal was captured using enhanced chemiluminescent reagent and the commercial imaging system (Bio-Rad). Data are expressed as mean ± SE. *P < 0.05. n = 4.
The NF-κB, a transcriptional factor, regulates the production of numerous proteins including matrix metalloproteinases,21,22 so we determined if SNC-121 regulated the expression levels and translocation of NF-κB to the nucleus. TNF-α treatment increased the expression of NF-κB to 179% ± 21% (P = 0.033) and 139% ± 6% (P = 0.006) at 6 and 24 hours, respectively (Figs. 4A, 4B). Pretreatment with SNC-121 reduced the expression of NF-κB to 114% ± 10% (P = 0.032) and 104% ± 3% (P = 0.001) at 6 and 24 hours, respectively (Figs. 4A, 4B). In parallel experiments, we also have observed an intense cytosolic staining of NF-κB in the resting ONH astrocytes. TNF-α treatment clearly translocated NF-κB to the nuclei, as determined by intense nuclear staining. In contrast, nuclear staining of NF-κB was reduced and dispersed to cytoplasm by SNC-121 treatment (Fig. 5). NF-κB resides mainly in the cytosol in its inactive state, where it is bound with IκBα.23,24 During activation, IκBα is phosphorylated and released from the complex and undergoes proteasome-dependent degradation. Subsequently, the freed NF-κB is translocated to the nucleus to induce the expression of target genes.23 To test if NF-κB activation is mediated via IκBα-dependent pathway, we measured the effects of SNC-121 on TNF-α–induced IκBα phosphorylation. As shown in Figure 6, TNF-α treatment increased the phosphorylation of IκBα to 344% ± 62% (Fig. 6A) and 268% ± 38% (Fig. 6B) over basal levels at 6 and 24 hours, respectively. TNF-α–induced IκBα phosphorylation was significantly reduced in the presence of SNC-121 (Figs. 6A, 6B).
Figure 4.
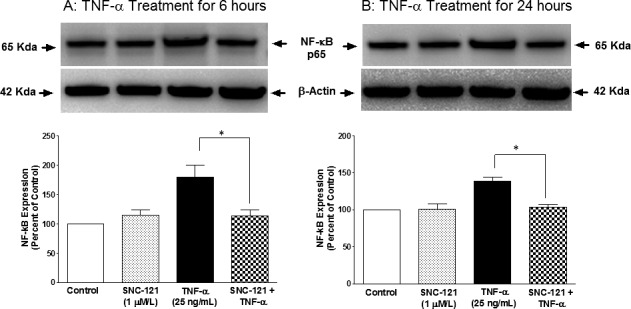
TNF-α–induced upregulation of NF-κB in ONH astrocytes. ONH astrocytes were starved overnight in serum-free medium. Cells were then pretreated with SNC-121 (1 μmol/L) for 15 minutes followed by TNF-α (25 ng/mL) treatment for 6 (A) or 24 (B) hours. Cell lysate (15 μg protein) was analyzed by Western blotting using selective anti–NF-κB antibodies followed by incubation with appropriate secondary antibodies (HRP-conjugated; dilution 1:3000). The signal was captured using enhanced chemiluminescent reagent and a commercial imaging system (Bio-Rad). Data are expressed as mean ± SE. *P < 0.05. n = 4.
Figure 5.

TNF-α–induced translocation of NF-κB in ONH astrocytes at 6 hours. ONH astrocytes were starved in serum-free medium overnight. Cells were then pretreated with SNC-121 (1 μmol/L) for 15 minutes followed by TNF-α (25 ng/mL) treatment for 6 hours. ONH astrocytes were fixed by 4% paraformaldehyde followed by immunostaining with anti–NF-κB antibodies. The signal was captured by using AlexaFluor 488 conjugated secondary antibodies. Green color indicated staining for NF-κB and nuclear staining by DAPI was indicated by blue. There was no positive staining when primary antibodies were omitted (not shown). Fluorescence microscopy; bar size is 20 microns. Data shown in this figure are a representation of at least four independent experiments.
Figure 6.
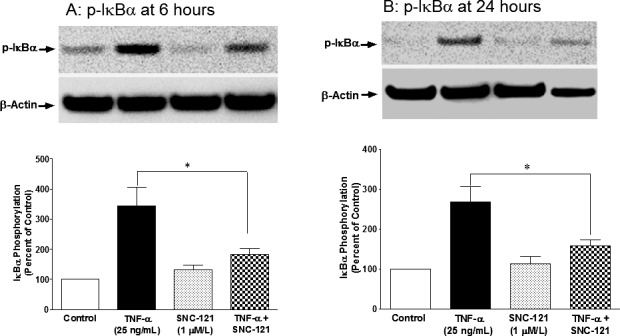
TNF-α–induced phosphorylation of IκBα in ONH astrocytes at 6 (A) and 24 (B) hours. ONH astrocytes were starved in serum-free medium overnight. Cells were then pretreated with SNC-121 (1 μmol/L) for 15 minutes followed by TNF-α (25 ng/mL) treatment for 6 or 24 hours. Cell lysate (15-μg protein) was analyzed by Western blotting using selective anti–phospho-IκBα antibodies followed by incubation with appropriate secondary antibodies (HRP-conjugated; dilution 1:3000). The signal was captured using enhanced chemiluminescent reagent and a commercial imaging system (Bio-Rad). Data are expressed as mean ± SE. *P < 0.05. n = 5 to 6.
Role of p38 MAP Kinase and NF-κB in MMP-2 Secretion From ONH Astrocytes
To determine the direct roles of p38 MAP kinase and NF-κB in TNF-α–induced MMP-2 secretion, ONH astrocytes were treated with a selective p38 MAP kinase inhibitor (SB-203580) and NF-κB inhibitor (Helenalin) prior to TNF-α treatment. TNF-α–induced MMP-2 secretion was completely inhibited (P = 0.004) when ONH astrocytes were pretreated with SB-203580 (Fig. 7A), whereas pretreatment of ONH astrocytes with Helenalin blocked TNF-α–induced MMP-2 secretion partially (78%), but significantly (P = 0.011; Fig. 7B) at 6 hours post-TNF-α treatment. To identify the sequence of events taking place in response to TNF-α treatment of ONH astrocytes, we determined if SB-203580 could inhibit the TNF-α–induced NF-κB expression. As shown in Figure 8A, TNF-α–induced NF-κB expression was fully blocked by SB-203580 treatment, suggesting that p38 MAP kinase lies upstream of NF-κB in this signaling cascade. As we expected, Helenalin had no inhibitory effect on p38 MAP kinase phosphorylation/activation (Fig. 8B) at 6 hours post–TNF-α treatment.
Figure 7.
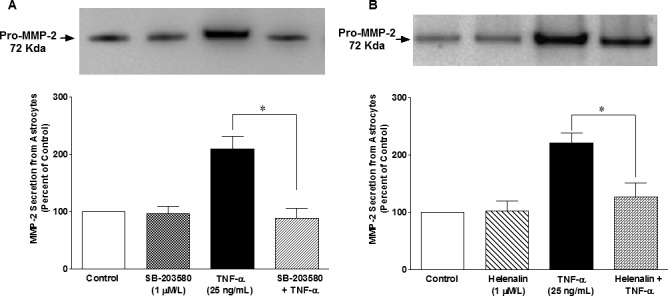
Effects of SB-203580 (A) and Helenalin (B) on TNF-α–induced MMP-2 secretion from ONH astrocytes. ONH astrocytes were starved overnight in serum-free medium. Cells were then pretreated with a p38 MAPK inhibitor, SB-203580 (1 μmol/L), or NF-κB inhibitor, Helenalin (1 μmol/L) for 15 minutes followed by TNF-α (25 ng/mL) treatment for 6 hours. Media was collected, concentrated 10-fold, and analyzed by Western blotting using selective anti-MMP-2 antibodies followed by incubation with appropriate secondary antibodies (HRP-conjugated; dilution 1:3000). The signal was captured using enhanced chemiluminescent reagent and a commercial imaging system (Bio-Rad). Data are expressed as mean ± SE. *P < 0.05. n = 7 to 8.
Figure 8.
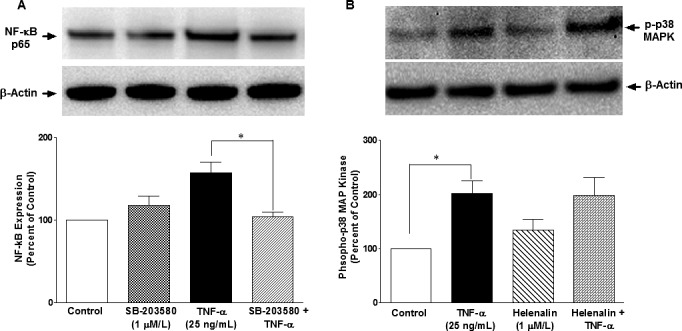
Effects of SB-203580 (A) and Helenalin (B) on TNF-α–induced upregulation of NF-κB and p38 MAPK activation, respectively, in ONH astrocytes. ONH astrocytes were starved overnight in serum-free medium. Cells were then pretreated with SB203580 (1 μmol/L) or Helenalin (1 μmol/L) for 15 minutes followed by TNF-α (25 ng/mL) treatment for 6 hours. Cell lysate (15 μg protein) was analyzed by Western blotting using selective anti–NF-κB or anti–phospho-p38 MAP kinase antibodies followed by incubation with appropriate secondary antibodies (HRP-conjugated; dilution 1:3000). The signal was captured using enhanced chemiluminescent reagent and a commercial imaging system (Bio-Rad). Data are expressed as mean ± SE. *P < 0.05. n = 7 to 8.
Discussion
ONH astrocytes interact and provide cellular support to the axons of RGCs by regulating the ionic balance of the region,25,26 providing neurotrophic support to RGCs, and maintaining the extracellular matrix.1,27,28 In normal individuals, astrocytes in the lamina cribrosa of the optic nerve head are exposed to a hydrostatic pressure gradient between the intraocular compartment and the retrolaminar tissue pressure.29 This hydrostatic pressure gradient normally fluctuates, due to the ocular pulse and the diurnal changes in IOP.30 In glaucoma, there is an elevated IOP as well as daily fluctuations. In some types of glaucoma, spikes of IOP also occur. Under such conditions, the lamina cribrosa undergoes significant deformation in response to changes in IOP, which generates additional biomechanical stress on the ONH astrocytes and other cells in the lamina cribrosa.31,32 Studies have shown that activation/reactivation of astrocytes leads to the initiation of changes in cribrosal physiology and biochemistry, alteration in matrix remodeling, and release of potential toxins (TNF-α, TGF-β1, nitric oxide, and synthesis of matrix metalloproteinases); all contributing to the characteristic glaucomatous optic neuropathy.1,9,33–35 This IOP-induced stress changes astrocytes to reactive astrocytes; however, mechanisms that regulate this process and the cellular consequences are not fully understood.
Recently, we have demonstrated the presence of functional opioid-receptor subtypes in the retina, optic nerve, and ONH astrocytes.15,36 Additionally, we have shown an upregulation of TNF-α and p38 mitogen-activated protein kinase (MAPK) in response to glaucomatous injury in a rodent glaucoma model.16 To dissect out downstream targets of TNF-α and p38 MAPK pathways, we determined the MMP secretion from ONH astrocytes. Based on our previous findings and current data, we hypothesized that the production of TNF-α in response to glaucomatous injury is an early event within the optic nerve that leads to the activation of downstream signaling molecules (e.g., p38 MAPK and NF-κB), which subsequently leads to excessive production of MMP-2. However, the signaling mechanisms that are actually involved in optic nerve destabilization and subsequent RGC death in response to glaucomatous injury remained largely unknown. To learn more about the signaling mechanisms, the main objective of this study was to determine the regulatory roles of p38 MAP kinase, NF-κB, and δ-opioid–receptor agonist (SNC-121) in the TNF-α–induced MMP-2 production from ONH astrocytes.
A significant increase in MMP-2 secretion was seen by TNF-α treatment as early as 6 hours. In contrast, we have not seen any significant changes in the secretion of MMP-1, MMP-3, and MMP-9 at 6 and 24 hours when treated with TNF-α. The data shown herein provided clues that TNF-α–induced MMP-2 secretion from ONH astrocytes was due to sustained p38 MAPK activation and not due to changes in protein expression of p38 MAPK. Additionally, we showed a significant upregulation of NF-κB expression by TNF-α treatment, which we believe was a downstream target of p38 MAPK for MMP-2 production. This was later confirmed by subsequent experiments in which TNF-α–induced NF-κB upregulation was blocked by SB-203580, a p38 MAPK inhibitor. In contrast, TNF-α–induced p38 MAPK phosphorylation/activation was not blocked by Helenalin, a NF-κB inhibitor.
We and others have shown that TNF-α activates p38 MAPK16 and upregulates NF-κB expression.37 In the eye, a pivotal role for NF-κB has been suggested in TNF-α–mediated optic nerve degeneration.37 Our data clearly demonstrated a translocation of NF-κB to the nucleus and phosphorylation of IκBα in response to TNF-α treatment, which was blocked by SNC-121 treatment. These data suggested that SNC-121 interfered in the dissociation mechanisms of IκBα and NF-κB, which subsequently attenuated the TNF-α–induced MMP-2 secretion from ONH astrocytes. Furthermore, it was important to emphasize that both p38 MAPK activation and NF-κB upregulation were significantly inhibited in the presence of SNC-121, suggesting that both p38 MAPK and NF-κB were the downstream targets of SNC-121.
This manuscript established a link between TNF-α and MMP-2 production within ONH astrocytes. Furthermore, both p38 MAPK and NF-κB regulated the MMP-2 secretion, because TNF-α–induced MMP-2 secretion was fully blocked in the presence of SB-203580 (p38 MAPK inhibitor) and Helenalin (NF-κB inhibitor). Studies have shown deleterious connective tissue remodeling of the lamina cribrosa in the early stages of glaucomatous damage.38 Studies have also shown an increase in MMP-2 immunostaining in ONH of primary open angle glaucoma and normal pressure glaucoma.9,10 Similar to our studies, He and colleagues 39 have shown that ET-1 increases the activity of MMP-2 in ONH astrocytes in an extracellular signal-reduced kinase–protein kinase C–dependent pathway. Additionally, the presence of MMP-1, MMP-2, MMP-3, MMP-9, MT1-MMP, and TIMPs1-3 is documented in human vitreous, interphotoreceptor matrix, retinal ganglion cells, and astrocytes.11,40,41 Studies have also shown that reactive astrocytes express increased levels of MT1-MMP and MMP-1 in glaucomatous human astrocytes and a glaucomatous monkey model.11
In summary, data shown herein support the idea that both p38 MAPK and NF-κB play central roles in TNF-α–induced MMP-2 secretion from ONH astrocytes, and provide novel information that both p38 MAPK and NF-κB are negatively regulated by δ-opioid–receptor activation.
Acknowledgments
The authors thank Luanna Bartholomew (Medical University of South Carolina-Storm Eye Institute) for review of the manuscript.
Supported in part by NIH/NEI Grant EY019081 (SH) and an unrestricted grant to the Medical University of South Carolina-Storm Eye Institute from Research to Prevent Blindness, New York, NY.
Disclosure: N. Akhter, None; M. Nix, None; Y. Abdul, None; S. Singh, None; S. Husain, None
References
- 1. Hernandez MR. The optic nerve head in glaucoma: role of astrocytes in tissue remodeling. Prog Retin Eye Res. 2000; 19: 297– 321 [DOI] [PubMed] [Google Scholar]
- 2. Hernandez MR, Pena JD. The optic nerve head in glaucomatous optic neuropathy. Arch Ophthalmol. 1997; 115: 389– 395 [DOI] [PubMed] [Google Scholar]
- 3. Morrison JC, Dorman-Pease ME, Dunkelberger GR, Quigley HA. Optic nerve head extracellular matrix in primary optic atrophy and experimental glaucoma. Arch Ophthalmol. 1990; 108: 1020– 1024 [DOI] [PubMed] [Google Scholar]
- 4. Toft-Hansen H, Fuchtbauer L, Owens T. Inhibition of reactive astrocytosis in established experimental autoimmune encephalomyelitis favors infiltration by myeloid cells over T cells and enhances severity of disease. Glia. 2011; 59: 166– 176 [DOI] [PubMed] [Google Scholar]
- 5. Woessner JF Jr. Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J. 1991; 5: 2145– 2154 [PubMed] [Google Scholar]
- 6. Mignatti P, Rifkin DB. Biology and biochemistry of proteinases in tumor invasion. Physiol Rev. 1993; 73: 161– 195 [DOI] [PubMed] [Google Scholar]
- 7. Matrisian LM. The matrix-degrading metalloproteinases. Bioessays. 1992; 14: 455– 463 [DOI] [PubMed] [Google Scholar]
- 8. Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003; 92: 827– 839 [DOI] [PubMed] [Google Scholar]
- 9. Yan X, Tezel G, Wax MB, Edward DP. Matrix metalloproteinases and tumor necrosis factor alpha in glaucomatous optic nerve head. Arch Ophthalmol. 2000; 118: 666– 673 [DOI] [PubMed] [Google Scholar]
- 10. Yuan L, Neufeld AH. Activated microglia in the human glaucomatous optic nerve head. J Neurosci Res. 2001; 64: 523– 532 [DOI] [PubMed] [Google Scholar]
- 11. Agapova OA, Kaufman PL, Lucarelli MJ, Gabelt BT, Hernandez MR. Differential expression of matrix metalloproteinases in monkey eyes with experimental glaucoma or optic nerve transection. Brain Res. 2003; 967: 132– 143 [DOI] [PubMed] [Google Scholar]
- 12. Agapova OA, Ricard CS, Salvador-Silva M, Hernandez MR. Expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in human optic nerve head astrocytes. Glia. 2001; 33: 205– 216 [DOI] [PubMed] [Google Scholar]
- 13. Shohami E, Ginis I, Hallenbeck JM. Dual role of tumor necrosis factor alpha in brain injury. Cytokine Growth Factor Rev. 1999; 10: 119– 130 [DOI] [PubMed] [Google Scholar]
- 14. Montgomery SL, Bowers WJ. Tumor necrosis factor-alpha and the roles it plays in homeostatic and degenerative processes within the central nervous system. J Neuroimmune Pharmacol. 2012; 7: 42– 59 [DOI] [PubMed] [Google Scholar]
- 15. Husain S, Liou GI, Crosson CE. Opioid receptor activation: suppression of ischemia/reperfusion-induced production of TNF-alpha in the retina. Invest Ophthalmol Vis Sci. 2011; 52: 2577– 2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Abdul Y, Akhter N, Husain S. Delta-opioid agonist SNC-121 protects retinal ganglion cell function in a chronic ocular hypertensive rat model. Invest Ophthalmol Visual Sci. 2013; 54: 1816– 1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Husain S, Abdul Y, Crosson CE. Preservation of retina ganglion cell function by morphine in a chronic ocular-hypertensive rat model. Invest Ophthalmol Vis Sci. 2012; 53: 4289– 4298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tezel G, Li LY, Patil RV, Wax MB. TNF-alpha and TNF-alpha receptor-1 in the retina of normal and glaucomatous eyes. Invest Ophthalmol Vis Sci. 2001; 42: 1787– 1794 [PubMed] [Google Scholar]
- 19. Yuan L, Neufeld AH. Tumor necrosis factor-alpha: a potentially neurodestructive cytokine produced by glia in the human glaucomatous optic nerve head. Glia. 2000; 32: 42– 50 [PubMed] [Google Scholar]
- 20. Roh M, Zhang Y, Murakami Y, et al. Etanercept, a widely used inhibitor of tumor necrosis factor-alpha (TNF-alpha), prevents retinal ganglion cell loss in a rat model of glaucoma. PloS One. 2012; 7: e40065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li W, Li H, Bocking AD, Challis JR. Tumor necrosis factor stimulates matrix metalloproteinase 9 secretion from cultured human chorionic trophoblast cells through TNF receptor 1 signaling to IKBKB-NFKB and MAPK1/3 pathway. Biol Reprod. 2010; 83: 481– 487 [DOI] [PubMed] [Google Scholar]
- 22. Fan Z, Yang H, Bau B, Soder S, Aigner T. Role of mitogen-activated protein kinases and NFkappaB on IL-1beta-induced effects on collagen type II, MMP-1 and 13 mRNA expression in normal articular human chondrocytes. Rheumatol Int. 2006; 26: 900– 903 [DOI] [PubMed] [Google Scholar]
- 23. Baeuerle PA, Baltimore D. NF-kappa B: ten years after. Cell. 1996; 87: 13– 20 [DOI] [PubMed] [Google Scholar]
- 24. Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005; 30: 43– 52 [DOI] [PubMed] [Google Scholar]
- 25. Johnson EC, Deppmeier LM, Wentzien SK, Hsu I, Morrison JC. Chronology of optic nerve head and retinal responses to elevated intraocular pressure. Invest Ophthalmol Vis Sci. 2000; 41: 431– 442 [PubMed] [Google Scholar]
- 26. Morgan JE. Optic nerve head structure in glaucoma: astrocytes as mediators of axonal damage. Eye. 2000; 14: 437– 444 [DOI] [PubMed] [Google Scholar]
- 27. Quigley HA. Gap junctions between optic nerve head astrocytes. Invest Ophthalmol Vis Sci. 1977; 16: 582– 585 [PubMed] [Google Scholar]
- 28. Hernandez MR, Wang N, Hanley NM, Neufeld AH. Localization of collagen types I and IV mRNAs in human optic nerve head by in situ hybridization. Invest Ophthalmol Vis Sci. 1991; 32: 2169– 2177 [PubMed] [Google Scholar]
- 29. Morgan WH, Yu DY, Alder VA, et al. The correlation between cerebrospinal fluid pressure and retrolaminar tissue pressure. Invest Ophthalmol Vis Sci. 1998; 39: 1419– 1428 [PubMed] [Google Scholar]
- 30. Zeimer RC, Wilensky JT, Gieser DK, Viana MA. Association between intraocular pressure peaks and progression of visual field loss. Ophthalmology. 1991; 98: 64– 69 [DOI] [PubMed] [Google Scholar]
- 31. Burgoyne CF, Quigley HA, Thompson HW, Vitale S, Varma R. Early changes in optic disc compliance and surface position in experimental glaucoma. Ophthalmology. 1995; 102: 1800– 1809 [DOI] [PubMed] [Google Scholar]
- 32. Zeimer RC, Ogura Y. The relation between glaucomatous damage and optic nerve head mechanical compliance. Arch Ophthalmol. 1989; 107: 1232– 1234 [DOI] [PubMed] [Google Scholar]
- 33. Tezel G, Wax MB. Increased production of tumor necrosis factor-alpha by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J Neurosci. 2000; 20: 8693– 8700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu B, Neufeld AH. Expression of nitric oxide synthase-2 (NOS-2) in reactive astrocytes of the human glaucomatous optic nerve head. Glia. 2000; 30: 178– 186 [DOI] [PubMed] [Google Scholar]
- 35. Gottschall PE, Yu X. Cytokines regulate gelatinase A and B (matrix metalloproteinase 2 and 9) activity in cultured rat astrocytes. J Neurochem. 1995; 64: 1513– 1520 [DOI] [PubMed] [Google Scholar]
- 36. Husain S, Potter DE, Crosson CE. Opioid-receptor-activation protects the retina from ischemic injury. Invest Ophthalmol Vis Sci. 2009; 50: 3853– 3859 [DOI] [PubMed] [Google Scholar]
- 37. Kitaoka Y, Kwong JM, Ross-Cisneros FN, et al. TNF-alpha-induced optic nerve degeneration and nuclear factor-kappaB p65. Invest Ophthalmol Vis Sci. 2006; 47: 1448– 1457 [DOI] [PubMed] [Google Scholar]
- 38. Roberts MD, Grau V, Grimm J, et al. Remodeling of the connective tissue microarchitecture of the lamina cribrosa in early experimental glaucoma. Invest Ophthalmol Vis Sci. 2009; 50: 681– 690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. He S, Prasanna G, Yorio T. Endothelin-1-mediated signaling in the expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in astrocytes. Invest Ophthalmol Vis Sci. 2007; 48: 3737– 3745 [DOI] [PubMed] [Google Scholar]
- 40. Brown D, Hamdi H, Bahri S, Kenney MC. Characterization of an endogenous metalloproteinase in human vitreous. Curr Eye Res. 1994; 13: 639– 647 [DOI] [PubMed] [Google Scholar]
- 41. Plantner JJ, Jiang C, Smine A. Increase in interphotoreceptor matrix gelatinase A (MMP-2) associated with age-related macular degeneration. Exp Eye Res. 1998; 67: 637– 645 [DOI] [PubMed] [Google Scholar]